

Background: Vinculin, an actin-binding protein, is essential for cytoskeletal organization.
Results: Vinculin deficiency in osteoclasts impairs osteoclast function, not differentiation, leading to increased bone mass in vivo.
Conclusion: Vinculin expression regulates osteoclast function in a talin-dependent, αvβ3 integrin-independent manner.
Significance: This is the first demonstration that vinculin regulates osteoclast function.
Keywords: Bone, Cell Biology, Cytoskeleton, Integrins, Osteoclast, Vinculin
Abstract
Osteoclastic bone resorption depends upon the cell's ability to organize its cytoskeleton. Because vinculin (VCL) is an actin-binding protein, we asked whether it participates in skeletal degradation. Thus, we mated VCLfl/fl mice with those expressing cathepsin K-Cre (CtsK-VCL) to delete the gene in mature osteoclasts or lysozyme M-Cre (LysM-VCL) to target all osteoclast lineage cells. VCL-deficient osteoclasts differentiate normally but, reflecting cytoskeletal disorganization, form small actin rings and fail to effectively resorb bone. In keeping with inhibited resorptive function, CtsK-VCL and LysM-VCL mice exhibit a doubling of bone mass. Despite cytoskeletal disorganization, the capacity of VCL−/− osteoclastic cells to normally phosphorylate c-Src in response to αvβ3 integrin ligand is intact. Thus, integrin-activated signals are unrelated to the means by which VCL organizes the osteoclast cytoskeleton. WT VCL completely rescues actin ring formation and bone resorption, as does VCLP878A, which is incapable of interacting with Arp2/3. As expected, deletion of the VCL tail domain (VCL1–880), which binds actin, does not normalize VCL−/− osteoclasts. The same is true regarding VCLI997A, which also prevents VCL/actin binding, and VCLA50I and VCL811–1066, both of which arrest talin association. Thus, VCL binding talin, but not Arp2/3, is critical for osteoclast function, and its selective inhibition retards physiological bone loss.
Introduction
The osteoclast is the unique cell responsible for removing the organic and inorganic components of bone in physiological and pathological circumstances (1). This polykaryon is the product of differentiation and fusion of monocyte-macrophage precursors. The magnitude of bone degradation by osteoclasts is dictated by their abundance and the resorptive capacity of the individual cell.
Osteoclast number is regulated by M-CSF and RANK ligand (RANKL).2 These key osteoclastogenic cytokines also stimulate the bone-degrading activity of the mature polykaryon (2, 3). In all circumstances, such enhanced resorption is the product of cytoskeletal organization to enable polarized secretion of bone-degrading molecules into an isolated extracellular microenvironment. Cytokine activation of osteoclasts, however, requires matrix-derived signals transmitted intracellularly by integrins, particularly αvβ3, which induces c-Src to stimulate a canonical, cytoskeleton-organizing complex (4). A major product of this signaling event is formation of actin rings (sealing zones) to maintain local acidification and accumulation of matrix-degrading enzymes between the cell and bone surface. This “gasket-like” structure is formed by the integration and reorganization of podosomal units (5, 6). Podosomes share many features with focal adhesions but are dynamic and actively engage in matrix remodeling (7). They consist of an F-actin core, which also contains actin-regulating molecules. The podosomal core is surrounded by a “cloud” of focal adhesion proteins (6). Bone-residing osteoclasts cluster podosomes to form numerous actin rings, each encompassing areas of bone degradation in a single cell (6, 8).
A number of podosome-associated proteins, such as the αvβ3 integrin, talin, paxillin, cortactin, and the Arp2/3 complex, are established regulators of the osteoclast cytoskeleton and the resorptive process. Although VCL is contained in podosomes (6, 8, 9), little is known about its role in osteoclasts, including its requirement for podosome assembly. In the present study, we address the role of VCL in the osteoclast's capacity to resorb bone.
VCL is an actin-binding cytoplasmic protein residing in membrane-associated adhesion complexes that tether cells to the extracellular matrix and adjacent cells (10–12). Fibroblasts isolated from VCL-null embryos contain smaller focal adhesions and exhibit reduced attachment to fibronectin, vitronectin, laminin, and collagen (13). In addition, VCL bridges extracellular matrix and F-actin in podosomes (7). Given that proteins such as talin and Arp2/3, which are central to organization of the osteoclast cytoskeleton and thus dictate bone resorption, interact with VCL, we hypothesized that it also participates in the cell's capacity to polarize and resorb bone. Although the absence of VCL does not retard osteoclast differentiation or number, VCL-deficient osteoclasts are incapable of forming normal actin rings. As such, resorption is compromised, resulting in a doubling of trabecular bone volume. Deletion of its Arp2/3 recognition sequence has no effect on the cytoskeleton-organizing properties of VCL. In contrast, the absence of F-actin and talin binding sites obviates the cytoskeleton- and resorption-promoting properties of VCL. Thus, VCL is necessary for optimal organization of the osteoclast cytoskeleton and its capacity to resorb bone in a talin-dependent manner. The maintenance of normal c-Src activation in VCL-deficient cells indicates that VCL organizes the osteoclast cytoskeleton and promotes bone resorption via a mechanism distinct from that induced by the αvβ3 integrin.
EXPERIMENTAL PROCEDURES
Mice
Generation of VCLfl/fl mice has been described (14). LysM-Cre mice were purchased from the Jackson Laboratory. CtsK-Cre mice were provided by Shigeaki Kato (University of Tokyo) (15). To obtain mice with VCL-deficient macrophages (LysM-VCL), VCLfl/fl LysMCre+/WT males were bred with VCLfl/fl LysMCre+/WT females. In all experiments, VCLfl/fl Cre− mice served as control. All mice were housed in the animal care unit of the Washington University School of Medicine and maintained according to the guidelines set by the Association for Assessment and Accreditation of Laboratory Animal Care. All animal experimentation was approved by the Animal Studies Committee of Washington University of Medicine.
Reagents
Glutathione S-transferase (GST)-RANKL was expressed in our laboratory as described previously (16). The sources of antibodies are follows: mouse anti-VCL and anti-β-actin antibodies were from Sigma; mouse anti-cathepsin K antibody was from Millipore (Temecula, CA); antiphosphotyrosine mAb 4G10 was from Upstate (Charlottesville, VA); mAb 327, directed against the c-Src protein, was a gift of Andrey Shaw (Department of Pathology, Washington University School of Medicine); mouse anti-NFATc1 antibody was from Santa Cruz Biotechnology, Inc.; rabbit anti-β3-integrin antibody was from ThermoScientific; mouse anti-hemagglutinin (HA) antibody was from Covance; rabbit anti-Src p-Y416 antibody was from Cell Signaling (Beverly, MA). Alexa-Flour-546-phalloidin was from Invitrogen. All other chemicals were obtained from Sigma.
Macrophage Isolation and Osteoclast Culture
Isolated bone marrow macrophages (BMMs) and splenocytes were differentiated into mature, multinucleated osteoclasts as described (17). Following 3–6 days of culture in 1:50 CMG (conditioned medium supernatant containing recombinant M-CSF) (18) and GST-RANKL (100 ng/ml), cells were stained for tartrate-resistant acid phosphatase (TRAP) activity (kit 387-A, Sigma).
For actin ring and pit resorptive assays, cells were cultured on bovine bone slices with M-CSF and RANKL for 5 or 6 days. After cells were fixed in 4% paraformaldehyde, permeabilized in 0.1% Triton X-100, and rinsed in PBS, actin rings were visualized using Alexa-Fluor-546 phalloidin (Invitrogen). To measure resorption, medium from osteoclasts cultured on bone slices was collected and analyzed using the CrossLaps for Culture enzyme-linked immunosorbent assay (ELISA) kit (Nordic Bioscience Diagnosis A/S). To assess resorptive pit formation, cells were removed from bone slices with mechanical agitation. Bone slices were incubated with peroxidase-conjugated wheat germ agglutinin (Sigma) for 1 h and stained with 3,3′-diaminobenzidine (Sigma).
Plasmids and Retroviral Transduction
Full-length mouse VCL1–1066 construct was kindly provided from Dr. Wolfgang H. Goldmann (Friedrich-Alexander-University of Erlangen-Nuremberg, Germany) (19) and subcloned into pMX-IRES-BSR by introducing the EcoRI 5′ and NotI 3′ restriction sites with a hemagglutinin (HA) tag at the 3′-end. All mutated constructs were generated using standard molecular biology techniques. 10 μg of the pMX retroviral vector was transfected transiently into the Plat-E packaging cell line (20) using calcium phosphate precipitation (21). Virus was collected 48 h after transfection. BMMs or splenocytes were infected with virus for 24 h in the presence of 100 ng/ml M-CSF and 4 μg/ml Polybrene (Sigma). Cells were selected in the presence of M-CSF and 1 μg/ml blasticidin (Calbiochem) for 3 days prior to use as osteoclast precursors.
Dynamic Imaging of Osteoclasts on Bone
WT or LysM-VCL macrophages, transduced with GFP-actin using standard culture conditions (37 °C and 5% CO2, 95% air atmosphere), were maintained for 10 days, with RANKL and M-CSF, in a bone powder-coated Bioptechs (non-liquid-perfused) Delta T culture system, consisting of a heated, indium-tin-oxide-coated glass dish attached to a calibrated Bioptechs microperfusion peristaltic pump. All cultures were observed with the ×20 objective (numerical aperture 0.4) of an inverted automated wide field epifluorescence differential interference contrast microscope (Leica DMIRE2, Leica Microsystems (Wetzlar, Germany)). An objective lens heater was used to improve temperature homogeneity. Images (608 × 512 pixels spatial and 12-bit intensity resolution) were recorded with a cooled Retiga 1300 camera (Qimaging, Burnaby, Canada) every 2 min in 2 × 2 binned acquisition mode, using 100–300-ms exposures. Dynamic images were composed using ImageJ.
Western Blotting
Cells were washed with ice-cold PBS and lysed in radioimmune precipitation assay buffer containing 20 mm Tris-HCl, pH 7.5, 150 mm NaCl, 1 mm EDTA, 1 mm EGTA, 1% Triton X-100, 2.5 mm sodium pyrophosphate, 1 mm β-glycerophosphate, 1 mm sodium orthovanadate, 1 mm sodium fluoride, and 1× protease inhibitor mixture (Roche Applied Science). After a 15-min incubation on ice, lysates were cleared of debris by a 15-min centrifugation at 21,100 × g. 30–40 μg of lysates were subjected to 8% SDS-PAGE and transferred onto polyvinylidene difluoride membranes. Filters were blocked in 0.1% casein in PBS for 1 h and incubated with primary antibodies at 4 °C overnight, followed by probing with fluorescence-labeled secondary antibodies (Jackson Laboratory). Proteins were detected with the Odyssey infrared imaging system (LI-COR Biosciences).
Microcomputed Tomography (μCT) Analysis
The trabecular volume in the distal femoral metaphysis was measured using a Scanco μCT40 scanner (Scanco Medical AG, Bassersdorf, Switzerland). A threshold of 184 was used for evaluation of all scans, and 50 slices were analyzed, starting with the first slice in which condyles and primary spongiosa were no longer visible.
Histology and Histomorphometry
Following sacrificing by CO2 inhalation, the tibiae were isolated and fixed in 10% phosphate-buffered formalin (Fisher), followed by decalcification in 14% EDTA, pH 7.2, for 10 days, paraffin embedding, and TRAP staining. Osteoclastic parameters were measured and analyzed using BioQuant OsteoII (BioQuant Image Analysis Corp., Nashville, TN) in blind fashion. Pit areas were measured with ImageJ.
Statistical Analysis
Statistical significances were determined using Student's t test. Data are represented as mean ± S.D.
RESULTS
VCL Expression Increases with Osteoclastogenesis
To determine the impact of osteoclast differentiation on VCL expression, we cultured WT bone marrow macrophages (BMMs) with M-CSF and RANKL. Immunoblot of cell lysates reveals that VCL increases with osteoclastogenesis (Fig. 1A). Consistent with previous reports (6, 8), VCL localizes around actin belts, which are circular structures formed in osteoclasts resident on non-mineralized matrix, as double rings on glass (Fig. 1B). These data suggest that VCL may regulate osteoclast differentiation and/or function.
FIGURE 1.

VCL is increased during osteoclastogenesis. A, WT BMMs were cultured in M-CSF and RANKL with time. VCL and the osteoclast differentiation markers, β3 integrin subunit, NFATc1, c-Src, and CtsK, were determined by immunoblot. β-Actin served as a loading control. B, WT BMMs were cultured with RANKL and M-CSF on glass for 5 days. The cells were stained for VCL (green) and F-actin (red). Scale bar, 25 μm.
Deletion of VCL in Osteoclast Lineage Cells Does Not Affect Osteoclast Differentiation
Global deletion of VCL causes embryonic lethality (13). To circumvent this problem, we first crossed VCLfl/fl mice with those expressing lysosome M Cre (LysM-Cre), which targets all myeloid cells, including those of the osteoclast lineage (22). (Mice and osteoclasts with conditional deletion of VCL by LysM-Cre are designated LysM-VCL.) Immunoblot confirms LysM-Cre-mediated deletion of VCL throughout osteoclastogenesis (Fig. 2A).
FIGURE 2.
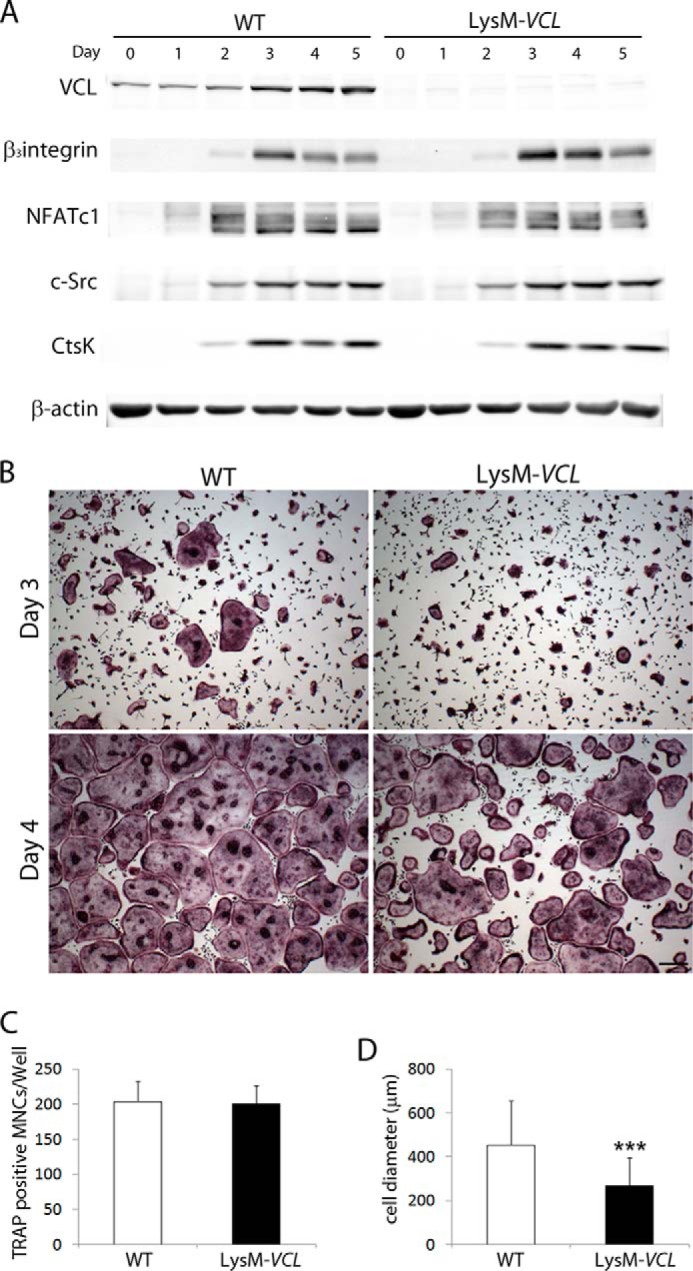
LysM-VCL osteoclasts differentiate into osteoclasts normally. A, WT (Cre-) and LysM-VCL BMMs were cultured in M-CSF and RANKL with time. Osteoclast differentiation markers were determined by immunoblot. β-Actin served as a loading control. B and C, WT and LysM-VCL BMMs were cultured in M-CSF and RANKL for 3 or 4 days. B, the cells were stained for TRAP activity. Scale bars, 200 μm. C, quantitation of TRAP-expressing multinucleated cells (MNCs) (more than 3 nuclei/cell) generated from WT and LysM-VCL BMMs. D, WT and LysM-VCL BMMs were cultured in M-CSF and RANKL for 4 days. The diameter of MNCs was measured. ***, p < 0.001. Error bars, S.D.
To assess the role of VCL in the bone-resorptive process, we cultured WT and LysM-VCL BMMs with M-CSF and RANKL. After a 3-day exposure to cytokines, WT BMMs differentiated into characteristic, multinucleated, TRAP-expressing osteoclasts (Fig. 2B). Although all cells in LysM-VCL cultures are also TRAP-expressing, few are multinucleated. By day 4, LysM-VCL cells are multinuclear and spread. They are also equal in number to WT (Fig. 2C). The mutant osteoclasts, however, are only one-half the size of their WT counterparts (Fig. 2D). Confirming that the phenotype of LysM-VCL osteoclasts does not reflect arrested differentiation, temporal expression of osteoclastogenic proteins mirrors WT (Fig. 2A).
VCL-deficient Osteoclasts Are Dysfunctional
The appearance of LysM-VCL osteoclasts is suggestive of cytoskeletal disorganization. To determine if this is so, we generated TRAP-expressing polykaryons on bone slices. After 6 days in M-CSF and RANKL, the actin cytoskeleton was visualized with fluorescein isothiocyanate-phalloidin. Although both WT and LysM-VCL osteoclasts have abundant actin rings, those lacking VCL are much smaller (Fig. 3A and supplemental Movie 1). These abnormalities of cytoskeletal organization translate into function as the resorptive capacity of LysM-VCL osteoclasts is markedly reduced, as measured by medium content of the bone-derived collagen degradation product C-terminal telopeptide (CTx) and pit formation (Fig. 3, B, C, and D).
FIGURE 3.
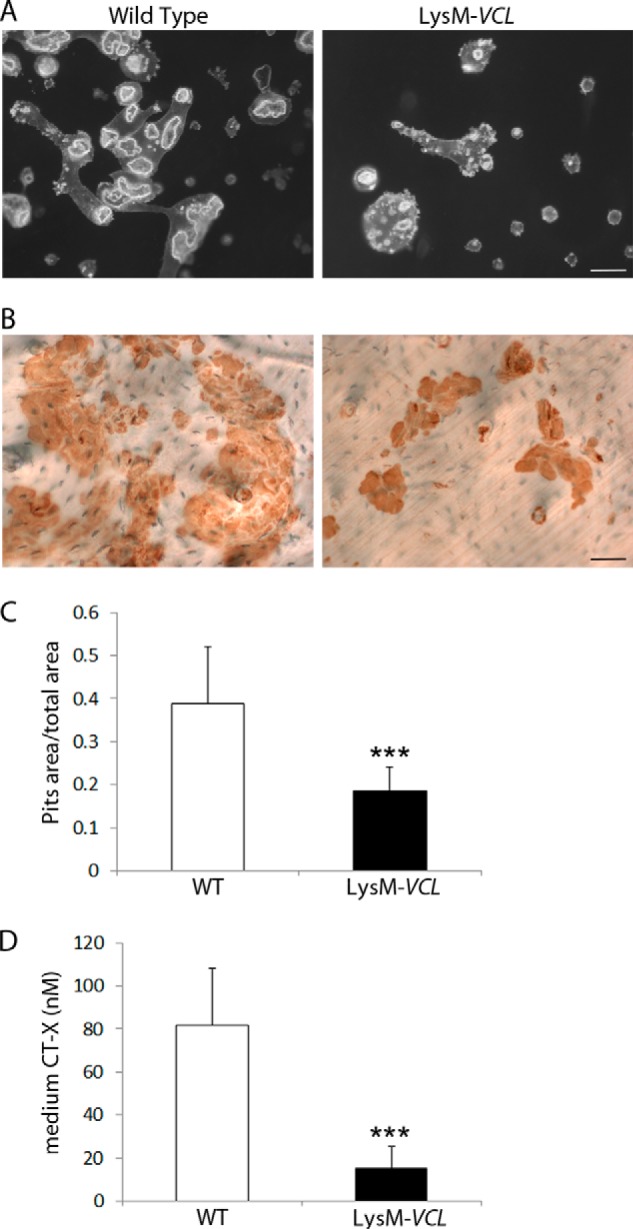
LysM-VCL osteoclasts fail to effectively resorb bone. Equal numbers of WT and LysM-VCL BMMs were cultured on bone slices for 6 days in the presence of M-CSF and RANKL. A, the actin cytoskeleton was visualized by Alexa-Fluor-546-phallodin staining. Scale bar, 50 μm. B, the cells were removed, and the resorption pits were visualized by wheat germ agglutinin lectin staining. Scale bar, 50 μm. C, histomorphometric analysis of pit area/total area. D, culture medium was analyzed for CTx content, a marker of global bone resorption. ***, p < 0.001. Error bars, S.D.
Because LysM-Cre targets all myeloid lineage cells, including immature osteoclast precursors, we determined the impact of deleting VCL exclusively in mature polykaryons. To this end, we crossed VCLfl/fl mice with those expressing cathepsin K Cre (CtsK-Cre) (15). (Mice and osteoclasts with conditional deletion of VCL by CtsK-Cre are referred to as CtsK-VCL.) As expected, CtsK-Cre does not appreciably alter VCL expression prior to 3-day exposure to RANKL and M-CSF, after which it prevents the increase extant in WT cells (Fig. 4A). Like LysM-VCL cells, osteoclast differentiation markers are unimpaired in those expressing CtsK-Cre. Further mirroring LysM-Cre-mediated deletion, day 3 CtsK-VCL cells are TRAP-expressing but show little evidence of multinucleation (Fig. 4B). Similarly, day 4 CtsK-VCL osteoclasts are spread and mirror WT in number (Fig. 4, B and C). The conditionally deleted osteoclasts, however, are reduced in size, have small actin rings, and ineffectively resorb bone (Fig. 4, D–H).
FIGURE 4.

CtsK-VCL osteoclasts fail to effectively resorb bone. A, WT (Cre-) and CtsK-VCL BMMs were cultured in M-CSF and RANKL for various times. Osteoclast differentiation markers were determined by immunoblot. β-Actin served as a loading control. B, WT and CtsK-VCL BMMs were cultured in M-CSF and RANKL for 3 or 4 days. The cells were stained for TRAP activity. Scale bars, 200 μm. C, quantitation of TRAP-expressing MNCs (more than 3 nuclei/cell) generated from WT and CtsK-VCL BMMs. D, WT and CtsK-VCL BMMs were cultured in M-CSF and RANKL for 4 days. The diameter of MNCs was measured. E–H, WT and CtsK-VCL BMMs were cultured on bone slices with M-CSF and RANKL. E, after 6 days, the cells were stained with Alexa-Fluor-546-phallodin to visualize the actin cytoskeleton. F, following removal of the cells, resorption pits were visualized by wheat germ agglutinin-lectin staining. G, histomorphometric analysis of pit area/total area. H, culture medium was analyzed for CTx content. **, p < 0.01; ***, p < 0.001. Scale bar, 50 μm. Error bars, S.D.
Absence of VCL in Osteoclast Lineage Cells Increases Bone Mass
To determine the physiological relevance of our in vitro observations, we assessed skeletal mass by μCT. Reflecting the impaired resorptive capacity of their osteoclasts, both LysM-VCL and CtsK-VCL mice exhibit a doubling of trabecular bone mass (Figs. 5 (A and B) and 6 (A and B)). This increased bone mass of is confirmed by histomorphometry, which also validates our in vitro data indicating that deletion of VCL in myeloid lineage cells or mature polykaryons does not alter osteoclast number (Figs. 5 (C and D) and 6 (C and D)).
FIGURE 5.

LysM-VCL mice are osteopetrotic. A, μCT image of distal femur of 9-week-old WT and LysM-VCL mice. B, μCT determination of BV/TV, trabecular number, trabecular thickness, and trabecular separation of distal femoral metaphysis of WT and LysM-VCL mice. C, TRAP-stained histological sections of proximal tibia of WT and LysM-VCL mice. Black boxes indicate the region of magnification shown in the bottom panels. D, BV/TV, total number of osteoclasts normalized to bone surface (Oc.N/BS) and the percentage of bone surface to which osteoclasts are juxtaposed (Oc.S/BS) in C were histomorphometrically determined. *, p < 0.05; **, p < 0.01. Scale bars, 1 mm (A), 500 μm (C, top panels), and 50 μm (C, bottom panels). Error bars, S.D.
FIGURE 6.

CtsK-VCL mice are osteopetrotic. A, μCT image of distal femur of WT and CtsK-VCL mice at 9 weeks of age. B, μCT determination of BV/TV, trabecular number, trabecular thickness, and trabecular separation of distal femoral metaphysis of WT and CtsK-VCL mice at 9 weeks. C, TRAP-stained histological sections of proximal tibia of WT and CtsK-VCL mice. Black boxes indicate the region of magnification shown in the bottom panels. D, BV/TV, total number of osteoclasts normalized to bone surface (Oc.N/BS), and the percentage of bone surface to which osteoclasts are juxtaposed (Oc.S/BS) in C were histomorphometrically determined. *, p < 0.05; **, p < 0.01. Scale bars, 1 mm (A), 500 μm (C, top panels), 50 μm (C, bottom panels). Error bars, S.D.
αvβ3 Integrin Activation Is Unimpaired in VCL-deficient Osteoclasts
Bone resorption is initiated by αvβ3 integrin recognition of matrix-residing ligand, which stimulates a canonical, cytoskeleton-organizing signaling complex. c-Src is the most proximal established member of this complex. This tyrosine kinase constitutively associates with the β3 cytoplasmic domain and is activated upon integrin ligation (4, 23). To determine if the cytoskeletal abnormalities of VCL-deficient osteoclasts reflect failure of αvβ3 signaling, we cultured WT and LysM-VCL splenocytes in RANKL and M-CSF. After 3 days, cytokine-starved, prefusion osteoclasts were lifted and replated on the αvβ3 ligand, vitronectin, or maintained in suspension. Integrin-induced c-Src activity, as manifest by phosphorylation of total protein and c-SrcY416, in LysM-VCL osteoclasts is indistinguishable from WT (Fig. 7). Thus, the abnormal cytoskeleton and consequent arrested bone resorption of VCL-deficient osteoclasts does not reflect failure to activate αvβ3.
FIGURE 7.

VCL deletion in osteoclast lineage cells does not alter c-Src activation. WT and LysM-VCL BMMs were cultured with M-CSF and RANKL for 3 days to generate prefusion osteoclasts. The cells were lifted, cytokine/serum-starved, and maintained in suspension (S) or plated on vitronectin (A) for 30 min. Lysates were immunoblotted for total phosphotyrosine (p-Y) and activated c-Src (p-Src). Actin served as a loading control.
VCL-mediated Organization of the Osteoclast Cytoskeleton Requires Interaction with Talin
The absence of talin compromises the osteoclast cytoskeleton and prompts severe osteopetrosis (24). In other cells, VCL interacts with talin and links it to actin. To explore this issue in osteoclasts, we transduced a series of HA-tagged VCL mutants into LysM-VCL BMMs and exposed them to RANKL and M-CSF (Fig. 8A). The initially transduced constructs consisted of WT VCL, a tail-deleted construct (VCL1–880) incapable of binding actin, and a head-deleted construct (VCL811–1066), which cannot bind talin. Because talin activates αvβ3, which in turn stimulates Rac to organize the osteoclast cytoskeleton via Arp2/3, we included VCLP878A, which does not recognize the Arp2/3 complex. Expression of the various forms of VCL mirrors WT, and there is no evidence of arrest of differentiation as manifest by quantity of osteoclastogenic proteins (Figs. 8, B and C). Confirming that they do not exert a dominant/negative effect, these constructs do not impair spreading or actin ring formation by WT osteoclasts (Fig. 9). As expected, WT VCL completely normalizes the appearance of LysM-VCL osteoclasts as well as actin ring size and bone-resorptive capacity (Fig. 8, D–F). The same holds true regarding VCLP878A, indicating that its interaction with the Arp2/3 complex does not mediate the effects of VCL on the osteoclast cytoskeleton. In contrast, deletion of the talin-recognizing head (VCL811–1066) or the actin-binding tail (VCL1–880) prevents rescue of the VCL-deficient osteoclast cytoskeleton and function. Confirming the importance of talin in VCL-mediated cytoskeletal organization, VCLA50I, which also abrogates talin association (25) fails to rescue VCL-deficient cells. Similarly, VCLI997A, which is incapable of actin binding (26), does not compensate for the absence of WT VCL. This observation establishes that the same lack of effect by tail-deleted VCL1–880 does not reflect conformational disruption (Fig. 10).
FIGURE 8.

Structure/function analysis of osteoclast VCL. A, WT and mutated VCL constructs. B and C, HA-tagged WT and mutated VCL constructs were retrovirally transduced into LysM-VCL splenic macrophages. The cells were maintained in M-CSF and RANKL for 4 days. B, expression of each transductant and osteoclast differentiation marker was determined by immunoblot. Empty vector (vector) transduced into WT and LysM-VCL splenic macrophages served as a control. C, expression of HA-tagged VCL811–1066 was determined by immunoblot with anti-HA antibody. D, left panels, transduced osteoclasts, generated on plastic for 3 days, stained for TRAP activity. Middle panels, transduced splenic macrophages cultured with M-CSF and RANKL on bone slices. After 5 days, the cells were stained with Alexa-Fluor-546-phallodin to visualize the actin cytoskeleton. Right panels, following removal of the transduced osteoclasts, resorption pits were visualized by wheat germ agglutinin-lectin staining. E and F, histomorphometric analysis of actin ring area/field (E) and pit area/total area (F). ****, p < 0.0001; ***, p < 0.001; **, p < 0.01 compared with LysM VCL vector. Scale bars, 50 μm. Error bars, S.D.
FIGURE 9.
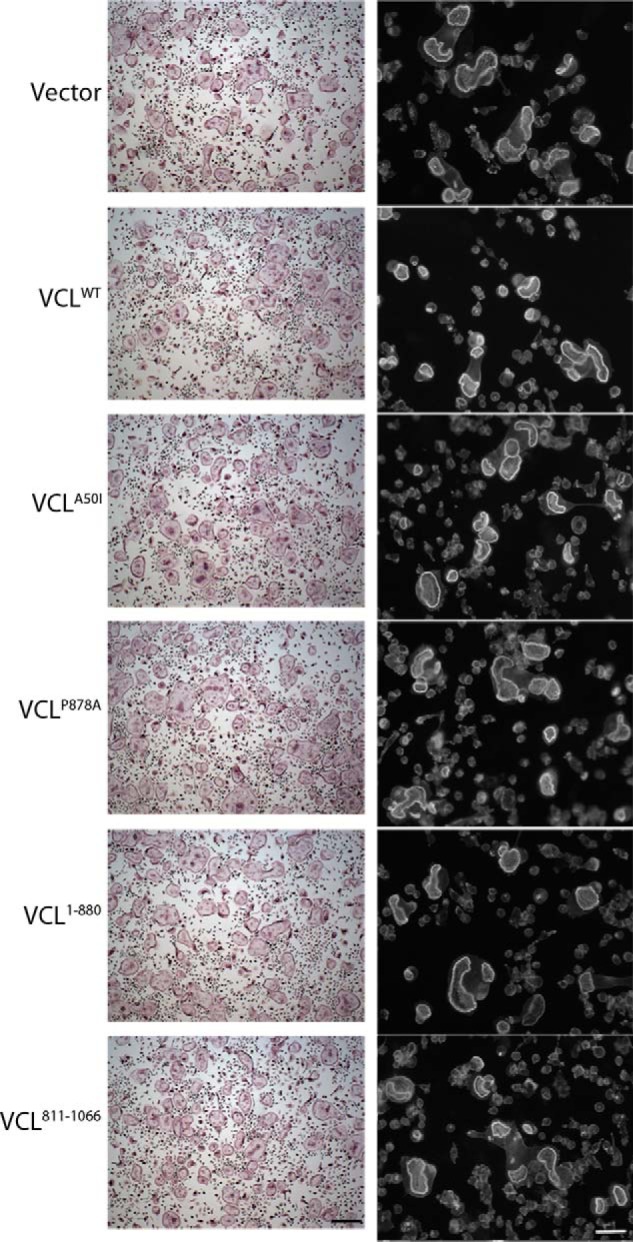
VCL mutants do not exert dominant negative effects on the osteoclast cytoskeleton. WT and mutated VCL constructs were retrovirally transduced into WT BMMs. Empty vector (Vector) served as a control. Left panels, osteoclasts generated on plastic were stained for TRAP activity. Right panels, the actin cytoskeleton of osteoclasts generated on bone slices was visualized by Alexa-Fluor-546-phallodin staining. Scale bar, 50 μm.
FIGURE 10.

Failure to bind F-actin obviates VCL function in osteoclasts. A, HA-tagged WT VCL or VCLI1997A was retrovirally transduced into LysM-VCL splenic macrophages. The cells were maintained in M-CSF and RANKL for 5 days. Expression of each transductant was determined by immunoblot. Empty vector served as a control. B, left panels, transduced osteoclasts, generated on plastic for 3 days, stained for TRAP activity. Middle panels, transduced splenic macrophages cultured with M-CSF and RANKL on bone slices. After 5 days, the cells were stained with Alexa-Fluor-546-phallodin to visualize the actin cytoskeleton. Right panels, following removal of the transduced osteoclasts, resorption pits were visualized by wheat germ agglutinin-lectin staining. C and D, histomorphometric analysis of actin ring area/field (C) and pit area/total area (D). ****, p < 0.0001; **, p < 0.01 compared with LysM VCL vector. Scale bars, 50 μm. Error bars, S.D.
DISCUSSION
The unique capacity of osteoclasts to resorb bone involves mobilization of its mineral phase by the creation of an acidic microenvironment and subsequent enzymatic degradation of its organic matrix (27). These events require the cell's polarization, wherein vesicles rich in bone-degrading molecules transit to the bone-apposed plasma membrane to which they fuse. Vesicle/membrane fusion eventuates in a unique plasmalemmal complex, the ruffled border, which delivers molecules, such as cathepsin K and HCl, into the resorptive microenvironment. This microenvironment is isolated from the general extracellular space by actin rings or sealing zones that encompass the ruffled border. Actin rings are the products of podosomal aggregation induced by adhesion to a mineralized surface. The absence of or structural abnormalities of actin rings are indicative of compromised skeletal degradation. Thus, organization of its cytoskeleton is a fundamental component of the osteoclast's bone-resorptive capacity.
VCL is a 1066-amino acid, ubiquitously expressed, actin-binding protein localized on the cytoplasmic face of integrin-containing podosomes in osteoclasts (10). It consists of a 90-kDa N-terminal globular head domain (VCL1–811), a flexible hinge region (VCL811–881), and a 27-kDa C-terminal tail domain (VCL811–1066), each participating in protein/protein interactions that dictate function of individual cells. In fibroblasts, VCL is critical for integrin-mediated attachment to matrix, and thus migration of these cells is enhanced in the protein's absence (28). To directly determine the role of VCL in osteoclast formation and function, we targeted the VCLfl/fl gene by CtsK-Cre, which is expressed in mature osteoclasts, and LysM-Cre, which is active throughout osteoclastogenesis. The fact that VCL expression is a late event in osteoclast differentiation suggested that the osteoclast phenotype of both strains of conditionally deleted mice would be similar, and such proved to be the case. Further supporting the conclusion that VCL regulates osteoclast function and not formation, CtsK-VCL and LysM-VCL mice are indistinguishable because their trabecular bone mass is twice that of WT, but the number of osteoclasts is unaffected.
Like VCL, β integrins, particularly αvβ3, do not regulate osteoclast differentiation but contribute to the cell's ability to optimally resorb bone (29). Also as in VCL deficiency, the compromised resorptive capacity of osteoclasts, lacking αvβ3, reflects cytoskeletal disorganization and, perhaps, the inability of the cell to achieve normal size. Although both αvβ3- and VCL-deficient osteoclasts form actin rings, these structures are abnormal (30). This observation is in keeping with VCL being a component of podosomes that aggregate to generate actin rings in osteoclasts residing on bone. Despite these similarities, αvβ3 probably does not regulate the impact of VCL on the osteoclast.
αvβ3 induces a cytoskeleton-organizing signaling complex, of which c-Src is the most proximal known constituent (4). Upon ligation by extracellular proteins, αvβ3 activates c-Src, which is constitutively associated with the integrin. Integrin-mediated phosphorylation of c-SrcY416 stimulates Syk, Dap12, Slp-76, Vav3, and, ultimately, Rac, which organizes the cytoskeleton (31). The absence of any component of this complex yields a common phenotype wherein the osteoclast cytoskeleton is disrupted, thereby compromising the cell's resorptive capacity. Whereas interruption of the integrin-initiated signals produces osteoclasts that fail to spread, such is not the case regarding VCL deficiency. The difference in appearance between cells lacking integrin-activated molecules and those without VCL suggests that the two processes differ. This posture is buttressed by maintenance of c-Src activation in VCL-deficient osteoclasts, which is disrupted in the absence of αvβ3. Thus, even in face of normal integrin-activated signals, VCL remains critical for optimal osteoclast cytoskeletal organization.
Paxillin is another actin-associated protein necessary for osteoclast cytoskeletal organization and function. Although the phenotypes of paxillin- and VCL-deficient osteoclasts differ, in that the former are larger and the latter smaller than WT, both spread (32). Thus, like VCL, paxillin is probably not regulated by the canonical αvβ3-stimulated signaling complex and, in fact, exerts its effects via delivery of myosin IIA to the cytoskeleton.
The Arp2/3 complex is an important regulator of actin polymerization (33) and a component of actin rings (34). In addition, knockdown of Arp2 in RAW 264.7 osteoclast-like cells disrupts these “gasket-like” structures (34). Therefore, actin ring formation requires Arp2/3. On the other hand, VCLP878A, which no longer binds Arp2/3, rescues the osteoclast phenotype of VCL-deficient cells as efficiently as WT. Because the Arp2/3 complex is the means by which αvβ3-activated Rac organizes the osteoclast cytoskeleton, this observation also indicates that VCL is not a component of the canonical integrin-activated signaling pathway (31).
In contrast to mutated Arp2/3 binding sites, VCL constructs that no longer recognize talin fail to rescue VCL−/− osteoclasts. Talin is a multifunctional protein whose head domain binds β integrins to promote inside-out enhancement of their ligand affinity. In fact, talin association is an essential component of integrin activation, thus enabling downstream signaling. As such, the absence of talin promotes severe osteopetrosis as multiple β integrins fail to signal when liganded (24). The fact that ligand-induced αvβ3 signaling is intact in VCL-deficient osteoclasts, as evidenced by total protein phosphorylation and, particularly, activation of c-Src, further establishes that VCL is not required for talin-mediated integrin activation.
Although binding of the talin head to β integrins is a final and essential step in their activation (34), talin's tail enhances actin recognition in a VCL-dependent manner. Thus, VCL links the integrin-talin complex to the actin cytoskeleton (10). Because the cytoskeleton is disorganized in VCL-deficient osteoclasts, it is likely that this adaptor function of talin is disturbed. If this model is correct, it would probably explain the phenotypic differences between VCL- and talin-deficient osteoclasts. Whereas osteoclasts lacking talin are small and “crenated” and thus resemble those in which the αvβ3-activated pathway is impaired, VCL-deficient cells spread but are smaller than their WT counterparts. These differences translate into conditional deletion of talin in osteoclastic cells, producing a more severe osteopetrotic phenotype than the absence of VCL. Thus, VCL or talin deficiency may impair integrin linkage to actin. Alternatively, the absence of talin, but not VCL, disrupts activation of integrin-induced cytoskeletal organizing signals.
In a process depending on myosin contractility, VCL contributes to adhesion strength to extracellular matrix as well as traction force (26, 35, 36). These biomechanical events require the VCL head and tail domains, both of which we find regulate osteoclast function (35, 36). These data suggest that the mechanical interaction of osteoclasts with bone is mediated by VCL, a hypothesis that remains to be proven.
Supplementary Material
This work was supported, in whole or in part, by National Institutes of Health Grants R01 AR032788, R01 AR057037, R01 AR046523, and P30 AR057235.

This article contains supplemental Movie 1.
- RANKL
- RANK ligand
- VCL
- vinculin
- TRAP
- tartrate-resistant acid phosphatase
- μCT
- microcomputed tomography
- CTx
- C-terminal telopeptide
- CtsK
- cathepsin K-Cre
- BV/TV
- bone volumetric density.
REFERENCES
- 1. Novack D. V., Teitelbaum S. L. (2008) The osteoclast: friend or foe? Annu. Rev. Pathol. 3, 457–484 [DOI] [PubMed] [Google Scholar]
- 2. Lacey D. L., Timms E., Tan H. L., Kelley M. J., Dunstan C. R., Burgess T., Elliott R., Colombero A., Elliott G., Scully S., Hsu H., Sullivan J., Hawkins N., Davy E., Capparelli C., Eli A., Qian Y. X., Kaufman S., Sarosi I., Shalhoub V., Senaldi G., Guo J., Delaney J., Boyle W. J. (1998) Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell 93, 165–176 [DOI] [PubMed] [Google Scholar]
- 3. Zou W., Reeve J. L., Liu Y., Teitelbaum S. L., Ross F. P. (2008) DAP12 couples c-Fms activation to the osteoclast cytoskeleton by recruitment of Syk. Mol. Cell 31, 422–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zou W., Kitaura H., Reeve J., Long F., Tybulewicz V. L., Shattil S. J., Ginsberg M. H., Ross F. P., Teitelbaum S. L. (2007) Syk, c-Src, the αvβ3 integrin, and ITAM immunoreceptors, in concert, regulate osteoclastic bone resorption. J. Cell Biol. 176, 877–888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Luxenburg C., Geblinger D., Klein E., Anderson K., Hanein D., Geiger B., Addadi L. (2007) The architecture of the adhesive apparatus of cultured osteoclasts: from podosome formation to sealing zone assembly. PLoS One 2, e179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Saltel F., Chabadel A., Bonnelye E., Jurdic P. (2008) Actin cytoskeletal organization in osteoclasts: a model to decipher transmigration and matrix degradation. Eur. J. Cell Biol. 87, 459–468 [DOI] [PubMed] [Google Scholar]
- 7. Linder S., Aepfelbacher M. (2003) Podosomes: adhesion hot-spots of invasive cells. Trends Cell Biol. 13, 376–385 [DOI] [PubMed] [Google Scholar]
- 8. Saltel F., Destaing O., Bard F., Eichert D., Jurdic P. (2004) Apatite-mediated actin dynamics in resorbing osteoclasts. Mol. Biol. Cell 15, 5231–5241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ziegler W. H., Liddington R. C., Critchley D. R. (2006) The structure and regulation of vinculin. Trends Cell Biol. 16, 453–460 [DOI] [PubMed] [Google Scholar]
- 10. Carisey A., Ballestrem C. (2011) Vinculin, an adaptor protein in control of cell adhesion signaling. Eur. J. Cell Biol. 90, 157–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Peng X., Nelson E. S., Maiers J. L., DeMali K. A. (2011) New insights into vinculin function and regulation. Int. Rev. Cell Mol. Biol. 287, 191–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Xu W., Baribault H., Adamson E. D. (1998) Vinculin knockout results in heart and brain defects during embryonic development. Development 125, 327–337 [DOI] [PubMed] [Google Scholar]
- 13. Zemljic-Harpf A. E., Miller J. C., Henderson S. A., Wright A. T., Manso A. M., Elsherif L., Dalton N. D., Thor A. K., Perkins G. A., McCulloch A. D., Ross R. S. (2007) Cardiac-myocyte-specific excision of the vinculin gene disrupts cellular junctions, causing sudden death or dilated cardiomyopathy. Mol. Cell Biol. 27, 7522–7537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nakamura T., Imai Y., Matsumoto T., Sato S., Takeuchi K., Igarashi K., Harada Y., Azuma Y., Krust A., Yamamoto Y., Nishina H., Takeda S., Takayanagi H., Metzger D., Kanno J., Takaoka K., Martin T. J., Chambon P., Kato S. (2007) Estrogen prevents bone loss via estrogen receptor α and induction of Fas ligand in osteoclasts. Cell 130, 811–823 [DOI] [PubMed] [Google Scholar]
- 15. Lam J., Takeshita S., Barker J. E., Kanagawa O., Ross F. P., Teitelbaum S. L. (2000) TNF-α induces osteoclastgenesis by direct stimulation of macrophages exposed to pemissive levels of RANK ligand. J. Clin. Invest. 106, 1481–1488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Faccio R., Zou W., Colaianni G., Teitelbaum S. L., Ross F. P. (2003) High dose M-CSF partially rescues the Dap12−/− osteoclast phenotype. J. Cell Biochem. 90, 871–883 [DOI] [PubMed] [Google Scholar]
- 17. Takeshita S., Kaji K., Kudo A. (2000) Identification and characterization of the new osteoclast progenitor with macrophage phenotypes being able to differentiate into mature osteoclasts. J. Bone Miner. Res. 15, 1477–1488 [DOI] [PubMed] [Google Scholar]
- 18. Diez G., Kollmannsberger P., Mierke C. T., Koch T. M., Vali H., Fabry B., Goldmann W. H. (2009) Anchorage of vinculin to lipid membranes influences cell mechanical properties. Biophys. J. 97, 3105–3112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Morita S., Kojima T., Kitamura T. (2000) Plat-E: an efficient and stable system for transient packaging of retroviruses. Gene Ther. 7, 1063–1066 [DOI] [PubMed] [Google Scholar]
- 20. Zou W., Croke M., Fukunaga T., Broekelmann T. J., Mecham R. P., Teitelbaum S. L. (2013) Zap70 inhibits Syk-mediated osteoclast function. J. Cell Biochem. 114, 1871–1878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Clausen B. E., Burkhardt C., Reith W., Renkawitz R., Förster I. (1999) Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 8, 265–277 [DOI] [PubMed] [Google Scholar]
- 22. Faccio R., Grano M., Colucci S., Villa A., Giannelli G., Quaranta V., Zallone A. (2002) Localization and possible role of two different αvβ3 integrin conformations in resting and resorbing osteoclasts. J. Cell Sci. 115, 2919–2929 [DOI] [PubMed] [Google Scholar]
- 23. Zou W., Izawa T., Zhu T., Chappel J., Otero K., Monkley S. J., Critchley D. R., Petrich B. G., Morozov A., Ginsberg M. H., Teitelbaum S. L. (2013) Talin1 and Rap1 are critical for osteoclast function. Mol. Cell Biol. 33, 830–844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Humphries J. D., Wang P., Streuli C., Geiger B., Humphries M. J., Ballestrem C. (2007) Vinculin controls focal adhesion formation by direct interactions with talin and actin. J. Cell Biol. 179, 1043–1057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Thievessen I., Thompson P. M., Berlemont S., Plevock K. M., Plotnikov S. V., Zemljic-Harpf A., Ross R. S., Davidson M. W., Danuser G., Campbell S. L., Waterman C. M. (2013) Vinculin-actin interaction couples actin retrograde flow to focal adhesions, but is dispensable for focal adhesion growth. J. Cell Biol. 202, 163–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Teitelbaum S. L. (2007) Osteoclasts: what do they do and how do they do it? Am. J. Pathol. 170, 427–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Xu W., Coll J. L., Adamson E. D. (1998) Rescue of the mutant phenotype by reexpression of full-length vinculin in null F9 cells: effects on cell locomotion by domain deleted vinculin. J. Cell Sci. 111, 1535–1544 [DOI] [PubMed] [Google Scholar]
- 28. McHugh K. P., Hodivala-Dilke K., Zheng M. H., Namba N., Lam J., Novack D., Feng X., Ross F. P., Hynes R. O., Teitelbaum S. L. (2000) Mice lacking β3 integrins are osteosclerotic because of dysfunctional osteoclasts. J. Clin. Invest. 105, 433–440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Faccio R., Novack D. V., Zallone A., Ross F. P., Teitelbaum S. L. (2003) Dynamic changes in the osteoclast cytoskeleton in response to growth factors and cell attachment are controlled by β3 integrin. J. Cell Biol. 162, 499–509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Croke M., Ross F. P., Korhonen M., Williams D. A., Zou W., Teitelbaum S. L. (2011) Rac deletion in osteoclasts causes severe osteopetrosis. J. Cell Sci. 124, 3811–3821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zou W., Deselm C. J., Broekelmann T. J., Mecham R. P., Vande Pol S., Choi K., Teitelbaum S. L. (2012) Paxillin contracts the osteoclast cytoskeleton. J. Bone Miner. Res. 27, 2490–2500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pollard T. D., Blanchoin L., Mullins R. D. (2001) Actin dynamics. J. Cell Sci. 114, 3–4 [DOI] [PubMed] [Google Scholar]
- 33. Hurst I. R., Zuo J., Jiang J., Holliday L. S. (2004) Actin-related protein 2/3 complex is required for actin ring formation. J. Bone Miner. Res. 19, 499–506 [DOI] [PubMed] [Google Scholar]
- 34. Tadokoro S., Shattil S. J., Eto K., Tai V., Liddington R. C., de Pereda J. M., Ginsberg M. H., Calderwood D. A. (2003) Talin binding to integrin β tails: a final common step in integrin activation. Science 302, 103–106 [DOI] [PubMed] [Google Scholar]
- 35. Plotnikov S. V., Pasapera A. M., Sabass B., Waterman C. M. (2012) Force fluctuations within focal adhesions mediate ECM-rigidity sensing to guide directed cell migration. Cell 151, 1513–1527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Dumbauld D. W., Lee T. T., Singh A., Scrimgeour J., Gersbach C. A., Zamir E. A., Fu J., Chen C. S., Curtis J. E., Craig S. W., García A. J. (2013) How vinculin regulates force transmission. Proc. Natl. Acad. Sci. U.S.A. 110, 9788–9793 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.