

Background: Inflammatory gene expression is reduced by anti-inflammatory glucocorticoids.
Results: With an inflammatory stimulus, glucocorticoids enhance expression of the phosphatase, DUSP1. This plays a transient role in the repression of some inflammatory genes.
Conclusion: Glucocorticoids repress inflammatory gene expression via DUSP1-dependent and -independent mechanisms.
Significance: Consideration of both DUSP1 and DUSP1-independent effectors is necessary to improve anti-inflammatory glucocorticoid action.
Keywords: Epithelial Cell, Gene Regulation, Glucocorticoid Receptor, Glucocorticoids, Inflammation, MAP Kinases (MAPKs), MAPK Phosphatase 1 (MKP-1), NR3C1, Anti-inflammatory, Corticosteroid
Abstract
Glucocorticoids act on the glucocorticoid receptor (NR3C1) to repress inflammatory gene expression. This is central to their anti-inflammatory effectiveness and rational improvements in therapeutic index depend on understanding the mechanism. Human pulmonary epithelial A549 cells were used to study the role of the mitogen-activated protein kinase (MAPK) phosphatase, dual-specificity phosphatase 1 (DUSP1), in the dexamethasone repression of 11 inflammatory genes induced, in a MAPK-dependent manner, by interleukin-1β (IL1B). Adenoviral over-expression of DUSP1 inactivated MAPK pathways and reduced expression of all 11 inflammatory genes. IL1B rapidly induced DUSP1 expression and RNA silencing revealed a transient role in feedback inhibition of MAPKs and inflammatory gene expression. With dexamethasone, which induced DUSP1 expression, plus IL1B (co-treatment), DUSP1 expression was further enhanced. At 1 h, this was responsible for the dexamethasone inhibition of IL1B-induced MAPK activation and CXCL1 and CXCL2 mRNA expression, with a similar trend for CSF2. Whereas, CCL20 mRNA was not repressed by dexamethasone at 1 h, repression of CCL2, CXCL3, IL6, and IL8 was unaffected, and PTGS2 repression was partially affected by DUSP1 knockdown. At later times, dexamethasone repression of MAPKs was unaffected by DUSP1 silencing. Likewise, 6 h post-IL1B, dexamethasone repression of all 11 mRNAs was essentially unaffected by DUSP1 knockdown. Qualitatively similar data were obtained for CSF2, CXCL1, IL6, and IL8 release. Thus, despite general roles in feedback inhibition, DUSP1 plays a transient, often partial, role in the dexamethasone-dependent repression of certain inflammatory genes. Therefore this also illustrates key roles for DUSP1-independent effectors in mediating glucocorticoid-dependent repression.
Introduction
Acting on the glucocorticoid receptor (NR3C1), glucocorticoids are central to the treatment of many inflammatory conditions (1). In asthma, where glucocorticoids are typically inhaled and referred to as inhaled corticosteroid, they act on multiple cells, in particular airway epithelial cells, to decrease the expression of cytokines, chemokines, adhesion molecules, and other inflammatory proteins (2). This reduces the recruitment and survival of many leukocytes and suppresses airways inflammation. However, clinical usage of glucocorticoids is limited by numerous side effects including increased gluconeogenesis, elevated blood glucose, amino acid and fatty acid mobilization, osteoporosis, and hypothalamic-pituitary-adrenal axis suppression (3). These effects arise from the normal endocrine roles of NR3C1 and indicate an unmet clinical need to improve the therapeutic index of NR3C1 ligands for anti-inflammatory therapies (4).
Unliganded NR3C1 is retained in the cytoplasm as a multiprotein complex. Binding of glucocorticoid to NR3C1 induces exchange of FK506-binding protein (FKBP)2 51 (FKBP5) for FKBP52 (FKBP4) and promotes nuclear translocation of the receptor (5). Although NR3C1 acts at glucocorticoid response elements to promote gene transcription (i.e. transactivation) (6), the principal anti-inflammatory effects of glucocorticoids are associated with the repression of inflammatory gene expression (1). One explanation for these repressive effects is interaction of NR3C1 with inflammatory transcription factors, such as nuclear factor (NF)-κB or activator protein-1 (7). This directly represses, or transrepresses, inflammatory gene transcription in part by recruiting histone deacetylases to inflammatory gene promoters (8). However, direct transcriptional repression does not address the fact that glucocorticoid-dependent repression of many inflammatory genes is only partially transcriptional and can involve post-transcriptional and translational mechanisms (9, 10). Furthermore, the repression of inflammatory gene expression by glucocorticoids can often be prevented by transcriptional or translational blockade (9, 11). For example, repression of cyclooxygenase-2 (PTGS2) expression by dexamethasone is prevented by transcriptional blockers in human fetal lung IMR-90 fibroblasts and A549 type II epithelial cells (12, 13). Such data imply that glucocorticoid-dependent gene expression plays a role in repression (9, 11). Indeed, many potentially repressive factors, for example the NF-κB inhibitor, inhibitor of κBα (NFKBIA), the transcriptional repressor, glucocorticoid-induced leucine zipper (TSC22D3), and the mRNA destabilizing protein, tristetraprolin (ZPF36), are all induced by glucocorticoids (14). Similarly, the ability of glucocorticoids to induce expression of the dual-specificity mitogen-activated protein kinase (MAPK) phosphatase 1 (DUSP1) explains the gene expression-dependent inhibition of MAPK signaling by glucocorticoids (15, 16). Because activation of NF-κB- and activator protein-1-dependent transcription, inflammatory gene mRNA stabilization and translation are all partly MAPK-dependent, this illustrates how glucocorticoids may profoundly repress inflammatory gene expression (14, 17). Indeed, DUSP1 expression is strongly, but transiently, induced by inflammatory stimuli and a firm role in feedback inhibition of MAPKs and the expression of many inflammatory genes is established (18–22). However, whereas the glucocorticoid-dependent repression of inflammatory gene expression is impaired for some genes in Dusp1-deficient mice, this does not hold for all repressed genes (23, 24). Similarly, DUSP1 is implicated in the glucocorticoid-dependent repression of CXCL1 and IL6 from human airway smooth muscle cells (25, 26), and IL8 from human airway epithelial cells (27). Despite this, and an often clear dependence on MAPKs, the demonstration of a role for glucocorticoid-induced DUSP1 in the repression of inflammatory gene expression can prove difficult (28, 29).
Because pulmonary A549 cells, like primary bronchial epithelial cells, show glucocorticoid-dependent repression of inflammatory gene expression, these cells were used to investigate the regulation of interleukin-1β (IL1B)-induced inflammatory mRNAs by dexamethasone (30). We previously identified 11 IL1B-induced mRNAs whose expression was not reduced by the translational blocker, cycloheximide, yet their repression by dexamethasone was cycloheximide-sensitive (30). As the dexamethasone-dependent repression of these mRNAs was attenuated by both a NR3C1 receptor antagonist and siRNA-mediated knock-down of NR3C1 (30), these genes represent prima facie targets for glucocorticoid-inducible anti-inflammatory genes. This model is therefore used to explore possible repressive roles of DUSP1 induced by IL1B and IL1B plus the synthetic glucocorticoid, dexamethasone.
EXPERIMENTAL PROCEDURES
Gene Nomenclature
To avoid ambiguity, all genes, and gene products, are referred to throughout by their official Human Genome Organization (HUGO) gene nomenclature committee gene symbols. In some cases, full common names are initially given as an aid to readers.
Cell Culture and Drugs
A549 cells were grown to confluence in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal calf serum and l-glutamine (all Invitrogen) in 6-well plates for all experiments except siRNA transfections, which were carried out using pre-confluent cells in 12-well plates. Cells were incubated in serum-free medium overnight prior to all experiments. Cells were changed to fresh serum-free medium containing cytokine and/or experimental drugs. IL1B (R&D systems) was dissolved in phosphate-buffered saline plus 0.1% bovine serum albumin (both Sigma). Dexamethasone (Sigma) was dissolved in Hanks' balanced salt solution (Sigma) and 203850 (Calbiochem), JNK inhibitor 8 (Calbiochem), and U0126 (Calbiochem) were dissolved in DMSO to final concentrations of <0.1%.
Enzyme-linked Immunosorbent Assay (ELISA)
Supernatants were harvested and ELISA for IL8, IL6, CSF2, or CXCL1 was performed using DuoSet ELISA kits (R&D Systems).
Western Blotting
Western blot analysis was as previously described (28). Following electrotransfer, Hybond-ECL membranes (GE Healthcare BioSciences) were probed with antisera against DUSP1 (M-18, sc-1102), PTGS2 (sc-1746), c-Jun (JUN) (sc-1694) (all Santa Cruz Biotechnology), phospho-p44/42, extracellular regulated kinase (ERK) 1/2 (number 9101), phospho-p38 MAPK (number 9211), phospho-JUN N-terminal kinase (JNK) (number 9251) (all Cell Signaling Technology), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (4699–9555(ST), AbD Serotec). After washing, membranes were incubated with horseradish peroxidase-linked secondary immunoglobulin (Dako or Jackson ImmunoResearch Laboratories) and immune complexes were detected by enhanced chemiluminescence (GE Healthcare).
RNA Isolation, cDNA Synthesis, and Real Time-PCR
Total RNA was extracted using the RNeasy mini kit (Qiagen) and 0.5 μg was reverse transcribed to cDNA (Quantas). Resultant cDNA was diluted 1:4 with RNase-free water and 2.5 μl were analyzed by SYBR Green ER (Invitrogen) real-time polymerase chain reaction (PCR) using ABI 7900HT or StepOnePlusTM instruments (Applied Biosynthesis). Relative cDNA concentrations were obtained from standard curves generated by serial dilution of an IL1B-treated sample. Amplifications were: 50 °C, 2 min; 95 °C, 10 min then 40 cycles of 95 °C, 15 s; 60 °C, 1 min. Primer specificity was assessed using dissociation (melt) curve analysis: 95 °C, 15 s; 60 °C, 20 s followed by ramping to 95 °C over 20 min. The specificity of primers was indicated by a single peak in the change of fluorescence with temperature.
Adenoviral Infection
A549 cells grown to ∼70% confluence were incubated for 24 h with the indicated multiplicity of infection of DUSP1-expressing adenoviral vector (Ad5-DUSP1) (Seven Hills Bioreagents) or a GFP (green fluorescent protein)-expressing vector (Ad5-GFP) (Qbiogene) as described (28).
siRNA-mediated Gene Silencing
A549 cells were grown to ∼60–70% confluence in 12-well plates. Cells were washed with serum-free medium and then incubated with 1 ml of serum-free medium containing 25 nm siRNA at 37 °C for 24 h prior to the addition of cytokine and drugs. Transfection medium was prepared by mixing each siRNA with Lipofectamine RNAiMAX (1 μl of 1 μg/μl) (Invitrogen) in 100 μl of serum-free DMEM at room temperature for 30 min prior to dilution to 1 ml and addition to cells.
Sequences for siRNA targeting were: DUSP1 siRNA 1 (SI00374801; 5′-TAG CGT CAA GAC ATT TGC TGA-3′); DUSP1 siRNA 2 (SI00374808; 5′-CTG TAC TAT CCT GTA AAT ATA-3′) (all Qiagen); and Lamin A/C (LMNA) siRNA (control siRNA) (5′-AAC TGG ACT TCC AGA AGA ACA-3′) (Qiagen).
Data Presentation and Statistical Analysis
All data are plotted as mean ± S.E. Statistical testing was performed using ANOVA with a Bonferroni post-test for five or fewer comparisons. Where greater than five comparisons are required, the Bonferroni post-test gives high and increasingly inappropriate false-negative rates (i.e. type II, or β, error). Therefore ANOVA with Newman-Keul multiple comparison test was used for greater than five comparisons as suggested for greater power in hypothesis testing (Prizm 5, Graphpad Software). For comparisons against a single control column ANOVA with a Dunnett's post-test was used.
RESULTS
Effect of Dexamethasone on MAPK Activation by IL1B
Activation of the p38 and ERK MAPK pathways by IL1B was previously found to be repressed by dexamethasone, in A549 cells (28, 29). We now show that IL1B robustly activated, as assessed by TXY motif phosphorylation, the p38, ERK, and JNK MAPK cascades and that dexamethasone co-treatment repressed all three pathways at 1, 2, and 6 h post-treatment (Fig. 1A). Although repression was primarily observed at, and after, 1 h, the peak in IL-1β-induced MAPK activity occurred at 30 min. At this time the effect of dexamethasone was variable, with no repression in some analyses and partial repression in others. Thereafter, IL1B-induced MAPK activation was reduced with time and the repression by dexamethasone became increasingly apparent.
FIGURE 1.
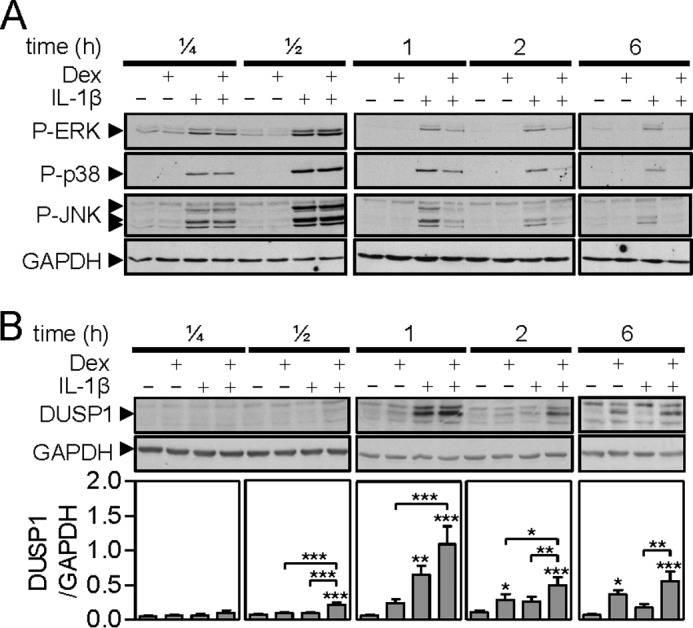
Effect of dexamethasone and IL1B on phospho-MAPKs and DUSP-1 expression. A, A549 cells were either not stimulated or stimulated with IL1B (1 ng/ml), dexamethasone (Dex, 1 μm) or a combination of the two as indicated. Cells were harvested after 0.15, 0.5, 1, 2, or 6 h, and total proteins were prepared for Western blot analysis of phospho-ERK (P-ERK), phospho-p38 (P-p38), phospho-JNK (P-JNK), and GAPDH. Blots representative of at least 4 such experiments are shown. B, as in A, cells were harvested at the times indicated for Western blot analysis of DUSP-1 and GAPDH. Following densitometric analysis, data (n = 9–11) were normalized to GAPDH and plotted as mean ± S.E. Significance, using ANOVA with a Bonferroni's multiple comparison test is indicated. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
DUSP1 Expression Is Induced by IL1B and Dexamethasone
The repression elicited by dexamethasone correlated with the expression of DUSP1 protein, which was strongly induced by IL1B at 1 h in a manner that was enhanced by dexamethasone at multiple time points (Fig. 1B). Following IL1B plus dexamethasone treatment, DUSP1 protein was detected at 30 min and dexamethasone alone induced DUSP1 expression at 1 h. At 2 h, dexamethasone-dependent expression of DUSP1 was also enhanced by dexamethasone with less effect of IL1B alone. At 6 h DUSP1 expression was primarily dexamethasone-dependent with some enhancement by IL1B.
Effect of Dexamethasone on IL1B-induced Gene Expression
Following treatment with IL1B, 11 mRNAs (official human genome nomenclature committee symbol with other commonly used gene symbols in parentheses), CCL2 (MCP1), CCL20 (MIP3α), CSF2 (GM-CSF), CXCL1 (GROα), CXCL2 (GROβ, MIP2α), CXCL3 (GROγ, MIP2β), IL6 (IL-6), IL8 (IL-8, CXCL8), ISG20 (CD25), OLR1 (LOX1), and PTGS2 (COX-2), which previously showed enhanced expression by IL1B that was reduced by dexamethasone in a cycloheximide-sensitive manner (30), were all strongly up-regulated (Fig. 2, A and B). Expression of CXCL1, IL8, and IL6 increased progressively over 6 h, whereas CXCL2 mRNA expression was highest at 1 h and was modestly reduced by 6 h. In the case of CCL2, CCL20, CXCL3, PTGS2, and CSF2 mRNAs, expression increased from 1 to 2 h, before remaining essentially static (CCL2, CCL20, CXCL3, and PTGS2) or reducing (CSF2) by 6 h (Fig. 2A). These kinetics are consistent with a rapid induction of mRNA expression due to pre-existing factors, i.e. these are immediate/early primary response genes whose expression induced by IL1B was not reduced by the translational blocker, cycloheximide (30).
FIGURE 2.

Effect of dexamethasone and IL1B on inflammatory gene expression. A and B, A549 cells were either not stimulated (NS) (○) or stimulated with IL1B (1 ng/ml) (●) or IL1B plus dexamethasone (1 μm) (IL1B + Dex) (■) as indicated. Cells were harvested after 1, 2, or 6 h for real-time PCR analysis of the indicated genes and GAPDH. Data (n = 11) were normalized to GAPDH and are plotted as mean ± S.E. Significance was tested relative to time-matched IL1B-treated samples using ANOVA with a Bonferroni post-test. *, p < 0.05; **, p < 0.01; ***, p < 0.001. Genes were grouped according to the expression pattern with apparent “early phase” mRNAs in A and “delayed response” mRNAs in B. C, the effect of IL1B + dexamethasone for each inflammatory mRNA in panel A is plotted as a percentage of IL1B for each time (left panel). The overall effect of dexamethasone in the presence of IL1B for the 9 genes combined is plotted as mean ± S.E. (right panel). D, cells were treated as in A and the supernatants harvested after 1, 2, or 6 h for cytokine/chemokine release measurement. Data (n = 6) expressed as picograms/ml are plotted as mean ± S.E. (left panel). Significance, relative to IL1B-treated samples was tested using ANOVA with a Bonferroni post-test. *, p < 0.05; **, p < 0.01; ***, p < 0.001. Cells were harvested at the times indicated for Western blot analysis of PTGS2 and GAPDH (right panel). Blots representing at least 4 such experiments are shown.
By contrast, the kinetics of IL1B-induced ISG20 and OLR1 mRNAs showed no, or low, enhancements at 1 h, with only modest increases at 2 h, prior to substantial increases in expression at 6 h (Fig. 2B). Although seemingly more consistent with late phase/secondary response genes, this is at odds with the previously shown insensitivity to cycloheximide (28). Nevertheless, the timing of induction for these two mRNAs is distinct from the nine showing classical primary response kinetics and we therefore refer to these as delayed response genes.
In the presence of dexamethasone, all 11 genes showed significant reductions in their mRNA expression induced by IL1B at 6 h (Fig. 2, A and B). This effect was more modest at 1 and 2 h with CCL20 showing no repression at 1 h. Excluding ISG20 and OLR1, where there was little mRNA accumulation at 1 or 2 h, expression of the remaining 9 primary response genes (Fig. 2A) showed a dexamethasone-dependent repression that increased progressively with time (Fig. 2C). This is consistent with the repression of these mRNAs being prevented or reduced by cycloheximide and the fact that time is necessary for the synthesis of glucocorticoid-induced gene products mediating repression (28).
Release of CSF2, CXCL1, IL6, and IL8 into the supernatant and cellular expression of PTGS2 was analyzed. In each case, this was time dependently increased by IL1B in a manner that was significantly reduced by dexamethasone (Fig. 2D).
Effect of MAPK Inhibitors on IL1B-induced Gene Expression
Recently, a highly selective, cell permeable inhibitor, JNK inhibitor 8, of the JNK MAPKs showing ∼1 μm potency on cellular JUN phosphorylation was described (31). In A549 cells, IL1B induced JUN phosphorylation by 11.9 ± 4.9-fold and this was prevented by JNK inhibitor 8 (EC50 = 0.8 μm) (data not shown). Near maximal (83%) repression was achieved at 10 μm and was used for subsequent studies.
To explore the role of p38 and ERK MAPK pathways in the expression of IL1B-induced mRNAs, the p38 inhibitor, SB203580, and the MAPK/ERK kinase (MEK) 1/2 inhibitor, U0126, were tested along with JNK inhibitor 8. Each inhibitor was used at 10 μm, i.e. maximally effective concentrations as determined by substrate phosphorylation and functional responses in A549 cells (32, 33). For most mRNAs, SB203580 produced a partial and time-variable inhibition of IL1B-induced gene expression (supplemental Table S1). CXCL2 and ISG20 were unaffected, and IL6 was strongly repressed. A generally lesser repressive effect was observed with U0126, with a further reduced inhibition observed for JNK inhibitor 8 (supplemental Table S1).
At the level of cytokine production, SB203580 significantly reduced CSF2, CXCL1, IL6, and IL8 release at 2 and 6 h (supplemental Table S1). Although robustly inhibiting CSF2 and CXCL1 release at 2 and 6 h, U0126 produced a lesser repressive effect on the release of IL6 and IL8 (supplemental Table S1). JNK inhibitor 8 was also effective at lowering CSF2 and CXCL1 release, but showed a reduced effect on IL6 and did not significantly inhibit IL8 at either time point (supplemental Table S1). PTGS2 protein expression was weakly induced by IL1B at 2 h and this appeared to be reduced by the individual kinase inhibitors (Fig. 3D). By 6 h, there was robust PTGS2 expression and this was reduced by each MAPK inhibitor.
FIGURE 3.

Effect of MAPK inhibitors on IL1B-induced inflammatory gene expression. A and B, A549 cells were not stimulated (NS) (○), treated with IL1B (1 ng/ml) (●), or pre-treated with a combination of UO126, SB203580 plus JNK inhibitor 8 each at 10 μm (UO + SB + J8) for 30 min prior to IL1B stimulation (■). Cells were harvested after 1, 2, or 6 h for real-time PCR analysis of the indicated genes and GAPDH. Data (n = 4) were normalized to GAPDH and plotted as mean ± S.E. Early phase genes are shown in A and delayed response genes in B. C, cells were treated as in A and the supernatants were harvested after 1, 2, or 6 h for measurement of cytokine/chemokine release. Data (n = 4) expressed in picograms/ml are plotted as mean ± S.E. D, cells were treated as in A and harvested at the times indicated for Western blot analysis of PTGS2 and GAPDH. Representative blots are shown. Following densitometric analysis, data (n = 4), normalized to GAPDH, are plotted as mean ± S.E. Significance, relative to time-matched IL1B-treated samples, was tested by ANOVA with a Bonferroni post-test. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Inflammatory stimuli induce the expression of DUSP1 in part via MAPK pathways, such that inhibition of a single MAPK pathway may reduce or prevent DUSP1 expression (34–36). Because DUSP1 acts on all three MAPK pathways, such inhibition could correspondingly enhance activation of the remaining two MAPK pathways. Equally, cross-feedback control by p38 MAPK down-regulates the ERK pathway, which may therefore be enhanced in the presence of a p38 inhibitor (37). Furthermore, the inhibitory effect of dexamethasone was not on any single pathway, but on all three MAPKs (Fig. 1A). Therefore the effect of concurrent inhibition of these three pathways was investigated on inflammatory gene expression (Fig. 3). IL1B strongly induced the expression of the 9 primary response genes and, in each case, this was significantly and substantially blocked by the combined MAPK inhibitors (Fig. 3A). A similar near total repression was observed for ISG20 and OLR1 (Fig. 3B), as well as for protein expression of CSF2, CXCL1, IL6, IL8, and PTGS2 (Fig. 3, C and D). There was no effect of the kinase inhibitors, alone or in combination on cell viability (data not shown).
Effect of DUSP1 Overexpression on MAPK Activation and Inflammatory Gene Expression
As previously described (28, 29), adenoviral overexpression of DUSP1 substantially reduced ERK and p38 phosphorylation following treatment with IL1B (Fig. 4A). A similar repressive effect is now reported with respect to JNK and, in each case, there was no effect of a GFP expressing adenovirus (Fig. 4A). With respect to the 11 IL1B-induced genes, a clear inhibition of mRNA, and where tested, protein, expression, by DUSP1 overexpression was shown for all genes (Fig. 4, B-D). There was no effect of Ad5-GFP. Thus, like dexamethasone, DUSP1 can inhibit activation of all three MAPK pathways and profoundly reduce IL1B-induced inflammatory gene expression.
FIGURE 4.

Effect of DUSP1 overexpression on IL1B-induced MAPK phosphorylation and inflammatory gene expression. A, A549 cells were either not infected or infected with Ad5-DUSP1 or Ad5-GFP at a multiplicity of infection of 10 for 24 h before IL1B treatment (1 ng/ml). After 1 h, the cells were harvested for Western blot analysis of DUSP1, phospho-ERK (P-ERK), phospho-p38 (P-p38), phospho-JNK (P-JNK), and GAPDH. Blots representative of at least 4 such experiments are shown. B and C, cells were treated as in A and harvested at 1, 2, or 6 h for real-time PCR analysis of the indicated genes and GAPDH. Data (n = 4) were normalized to GAPDH and plotted as mean ± S.E. Significance relative to time-matched IL1B and Ad5-GFP-treated samples was tested by ANOVA with a Bonferroni post-test. *, p < 0.05; **, p < 0.01; ***, p < 0.001. Early phase genes are shown in B and delayed response genes in C. D, cells were treated as in A and the supernatants were harvested after 2 or 6 h for cytokine/chemokine release measurement. Data (n = 4) expressed in picograms/ml are plotted as mean ± S.E. (top panel). Significance, relative to time-matched IL1B and Ad5-GFP-treated samples, was tested by ANOVA with a Bonferroni post-test. *, p < 0.05; **, p < 0.01; ***, p < 0.001. Cells were also harvested at the times indicated for Western blot analysis of PTGS2 and GAPDH (lower panel). Blots representative of at least 4 such experiments are shown.
Effect of DUSP1 siRNA on MAPK Activation by IL1B in the Absence and Presence of Dexamethasone
Whereas a control siRNA targeted to LMNA revealed no effect on DUSP1 expression (Fig. 5A), targeting of DUSP1 by two separate siRNAs resulted in a substantial and significant losses of DUSP1 expression induced by IL1B or by IL1B plus dexamethasone (Fig. 5B). This occurred at all times tested.
FIGURE 5.
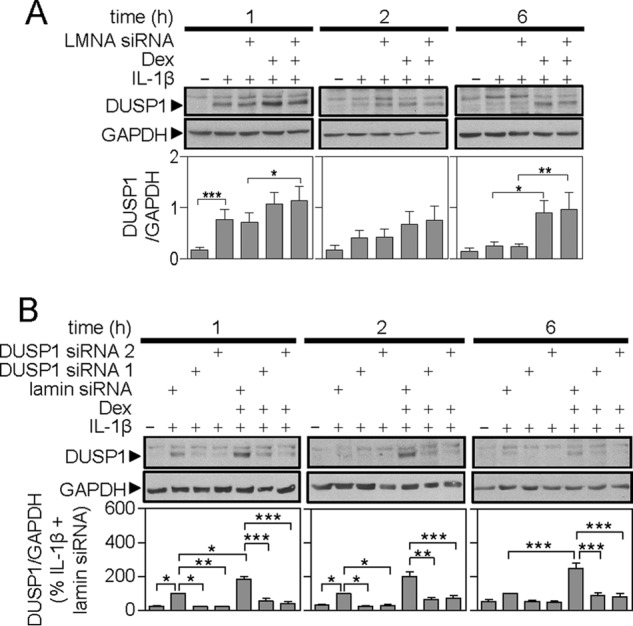
Effect of LMNA and DUSP1 targeting siRNAs on IL1B and IL1B plus dexamethasone-induced DUSP1 expression. A549 cells were: A, incubated without or with LMNA-specific siRNA; or B, incubated with LMNA (control) or DUSP1-specific siRNAs. After 24 h, cells were treated with IL1B (1 ng/ml) or IL-1β plus dexamethasone (1 μm) (Dex) as indicated. Cells were harvested at 1, 2, or 6 h for Western blot analysis of DUSP1 and GAPDH. Representative blots are shown. Following densitometric analysis, data in A (n = 7) were normalized to GAPDH and are plotted as mean ± S.E. In B, data (n = 10), normalized to GAPDH were expressed as a percentage of LMNA siRNA plus IL1B-stimulated cells for each time and plotted as mean ± S.E. In each case, significance was tested using ANOVA with Bonferroni's multiple comparison test is indicated. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
As described above, the three MAPK pathways were activated by IL1B and this was attenuated at 1, 2, and 6, by dexamethasone (Fig. 6). There was no effect of the LMNA siRNA on MAPK activation induced by IL1B or IL1B plus dexamethasone (Fig. 6A, supplemental Table S2). However, 1 h post-IL1B treatment, the DUSP1 targeting siRNAs significantly increased TXY phosphorylation of all three MAPKs relative to LMNA control (Fig. 6, B and C). This effect was not apparent 2 or 6 h post-IL1B treatment. Thus DUSP1 plays a significant role in feedback control of MAPKs at 1 h, but not at later times.
FIGURE 6.

Effect of LMNA- and DUSP1-targeting siRNA on IL1B-induced MAPK phosphorylation. A549 cells were: A, incubated without or with LMNA-specific siRNA, or B, incubated with LMNA (control) or DUSP1-specific siRNAs. After 24 h, cells were treated with IL1B (1 ng/ml) or IL1B plus dexamethasone (1 μm) (Dex) as indicated. Cells were then harvested at 1, 2, or 6 h, and total proteins were prepared for Western blot analysis of phospho-ERK (P-ERK), phospho-p38 (P-p38), phospho-JNK (P-JNK), and GAPDH. Representative blots are shown. Blots representative of at least 6–9 such experiments are shown. Densitometric data from A appear in supplemental Table S2. C, following densitometric analysis, data from B were normalized to GAPDH, expressed as a percentage of LMNA siRNA plus IL1B-stimulated for each time and plotted as mean ± S.E. Significance was tested using ANOVA with a Newman-Keul multiple comparison test. Significance between: LMNA control siRNA plus IL1B and each of the DUSP1 targeting siRNAs plus IL1B, and the LMNA control plus IL1B plus Dex is shown. Other comparisons are specifically indicated. *, p < 0.05; **, p < 0.01; ***, p < 0.001. D, for each P-MAPK at each time, the effect of IL1B plus Dex expressed as a percentage of IL1B for each of the three individual siRNAs is plotted as a mean ± S.E. The percent of IL1B plus dexamethasone/IL1B for the LMNA siRNA is compared with that for each DUSP1-specific siRNAs using ANOVA with a Dunnett's post-test. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
In the presence of dexamethasone, activation of ERK, p38, and JNK, as indicated by TXY phosphorylation, was reduced significantly at each time (Fig. 6, A–C). The two DUSP1 targeting siRNAs both significantly increased MAPK phosphorylation at 1 h relative to the IL1B plus dexamethasone-treated control (Fig. 6, B and C). Thus, DUSP1 expression induced by IL1B plus dexamethasone does act to reduce MAPK activation. However, in evaluating the role of DUPS1 in the repressive effects of dexamethasone, it is essential to account for the role of DUSP1 in feedback inhibition. Therefore in Fig. 6D, the MAPK phosphorylation produced by IL1B plus dexamethasone was expressed as a percentage of the IL1B-induced MAPK phosphorylation for each siRNA treatment. This allows the repressive effect of dexamethasone to be assessed and compared in the presence of DUSP1 or DUSP1 knockdown.
At 1 h, dexamethasone reduced the phosphorylation of each MAPK to ∼50% of the IL1B-treated level in the presence of LMNA control siRNA (Fig. 6, C and D). However, this repressive effect was significantly and substantially attenuated by the DUSP1-targeting siRNAs (Fig. 6D), i.e. the percentage of MAPK phosphorylation for IL1B plus dexamethasone/IL1B was close to 100%. Thus DUSP1 knock-down prevented the dexamethasone-dependent repression of IL1B-induced MAPK phosphorylation. We therefore conclude that DUSP1 not only plays a key role in feedback control of ERK, p38, and JNK MAPKs, but is responsible for their repression by dexamethasone at 1 h.
These data contrast markedly with effects observed 6 h post-IL1B treatment (Fig. 6, B–D). Although the overall level of MAPK activation was reduced relative to earlier times, there was still significant dexamethasone-dependent repression (Fig. 6, A and B). However, this was unaffected by the DUSP1-targeting siRNAs suggesting that the later repressive effects of dexamethasone were not due to DUSP1 (Fig. 6, B–D). At 2 h post-IL1B plus dexamethasone treatment, the effect of the two DUSP1 siRNAs was intermediate between the 1 and 6 h data. There were non-significant trends toward increased MAPK phosphorylation (Fig. 6C) and concomitant, but variable, losses of dexamethasone-dependent repression (Fig. 6D).
Effect of DUSP1 Knock-down on IL1B-induced mRNA Expression in the Absence and Presence of Dexamethasone
IL1B induced the expression of all 11 inflammatory mRNAs and this was unaffected by the LMNA siRNA (supplemental Table S2). Similarly, dexamethasone significantly repressed the IL1B-induced expression of all 11 mRNAs and this was also unaffected by LMNA siRNA (supplemental Table S2). Therefore, in subsequent analyses, the effects of DUSP1-targeting siRNAs were compared solely to the LMNA control siRNA.
Following IL1B treatment, DUSP1 knock-down increased the expression of all 9 acute-phase mRNAs at 1 h (Fig. 7A). This effect was significant with respect to one or both DUSP1-targeting siRNAs for CCL2, CSF2, CXCL3, IL6, IL8, and PTGS2. By 2 h post-IL1B treatment, and with the exception of IL6, this trend was markedly reduced or absent. Conversely, at 6 h post-IL1B treatment, DUSP1 knock-down produced a general trend toward lowering the expression of the 9 acute-phase genes. This was significant for CCL2, CCL20, CSF2, CXCL1, CXCL2, CXCL3, and PTGS2 with respect to one or both DUSP1 siRNAs (Fig. 7A). DUSP1 knockdown also enhanced IL1B-induced expression of the two delayed response genes (Fig. 7C). Although this was significant for OLR1 at 1 h, there was no effect on either gene at 6 h.
FIGURE 7.

Effect of DUSP1 targeting siRNAs on IL1B-induced inflammatory gene mRNA expression. A and C, A549 cells were incubated with LMNA (control) or DUSP1-specific siRNAs for 24 h before being treated with IL1B (1 ng/ml) or IL1B plus dexamethasone (1 μm) (Dex) as indicated. Cells were harvested after 1, 2, or 6 h for real time-PCR analysis of the indicated genes and GAPDH. Data (n = 5–13) normalized to GAPDH, were expressed as a percentage of LMNA siRNA plus IL1B stimulated for each time and plotted as mean ± S.E. Significance was tested using ANOVA with a Newman-Keul multiple comparison test. Significance between: LMNA control siRNA plus IL1B and each of the DUSP1 targeting siRNAs plus IL1B, and the LMNA control plus IL1B plus Dex is shown. Other comparisons are specifically indicated. *, p < 0.05; **, p < 0.01; ***, p < 0.001. Early phase genes are shown in A and delayed response genes in C. B and D, for each inflammatory gene at each time, the effect of IL1B plus Dex expressed as a percentage of IL1B for each of the three individual siRNAs is plotted as a mean ± S.E. The percent of IL1B plus dexamethasone/IL1B for the LMNA siRNA is compared with that for each of the DUSP1-specific siRNAs using ANOVA with a Dunnett's post-test. *, p < 0.05; **, p < 0.01; ***, p < 0.001. Early phase genes are shown in B and delayed response genes in D.
In the presence of dexamethasone, IL1B-induced expression of all mRNAs was repressed at 2 and 6 h (Fig. 7A). At 1 h all the mRNAs, with the exception of CCL20, were repressed by dexamethasone (Fig. 7A). Although not significant for CSF2, these data are consistent with Fig. 2. The DUSP1 targeting siRNAs again produced a general trend toward higher levels of inflammatory gene expression at 1 h post-treatment. Indeed relative to IL1B plus dexamethasone-treated (plus LMNA siRNA), there was significantly increased mRNA expression of CCL2, CCL20, CSF2, CXCL1, CXCL2, CXCL3, IL8, and PTGS2 with at least one DUSP1 siRNA (Fig. 7A). Although not induced by IL1B at 1 h, there was a significant repression by dexamethasone of ISG20 and this was prevented by the DUSP1 siRNAs (Fig. 7C). IL1B-induced OLR1 expression was also repressed by dexamethasone, but this was unaffected by DUSP1 knockdown (Fig. 7C). Thus, with the exception of OLR1, DUSP1 expression induced by IL1B plus dexamethasone does exert negative regulation of these inflammatory genes. However, this effect was completely lost by 6 h with no genes showing significant effects of DUSP1 knockdown relative to IL1B plus dexamethasone (plus LMNA siRNA) (Fig. 7, A and C). At 2 h, the effect of DUSP1 knock-down was variable and intermediate with the effects at 1 and 6 h.
To correctly assess the role of DUSP1 in the repressive effects of dexamethasone, it is necessary to consider the feedback control exerted by IL1B-induced DUSP1. For example, at 1 h post-IL1B treatment, DUSP1 exerts a significant feedback inhibition of CCL2 mRNA (Fig. 7A). At this time, dexamethasone significantly represses IL1B-induced CCL2 expression and this was modestly reversed by the DUSP1 siRNAs (Fig. 7A). However, expressing the IL1B plus dexamethasone-induced expression of CCL2 as a percentage of the IL1B-induced CCL2 for the LMNA and DUSP1 targeting siRNAs shows no difference in the repressive effect of dexamethasone (Fig. 7B, top left panel). This effect is more striking with respect to CXCL3, IL6, and IL8 at 1 h. In each case, there was significant repression of IL1B-induced expression by dexamethasone, and the DUSP1 targeting siRNAs increased this mRNA expression relative to IL1B plus the dexamethasone (plus LMNA siRNA)-treated (Fig. 7A). However, comparing the effect of dexamethasone in the presence of each siRNA (LMNA versus DUSP1 si1 and DUSP1 si2) showed no differences in the overall repression (Fig. 7B). Thus, despite a clear role in feedback inhibition of mRNA expression for these genes, there was no additional role for DUSP1 in the repression exerted by dexamethasone on CCL2, CXCL3, IL6, and IL8 mRNAs at 1 h. A similar result was observed for OLR1 (Fig. 7D).
This contrasts with the effects observed for CXCL1, CXCL2, PGTS2 and, although not significant, CSF2. For each of these genes, dexamethasone significantly repressed the IL1B-induced gene expression at 1 h and in each case their mRNA expression was increased by DUSP1 knock-down (Fig. 7A). Thus in the context of IL1B plus dexamethasone, DUSP1 exerts a negative regulatory effect on these mRNAs. The relative repressive effect of dexamethasone was next considered in the context of each siRNA treatment (Fig. 7B). For CXCL1 and CXCL2, the level of repression achieved by dexamethasone in the presence of LMNA siRNA was significantly greater than with the two DUSP1 targeting siRNAs (Fig. 7B). Indeed, there was no significant repression by dexamethasone in the presence of the DUSP1 targeting siRNAs. A similar trend was observed for CSF2. Thus, at 1 h DUSP1 accounts for virtually all of the repressive effect of dexamethasone on CXCL1 and CXCL2, and possibly CSF2. Similarly, ISG20 was also repressed by dexamethasone at 1 h and this repression was absent in the presence of the DUSP1 targeting siRNAs (Fig. 7, C and D). With respect to PTGS2 mRNA, dexamethasone produced a significant repression at 1 h that was significantly reversed by the two DUSP1 targeting siRNA (Fig. 7, A and B). However, even in the presence of the DUSP1 siRNAs, there was still significant repression by dexamethasone (Fig. 7, A and B). Thus, whereas establishing a role for DUSP1 in the dexamethasone-dependent repression of PTGS2, this does not account for the full repressive effect of dexamethasone.
Analysis of IL1B-induced mRNA expression at 6 h showed a very profound dexamethasone-dependent repression for all 9 primary response genes and the two delayed response genes (Fig. 7, A and C). However, in all cases this was unaffected by DUSP1 knockdown. This contrasts with 2 h, where often intermediate effects, between 1 and 6 h, were observed (Fig. 7A). It is important to note that the percent of effect of dexamethasone for the 9 primary response genes at 6 h was apparently reduced with DUSP1 knock-down relative to the repression achieved in the presence of LMNA siRNA (Fig. 7B). However, this effect is driven by the lower expression of each mRNA produced following DUSP1 knock-down in the presence of IL1B (Fig. 7A). As a consequence, these data do not support a role for DUSP1 in the dexamethasone-dependent repression of the 9 primary response genes at 6 h. Conversely, whereas IL1B plus dexamethasone-induced ISG20 mRNA expression was unaffected by DUSP1 knockdown (Fig. 7, C and D), the dexamethasone-dependent repression of OLR1 was modestly reduced by the DUSP1 targeting siRNA (Fig. 7D).
Effect of DUSP1 Knock-down on IL1B-induced Protein Expression in the Absence and Presence of Dexamethasone
Supernatants from the experiments in Figs. 5–7 were analyzed for cytokine release. IL1B stimulated the release of CSF2, CXCL1, IL6, and IL8 at 6 h and this was significantly repressed by dexamethasone in a manner that was unaffected by the LMNA control siRNA (supplemental Table S2). However, because of the need to use subconfluent cells for optimal siRNA transfections, the release of CSF2 at 2 h was below the detection limit of the assay (Fig. 8A). The two DUSP1-targeting siRNAs had no effect on the IL1B-induced release of CSF2 or CXCL1, whereas IL6 and IL8 release appeared to be enhanced (Fig. 8A). This reached significance for IL6 at 6 h with one DUSP1 siRNA. With respect to PTGS2, there was no effect of the LMNA siRNA, but DUSP1 targeting enhanced IL1B-induced expression (Fig. 8C).
FIGURE 8.

Effect of DUSP1 targeting siRNA on IL1B-induced inflammatory gene protein release/expression. A, A549 cells were incubated with LMNA (control) or DUSP1-specific siRNAs for 24 h before being treated with IL1B (1 ng/ml) or IL1B plus dexamethasone (1 μm) (Dex) as indicated. Supernatants were harvested after 2 or 6 h for cytokine/chemokine release measurement. Data (n = 9–15) expressed as picograms/ml are plotted as mean ± S.E. Note: release of CSF2 at 2 h was below the detection limit of the assay. Significance was tested between the LMNA control siRNA plus IL1B and each of the DUSP1 targeting siRNAs plus IL1B, and LMNA control plus IL1B plus Dex using ANOVA with a Newman-Keul multiple comparison test. Other comparisons are specifically indicated. *, p < 0.05; **, p < 0.01; ***, p < 0.001. B, for each cytokine/chemokine at each time, the effect of IL1B plus Dex was expressed as a percentage of IL1B for each of the three individual siRNAs and is plotted as a mean ± S.E. The percent of IL1B plus dexamethasone/IL1B for the LMNA siRNA is compared with that for each DUSP1-specific siRNAs using ANOVA with a Dunnett's post-test. *, p < 0.05; **, p < 0.01; ***, p < 0.001. C, Cells were treated as in A and harvested for Western blot analysis of PTGS2 and GAPDH. Blots representative of at least 4 such experiments are shown.
Dexamethasone significantly reduced the release of each cytokine/chemokine and the DUSP1 targeting siRNAs generally increased this. This was significant for CXCL1 at 2 and 6 h and IL8 at 2 h (Fig. 8A). When expressed as a percentage of IL1B for each siRNA (Fig. 8B), the dexamethasone-dependent repression at 6 h was significantly reduced by both DUSP1 targeting siRNAs for CSF2, CXCL1, and IL8 and by one DUSP1 targeting siRNA for IL6. Thus a role for DUSP1 in the dexamethasone-dependent repression of CSF2, CXCL1, IL6, and IL8 protein release is indicated. However, this reversal of repression was very partial at 6 h and indicates the existence of DUSP1-independent mechanisms of repression (Fig. 8B). In contrast, the reversal of repression by the DUSP1 siRNAs at 2 h was complete for CXCL1 and near complete for IL8 at 2 h (Fig. 8, A and B). Thus, DUSP1 provides all, or the greater part, of the repressive effect of dexamethasone on CXCL1 and IL8 at 1 h, but a lesser role at 6 h.
Analysis of PTGS2 protein revealed similar data to that for IL6 release. The LMNA siRNA had no effect on IL1B + dexamethasone-induced PTGS2 expression (Fig. 8C). However, despite a clear feedback role, independent effects of DUSP1 in dexamethasone-dependent repression of the PTGS2 protein were not evident (Fig. 8C).
DISCUSSION
Glucocorticoids repress inflammatory gene expression via mechanisms that may include direct repression of transcription (transrepression) and the induction of glucocorticoid-induced effector genes that then reduce inflammatory gene expression (10, 14). Importantly, inflammatory gene expression frequently involves MAPK activation and there is considerable interest in DUSP1 as a major effector of glucocorticoid repression (17, 38). However, even where inflammatory gene expression is MAPK-dependent and repressed by DUSP1 overexpression, simple knockdown or loss of DUSP1 does not necessarily prevent, or even attenuate, glucocorticoid-dependent repression (23, 24). To explore this, we turned to 11 IL1B-induced genes whose mRNA expression was repressed by dexamethasone in a cycloheximide-sensitive manner (30). This implies that gene expression is necessary for repression and represents the starting point for the current study.
Inhibitors of p38, MEK1/2, and JNK variably, but generally only partially, inhibited mRNA expression of the 11 inflammatory genes. However, by combining all three inhibitors together, a striking and near complete inhibition of the mRNA and protein expression of all 11 genes was observed. Explanations for this effect are multiple, but may include pathway redundancy in the induction of these genes. Equally, cross-regulatory control between MAPK pathways (see above) could substantially under-represent the role of any one pathway following its inhibition (38, 39). Conversely, simultaneous inhibition of all three MAPK pathways prevents this issue and represents a highly effective means of inhibiting inflammatory gene expression, one that is also adopted by glucocorticoids. Likewise DUSP1 over-expression inhibited each MAPK pathway and reduced inflammatory gene expression. These 11 genes are therefore induced by IL1B in a MAPK-dependent manner that is prevented by DUSP1 overexpression. As DUSP1 expression is induced by IL1B and enhanced by dexamethasone, these genes represent targets to explore DUSP1 function.
To understand regulation of inflammatory gene expression by glucocorticoids, it is essential to consider the feedback role of DUSP1 (38, 39). In the presence of IL1B, DUSP1 provides transient feedback control of the ERK, p38, and JNK MAPKs. Loss of DUSP1 elevated MAPK phosphorylation at 1 h, but had no effect at 2 or 6 h post-IL1B treatment. Similarly, at 1 h the 9 primary response genes, plus ISG20 and OLR1, showed a trend, significant for 7 genes, toward increased expression following DUSP1 knockdown. This is consistent with feedback control by DUSP1 for multiple genes (20, 21), including, in A549 cells, CSF2, IL6, IL8, and PTGS2 (40). However, by 2 h post-IL1B this enhancement had largely disappeared. Indeed, by 6 h post-IL1B, DUSP1 knockdown significantly reduced the IL1B-induced expression of many of the primary response genes, but not the two delayed response genes.
One possible candidate to explain this effect is tristetraprolin (ZFP36). This binds AUUUA-containing mRNAs, such as CSF2, IL8, and PTGS2, to promote mRNA destablization and translational silencing (40, 41). ZFP36 is strongly and transiently induced following proinflammatory stimuli (41), including IL1B in A549 cells, where its expression is MAPK-dependent (42). Furthermore, loss of DUSP1, by increasing MAPK activation, is shown to increase ZFP36 expression (43). This is predicted to enhance feed-forward control of AUUUA-containing inflammatory genes. Because all 9 of the primary response genes in the current study have one, but typically more, AUUUA motifs in their 3′ untranslated regions, this offers some explanation for the observed reductions in inflammatory gene expression at 6 h.
In terms of the repression elicited by dexamethasone, we document a non-redundant role for DUSP1 as the primary effector in the early onset repression of all three MAPK pathways following IL1B plus dexamethasone co-treatment. This coincides with the greatest levels of DUSP1 expression induced by IL1B plus dexamethasone. However, by 2 h post-treatment the role of DUSP1 in MAPK repression had waned and by 6 h, there was no effect of DUSP1 knockdown. This suggests the existence of additional mechanisms by which dexamethasone must repress MAPKs. Such data are supported by studies showing only a partial effect of DUSP1 knockdown on the repression of MAPK phosphorylation following dexamethasone pre-treatment (28). However, whereas up-regulation of other DUSPs is not indicated (16, 38), glucocorticoid-induced genes such as TSC22D3, which may inhibit Ras-Raf activation of MAPKs (44), or the cyclin-dependent kinase inhibitor 1C (CDKN1C), which may inhibit JNK signaling (45), are both induced by dexamethasone in A549 cells (46–48).
Dealing with the dexamethasone-dependent control of gene expression, DUSP1 knockdown almost totally prevented the repression of IL1B-induced CXCL1 and CXCL2 mRNAs at 1 h, with a similar trend for CSF2. Conversely, and aside from roles in feedback inhibition, dexamethasone repression of CCL2, CXCL3, IL6, IL8, and OLR1 at 1 h was unaltered by loss of DUSP1. One explanation for this would be a failure to sufficiently knock-down DUSP1 expression. However, the fact that dexamethasone-dependent inhibition of MAPK phosphorylation and repression of CXCL1, CXCL2 mRNA at 1 h was prevented by the DUSP1-targeting siRNAs suggest sufficiency of the DUSP1 knock-down. DUSP1-independent mechanisms are therefore likely to be responsible for this repression. With PTGS2, DUSP1 was involved in feedback control and at 1 h, dexamethasone-dependent repression was partly prevented by DUSP1 knockdown. This supports both DUSP1-dependent and DUSP1-independent mechanisms of repression. Moving on to 6 h, the dexamethasone-dependent repression of all mRNAs was enhanced relative to earlier times. However, in this case repression was essentially unaffected by DUSP1 knockdown. Therefore (non-redundant) roles for DUSP1 are not supported (notwithstanding a modest role in the repression of OLR1 at 6 h). Thus, following the early phase of DUSP1-dependent repression, there must be an induction of additional effector processes that either become the predominant effectors or act redundantly with DUSP1. For example, whereas expression of the DUSP1 protein was evident within 30 min to 1 h of IL1B plus dexamethasone treatment, the expression of TSC22D3, which reduces activation of NF-κB, activator protein-1, and possibly MAPKs (44), is detected 1–2 h following IL1B plus dexamethasone treatment (48).
At the protein level, DUSP1 knockdown tended to increase IL6, IL8, and PTGS2 expression and this supports the previously described feedback role on these genes (40). Two hours post-IL1B, dexamethasone-dependent repression of CXCL1 release was totally prevented by loss of DUSP1. This confirms DUSP1 as the major effector of repression at this time. However, by 6 h post-IL1B, DUSP1 only accounted for around half of the dexamethasone-dependent repression of CXCL1 release. Similarly, there was clear evidence of a role for DUSP1 in dexamethasone repression of IL8 release at 2 h, whereas by 6 h, both IL8 and CSF2 release only showed very modest losses of repression following DUSP1 knockdown. Thus, the release of CXCL1 and IL8 confirm key roles for DUSP1 in the repression by dexamethasone at 2 h, yet more modest, if any, roles at 6 h. Conversely, DUSP1-independent mechanisms of repression appear to play dominant roles in the dexamethasone repression of IL6 and PTGS2 at both 2 and 6 h.
In conclusion, we use pulmonary A549 cells to document a central role for glucocorticoid-induced DUSP1 in the early repression of selected inflammatory mRNAs and proteins. However, this role is transitory, and by 6 h DUSP1-independent mechanisms of repression dominate with respect to the 11 genes tested. As these genes were selected based on the cycloheximide sensitivity of their repression by dexamethasone (30), these data point to the existence of additional dexamethasone-dependent gene expression events that are important for repression. These should be required for the repression of some genes at early times and for all 11 genes at later times. This finding explains previous difficulties in establishing roles for DUSP1 in the dexamethasone-dependent repression of IL8 and CSF2 (28, 29). Importantly, if widespread in relevant human systems, and this now requires testing, these data may have repercussions for the development of improved anti-inflammatory NR3C1 agonists. Contrary to DUSP1 being a major effector of glucocorticoid action (4, 38), the current data suggests a transient, more minor, role for DUSP1 compared with DUSP1-independent mechanisms of repression. Therefore, whereas the up-regulation of DUSP1 by novel NR3C1 receptor modulators has correlated with repressive or anti-inflammatory effects (49, 50), this may not hold for all such agonists. The current data emphasize the need to continue the characterization of glucocorticoid-induced effector responses and to explore their roles relative to known effectors such as DUSP1. These findings are currently restricted to a specific number of genes in A549 cells. Therefore further testing with additional inflammatory genes, in different cell types and different inducing agents is required. Only by fully understanding the detailed nature of the anti-inflammatory response can we hope to achieve rational improvements in anti-inflammatory NR3C1 agonist therapies.
Supplementary Material
Acknowledgments
Real-time PCR was performed by virtue of an equipment and infrastructure grant from the Canadian Fund of Innovation (CFI) and the Alberta Science and Research Authority.
This work was supported in part by Canadian Institutes of Health Research Grants MOP 68828 and 125918 (to R. N.), a studentship from the Lung Association of Alberta and the North West Territories (to S. S.), and AstraZeneca and GlaxoSmithKline (to R. N.).

This article contains supplemental Tables S1 and S2.
- FKBP
- FK506-binding protein
- Ad5
- adenovirus serotype 5
- NF
- nuclear factor
- DUSP1
- dual-specificity phosphatase 1
- ANOVA
- analysis of variance.
REFERENCES
- 1. Barnes P. J. (2006) Corticosteroids: the drugs to beat. Eur. J. Pharmacol. 533, 2–14 [DOI] [PubMed] [Google Scholar]
- 2. Barnes P. J. (2008) Immunology of asthma and chronic obstructive pulmonary disease. Nat. Rev. Immunol. 8, 183–192 [DOI] [PubMed] [Google Scholar]
- 3. Schäcke H., Döcke W. D., Asadullah K. (2002) Mechanisms involved in the side effects of glucocorticoids. Pharmacol. Ther. 96, 23–43 [DOI] [PubMed] [Google Scholar]
- 4. Newton R. (2014) Eur. J. Pharmacol. 724, 231–236 [DOI] [PubMed] [Google Scholar]
- 5. Stechschulte L. A., Sanchez E. R. (2011) FKBP51-a selective modulator of glucocorticoid and androgen sensitivity. Curr. Opin. Pharmacol. 11, 332–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Beato M., Chávez S., Truss M. (1996) Transcriptional regulation by steroid hormones. Steroids 61, 240–251 [DOI] [PubMed] [Google Scholar]
- 7. De Bosscher K., Vanden Berghe W., Haegeman G. (2003) The interplay between the glucocorticoid receptor and nuclear factor-κB or activator protein-1: molecular mechanisms for gene repression. Endocr. Rev. 24, 488–522 [DOI] [PubMed] [Google Scholar]
- 8. Ito K., Barnes P. J., Adcock I. M. (2000) Glucocorticoid receptor recruitment of histone deacetylase 2 inhibits interleukin-1β-induced histone H4 acetylation on lysines 8 and 12. Mol. Cell. Biol. 20, 6891–6903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Newton R. (2000) Molecular mechanisms of glucocorticoid action: what is important? Thorax 55, 603–613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Clark A. R., Belvisi M. G. (2012) Maps and legends: the quest for dissociated ligands of the glucocorticoid receptor. Pharmacol. Ther. 134, 54–67 [DOI] [PubMed] [Google Scholar]
- 11. Stellato C. (2004) Post-transcriptional and nongenomic effects of glucocorticoids. Proc. Am. Thorac. Soc. 1, 255–263 [DOI] [PubMed] [Google Scholar]
- 12. Ristimäki A., Narko K., Hla T. (1996) Down-regulation of cytokine-induced cyclooxygenase-2 transcript isoforms by dexamethasone: evidence for post-transcriptional regulation. Biochem. J. 318, 325–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Newton R., Seybold J., Kuitert L. M., Bergmann M., Barnes P. J. (1998) Repression of cyclooxygenase-2 and prostaglandin E2 release by dexamethasone occurs by transcriptional and post-transcriptional mechanisms involving loss of polyadenylated mRNA. J. Biol. Chem. 273, 32312–32321 [DOI] [PubMed] [Google Scholar]
- 14. Newton R., Holden N. S. (2007) Separating transrepression and transactivation: a distressing divorce for the glucocorticoid receptor? Mol. Pharmacol. 72, 799–809 [DOI] [PubMed] [Google Scholar]
- 15. Kassel O., Sancono A., Krätzschmar J., Kreft B., Stassen M., Cato A. C. (2001) Glucocorticoids inhibit MAP kinase via increased expression and decreased degradation of MKP-1. EMBO J. 20, 7108–7116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lasa M., Abraham S. M., Boucheron C., Saklatvala J., Clark A. R. (2002) Dexamethasone causes sustained expression of mitogen-activated protein kinase (MAPK) phosphatase 1 and phosphatase-mediated inhibition of MAPK p38. Mol. Cell Biol. 22, 7802–7811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Clark A. R., Martins J. R., Tchen C. R. (2008) Role of dual specificity phosphatases in biological responses to glucocorticoids. J. Biol. Chem. 283, 25765–25769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhao Q., Shepherd E. G., Manson M. E., Nelin L. D., Sorokin A., Liu Y. (2005) The role of mitogen-activated protein kinase phosphatase-1 in the response of alveolar macrophages to lipopolysaccharide: attenuation of proinflammatory cytokine biosynthesis via feedback control of p38. J. Biol. Chem. 280, 8101–8108 [DOI] [PubMed] [Google Scholar]
- 19. Chi H., Barry S. P., Roth R. J., Wu J. J., Jones E. A., Bennett A. M., Flavell R. A. (2006) Dynamic regulation of pro- and anti-inflammatory cytokines by MAPK phosphatase 1 (MKP-1) in innate immune responses. Proc. Natl. Acad. Sci. U.S.A. 103, 2274–2279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Salojin K. V., Owusu I. B., Millerchip K. A., Potter M., Platt K. A., Oravecz T. (2006) Essential role of MAPK phosphatase-1 in the negative control of innate immune responses. J. Immunol. 176, 1899–1907 [DOI] [PubMed] [Google Scholar]
- 21. Zhao Q., Wang X., Nelin L. D., Yao Y., Matta R., Manson M. E., Baliga R. S., Meng X., Smith C. V., Bauer J. A., Chang C. H., Liu Y. (2006) MAP kinase phosphatase 1 controls innate immune responses and suppresses endotoxic shock. J. Exp. Med. 203, 131–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hammer M., Mages J., Dietrich H., Servatius A., Howells N., Cato A. C., Lang R. (2006) Dual specificity phosphatase 1 (DUSP1) regulates a subset of LPS-induced genes and protects mice from lethal endotoxin shock. J. Exp. Med. 203, 15–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Abraham S. M., Lawrence T., Kleiman A., Warden P., Medghalchi M., Tuckermann J., Saklatvala J., Clark A. R. (2006) Antiinflammatory effects of dexamethasone are partly dependent on induction of dual specificity phosphatase 1. J. Exp. Med. 203, 1883–1889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Maier J. V., Brema S., Tuckermann J., Herzer U., Klein M., Stassen M., Moorthy A., Cato A. C. (2007) Dual specificity phosphatase 1 knockout mice show enhanced susceptibility to anaphylaxis but are sensitive to glucocorticoids. Mol. Endocrinol. 21, 2663–2671 [DOI] [PubMed] [Google Scholar]
- 25. Issa R., Xie S., Khorasani N., Sukkar M., Adcock I. M., Lee K. Y., Chung K. F. (2007) Corticosteroid inhibition of growth-related oncogene protein-α via mitogen-activated kinase phosphatase-1 in airway smooth muscle cells. J. Immunol. 178, 7366–7375 [DOI] [PubMed] [Google Scholar]
- 26. Quante T., Ng Y. C., Ramsay E. E., Henness S., Allen J. C., Parmentier J., Ge Q., Ammit A. J. (2008) Corticosteroids reduce IL-6 in ASM cells via up-regulation of MKP-1. Am. J. Respir. Cell Mol. Biol. 39, 208–217 [DOI] [PubMed] [Google Scholar]
- 27. Dauletbaev N., Eklove D., Mawji N., Iskandar M., Di Marco S., Gallouzi I. E., Lands L. C. (2011) Down-regulation of cytokine-induced interleukin-8 requires inhibition of p38 mitogen-activated protein kinase (MAPK) via MAPK phosphatase 1-dependent and -independent mechanisms. J. Biol. Chem. 286, 15998–16007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. King E. M., Holden N. S., Gong W., Rider C. F., Newton R. (2009) Inhibition of NF-κB-dependent transcription by MKP-1: transcriptional repression by glucocorticoids occurring via p38 MAPK. J. Biol. Chem. 284, 26803–26815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Newton R., King E. M., Gong W., Rider C. F., Staples K. J., Holden N. S., Bergmann M. W. (2010) Glucocorticoids inhibit IL-1β-induced GM-CSF expression at multiple levels: roles for the ERK pathway and repression by MKP-1. Biochem. J. 427, 113–124 [DOI] [PubMed] [Google Scholar]
- 30. King E. M., Chivers J. E., Rider C. F., Minnich A., Giembycz M. A., Newton R. (2013) Glucocorticoid repression of inflammatory gene expression shows differential responsiveness by transactivation- and transrepression-dependent mechanisms. PLoS ONE 8, e53936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Szczepankiewicz B. G., Kosogof C., Nelson L. T., Liu G., Liu B., Zhao H., Serby M. D., Xin Z., Liu M., Gum R. J., Haasch D. L., Wang S., Clampit J. E., Johnson E. F., Lubben T. H., Stashko M. A., Olejniczak E. T., Sun C., Dorwin S. A., Haskins K., Abad-Zapatero C., Fry E. H., Hutchins C. W., Sham H. L., Rondinone C. M., Trevillyan J. M. (2006) Aminopyridine-based c-Jun N-terminal kinase inhibitors with cellular activity and minimal cross-kinase activity. J. Med. Chem. 49, 3563–3580 [DOI] [PubMed] [Google Scholar]
- 32. Newton R., Cambridge L., Hart L. A., Stevens D. A., Lindsay M. A., Barnes P. J. (2000) The MAP kinase inhibitors, PD098059, UO126 and SB203580, inhibit IL-1β-dependent PGE2 release via mechanistically distinct processes. Br. J. Pharmacol. 130, 1353–1361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Holden N. S., Catley M. C., Cambridge L. M., Barnes P. J., Newton R. (2004) ICAM-1 expression is highly NF-κB-dependent in A549 cells: no role for ERK and p38 MAPK. Eur. J. Biochem. 271, 785–791 [DOI] [PubMed] [Google Scholar]
- 34. Brondello J. M., Brunet A., Pouysségur J., McKenzie F. R. (1997) The dual specificity mitogen-activated protein kinase phosphatase-1 and -2 are induced by the p42/p44MAPK cascade. J. Biol. Chem. 272, 1368–1376 [DOI] [PubMed] [Google Scholar]
- 35. Byon J. C., Dadke S. S., Rulli S., Kusari A. B., Kusari J. (2001) Insulin regulates MAP kinase phosphatase-1 induction in Hirc B cells via activation of both extracellular signal-regulated kinase (ERK) and c-Jun-N-terminal kinase (JNK). Mol. Cell. Biochem. 218, 131–138 [DOI] [PubMed] [Google Scholar]
- 36. Hu J. H., Chen T., Zhuang Z. H., Kong L., Yu M. C., Liu Y., Zang J. W., Ge B. X. (2007) Feedback control of MKP-1 expression by p38. Cell Signal. 19, 393–400 [DOI] [PubMed] [Google Scholar]
- 37. Junttila M. R., Li S. P., Westermarck J. (2008) Phosphatase-mediated crosstalk between MAPK signaling pathways in the regulation of cell survival. FASEB J. 22, 954–965 [DOI] [PubMed] [Google Scholar]
- 38. Korhonen R., Moilanen E. (2014) Basic Clin. Pharmacol. Toxicol. 114, 24–36 [DOI] [PubMed] [Google Scholar]
- 39. Arthur J. S., Ley S. C. (2013) Mitogen-activated protein kinases in innate immunity. Nat. Rev. Immunol. 13, 679–692 [DOI] [PubMed] [Google Scholar]
- 40. Turpeinen T., Nieminen R., Moilanen E., Korhonen R. (2010) Mitogen-activated protein kinase phosphatase-1 negatively regulates the expression of interleukin-6, interleukin-8, and cyclooxygenase-2 in A549 human lung epithelial cells. J. Pharmacol. Exp. Ther. 333, 310–318 [DOI] [PubMed] [Google Scholar]
- 41. Brooks S. A., Blackshear P. J. (2013) Tristetraprolin (TTP): interactions with mRNA and proteins, and current thoughts on mechanisms of action. Biochim. Biophys. Acta 1829, 666–679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. King E. M., Kaur M., Gong W., Rider C. F., Holden N. S., Newton R. (2009) Regulation of tristetraprolin expression by interleukin-1β and dexamethasone in human pulmonary epithelial cells: roles for nuclear factor-κB and p38 mitogen-activated protein kinase. J. Pharmacol. Exp. Ther. 330, 575–585 [DOI] [PubMed] [Google Scholar]
- 43. Huotari N., Hömmö T., Taimi V., Nieminen R., Moilanen E., Korhonen R. (2012) Regulation of tristetraprolin expression by mitogen-activated protein kinase phosphatase-1. APMIS 120, 988–999 [DOI] [PubMed] [Google Scholar]
- 44. Ayroldi E., Riccardi C. (2009) Glucocorticoid-induced leucine zipper (GILZ): a new important mediator of glucocorticoid action. FASEB J. 23, 3649–3658 [DOI] [PubMed] [Google Scholar]
- 45. Chang T. S., Kim M. J., Ryoo K., Park J., Eom S. J., Shim J., Nakayama K. I., Nakayama K., Tomita M., Takahashi K., Lee M. J., Choi E. J. (2003) p57KIP2 modulates stress-activated signaling by inhibiting c-Jun NH2-terminal kinase/stress-activated protein kinase. J. Biol. Chem. 278, 48092–48098 [DOI] [PubMed] [Google Scholar]
- 46. Kaur M., Chivers J. E., Giembycz M. A., Newton R. (2008) Long-acting β2-adrenoceptor agonists synergistically enhance glucocorticoid-dependent transcription in human airway epithelial and smooth muscle cells. Mol. Pharmacol. 73, 203–214 [DOI] [PubMed] [Google Scholar]
- 47. Rider C. F., King E. M., Holden N. S., Giembycz M. A., Newton R. (2011) Inflammatory stimuli inhibit glucocorticoid-dependent transactivation in human pulmonary epithelial cells: rescue by long-acting β2-adrenoceptor agonists. J. Pharmacol. Exp. Ther. 338, 860–869 [DOI] [PubMed] [Google Scholar]
- 48. Kelly M. M., King E. M., Rider C. F., Gwozd C., Holden N. S., Eddleston J., Zuraw B., Leigh R., O'Byrne P. M., Newton R. (2012) Corticosteroid-induced gene expression in allergen-challenged asthmatic subjects taking inhaled budesonide. Br. J. Pharmacol. 165, 1737–1747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Joanny E., Ding Q., Gong L., Kong P., Saklatvala J., Clark A. R. (2012) Anti-inflammatory effects of selective glucocorticoid receptor modulators are partially dependent on up-regulation of dual specificity phosphatase 1. Br. J. Pharmacol. 165, 1124–1136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Vollmer T. R., Stockhausen A., Zhang J. Z. (2012) Anti-inflammatory effects of mapracorat, a novel selective glucocorticoid receptor agonist, is partially mediated by MAP kinase phosphatase-1 (MKP-1). J. Biol. Chem. 287, 35212–35221 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.