

Abstract
An exhaustive search for shortly spaced repeats in 74 bacterial chromosomes reveals that they are much more numerous than is usually acknowledged. These repeats were divided into five classes: close repeats (CRs), tandem repeats (TRs), simple sequence repeats (SSRs), spaced interspersed direct repeats, and “others.” CRs are widespread and constitute the most abundant class, particularly in coding sequences. The other classes are less frequent, but each individual element shows a higher potential for recombination, when the number of repeats and their distances are taken into account. SSRs and TRs are more frequent in pathogens, as expected given their role in contingency loci, but are also widespread in the other bacteria. The analysis of CRs shows that they have an important role in the evolution of genomes, namely by generating duplications and deletions. Several cases compatible with a significant role of small CRs in the formation of large repeats were detected. Also, gene deletion in Buchnera correlates with repeat density, suggesting that CRs may lead to sequence deletion in general and genome reductive evolution of obligatory intracellular bacteria in particular. The assembly of these results indicates that shortly spaced repeats are key players in the dynamics of genome evolution.
Genotypic variation in bacteria arises in many different ways, such as duplications or deletions of genetic material, horizontal transfer, or point mutations. Recombination plays a major role in many such events, as it is related to phage integration (King and Richardson 1986), horizontal transfer (Rayssiguier et al. 1989; Rocha et al. 1999), major chromosomal rearrangements (Hill and Gray 1988; Hughes 1999), fast adaptation of outer membrane proteins in pathogens (Moxon et al. 1994), and repair of stalled replication forks (Kuzminov 2001). One typically classifies intrachromosomal recombination into homologous recombination (dependent of RecA), site-specific recombination (dependent on specific recombinases), and illegitimate recombination (for reviews, see Michel 1999; Bzymek and Lovett 2001). Both homologous and illegitimate recombination depend to a certain extent on the similarity between the two copies of the repeat that pair in the recombination process. However, the exact mechanisms involved are quite different. Homologous recombination occurs between large repeats that may be very far apart in the chromosome by the action of RecA and a set of alternative pathways (for review, see Smith 1988). Illegitimate recombination is RecA-independent, but acts almost only between closely spaced repeats (Bzymek and Lovett 2001).
Different types of recombination have different effects on the genome dynamics. Because the frequency of homologous recombination does not seem to depend on the distance between the repeated sequences, it frequently results in large changes of the chromosomal structure (e.g., rearrangements), which may or may not be counterselected (Hughes 1999). Illegitimate recombination, by taking place between small repeats at short distances changes the chromosomal structure at a more local level (although this can have global consequences; e.g., see Rocha et al. 2002). Here, I concentrate on the analysis of the potential for illegitimate recombination in bacterial genomes.
Illegitimate recombination is thought to occur by at least two different mechanisms: slipped-mispair and single-strand annealing. Slipped-mispair occurs at replication pauses, which may lead to the dissociation of the newly synthesized strand from its template and pairing with the other close repeat sequence. Upon continuation of replication, such an event can lead to restoration of the original sequence, to conversion, or to deletion or duplication of one copy of the repeat and the intervening sequence (Levinson and Gutman 1987). Single-strand annealing starts by a DNA double-strand break. The following exonucleolytic degradation of the double-stranded ends allows the pairing of exposed complementary single-stranded sequences. Subsequent ligation leads to a deleted sequence (Michel 1999). Probably because these mechanisms are only favorable at short distances between the repeats, there is a strong negative dependence of the recombination frequency on the distance between the repeats (Chédin et al. 1994; Lovett et al. 1994). Also, because in both cases there should be a stable pairing between the two repeats, there is a strong dependence of recombination frequency on the length of the repeats (Peeters et al. 1988; Pierce et al. 1991). Experimental studies on different bacteria and phages have shown a significant number of recombination events for 8-nt repeats at a distance of 987 bp (Albertini et al. 1982), for 18-nt repeats at 2313 bp (Chédin et al. 1994), for 24-nt repeats at 1741 bp (Singer and Westlye 1988), and for 100-nt repeats up to 7000 bp (Lovett et al. 1994).
Repeats capable of engaging in illegitimate recombination have been typically divided into four classes of repeats. Close repeats (CRs) are short repeats (>8–10 nt) separated by a spacer of several nucleotides (Rocha and Blanchard 2002). Spacer interspersed direct repeats (SPIDRs) are multiple-copy CRs, separated by spacers that are not repeated (Jansen et al. 2002). Simple sequence repeats (SSRs) are tandem repeats of small motifs, typically from 1 to 5 nt (van Belkum et al. 1998). TRs are tandem repeats of larger elements (i.e., with motifs larger than 5 nt). Although the distinction between TRs and SSRs is somewhat artificial, it is frequently found in the literature. It reflects the emphasis that has been given to the study of SSRs and their role in pathogenicity (De Bolle et al. 2000) and bacterial typing (van Belkum et al. 1998). No equivalent focus has been made on the study of the other types of shortly spaced repeats, although recent reports suggest the widespread existence of these elements and their importance (Jansen et al. 2002; Oliver et al. 2002; Pericone et al. 2002; Rocha and Blanchard 2002; Rocha et al. 2002). Thus, I undertook an analysis aiming at identifying these types of repeats in bacterial genomes and their relative abundance.
A substantial number of methods have been developed to analyze different types of shortly spaced repeats, given some criteria of similarity, length, and repeatability (Karlin et al. 1988; Hancock and Armstrong 1994; Dsouza et al. 1997; Rivals et al. 1997; Sagot and Myers 1998; Benson 1999). These methods work well, but are too specific of a given type of repeat and rely on different algorithmic, statistical, and probabilistic bases. Because it is difficult to compare their out-puts in a single comparative analysis, their usage is impracticable in the framework of this study. Hence, I opted to perform an analysis that groups together all types of repeats in the same statistical formalism, allowing the comparison of their relative abundance.
I started by defining a conservative threshold of 1000 nt as the maximal allowed distance between repeats to engage at a significant rate into illegitimate recombination. The minimal length of repeats capable of recombining seems to depend on the sequence composition and the genome. Thus, I decided to use a statistical threshold, regarding repeats as significant if their probability of occurrence in a window of 1000 nt is less than 0.001. Because this minimal length is always larger than 12 nt, this probably leads to a small underestimation of the potential for illegitimate recombination. Then I used a set of procedures aimed at classifying these repeats into the four classes cited above. Some elements, for example, highly degenerated SSRs or TRs, are difficult to group, and therefore they were classed in a fifth class named “others.” The frequencies of these elements were then analyzed, compared between genomes, and put into relation with duplications and deletions of genetic material.
RESULTS AND DISCUSSION
Distribution of Shortly Spaced Repeats in Bacteria
Methodological Remarks
Repeats were filtered by applying a statistical test providing the minimal significant length beyond which one does not expect to find repeated sequences in a 1000-nt window (P < 0.001, Karlin and Ost 1985; see Methods). This test accounts for the sequence length and nucleotide composition, but not for multiple testing nor for higher-order biases, such as dinucleotide and codon usage (Karlin et al. 1997). To test whether these effects were causing an important systematic error in the analysis, I validated the method by simulation. For this, I selected three genomes with peculiar G+C contents—U. urealyticum (26% G+C), E. coli (51% G+C), and C. crescentus (67% G+C)—and containing the smaller density of observed repeats for their class of G+C content (thus rendering the test conservative). For each sliding window in each genome I counted the number of repeats (O), then I simulated 1000 random sequences with the same length and composition in trinucleotides and counted the number of occurring repeats (E). The results of these simulations indicate that the ratio of observed/expected (O/E) number of repeats is much larger than 1 (7.18 for U. urealyticum, 10.7 for E. coli, and 9.3 for C. crescentus), confirming a relatively small number of false positives. Also, from a biological point of view, since illegitimate recombination was found to proceed with repeats larger than 8 nt, the “false positives” repeats of 12 or 13 nt are still expected to engage in illegitimate recombination.
Abundance of Repeats
Here, 116,034 of two-copy strictly identical repeats were identified in the set of 74 chromosomes. After the clustering of multiple repeats and their classification, this represents a total of 32,500 repeated elements (Table 1). Each element may contain one or more repeats. The numbers of elements and of repeats are extremely variable among genomes, although in general the relative abundance of the different classes correlates well with each other and with the genome length (Table 2). The sole exception concerns SPIDRs, probably because most genomes have few or none of these elements.
Table 1.
General Results of the Analysis of 74 Bacterial Chromosomes
| SSR
|
TR
|
SPIDR
|
Others
|
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Chromosome | Length (kb) | CR E = R | E | R | E | R | E | R | E | R |
| A. pernix | 1670 | 224 | 3 | 3 | 9 | 9 | 3 | 953 | 8 | 22 |
| A. tumefaciens 1 | 2842 | 342 | 1 | 1 | 12 | 12 | 0 | 0 | 6 | 27 |
| A. tumefaciens 2 | 2075 | 239 | 4 | 4 | 11 | 12 | 0 | 0 | 8 | 115 |
| A. tumefaciens 3 | 543 | 26 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | |
| A. aeolicus | 1551 | 177 | 3 | 4 | 8 | 7 | 0 | 0 | 14 | 79 |
| A. fulgidus | 2178 | 222 | 3 | 4 | 8 | 8 | 3 | 1783 | 5 | 25 |
| B. halodurans | 4202 | 332 | 11 | 20 | 22 | 92 | 3 | 676 | 20 | 279 |
| B. subtilis | 4215 | 330 | 2 | 4 | 16 | 68 | 0 | 0 | 15 | 66 |
| B. burgdorferi | 911 | 71 | 2 | 3 | 5 | 94 | 0 | 0 | 1 | 3 |
| B. melitensis 1 | 2117 | 243 | 8 | 12 | 17 | 51 | 0 | 0 | 5 | 16 |
| B. melitensis 2 | 1178 | 131 | 1 | 3 | 10 | 21 | 0 | 0 | 0 | 0 |
| B. aphidicola Sg | 641 | 71 | 4 | 6 | 3 | 5 | 0 | 0 | 4 | 10 |
| B. aphidicola Ap | 641 | 64 | 7 | 11 | 2 | 3 | 0 | 0 | 4 | 9 |
| C. crescentus | 4017 | 665 | 26 | 34 | 37 | 78 | 0 | 0 | 41 | 280 |
| C. jejuni | 1641 | 229 | 3 | 3 | 10 | 17 | 0 | 0 | 7 | 31 |
| C. muridarum | 1069 | 54 | 3 | 32 | 4 | 17 | 0 | 0 | 3 | 19 |
| C. pneumoniae | 1230 | 105 | 5 | 6 | 6 | 18 | 0 | 0 | 5 | 19 |
| C. tepidum | 2155 | 246 | 6 | 38 | 22 | 161 | 1 | 720 | 7 | 42 |
| C. trachomatis | 1046 | 79 | 1 | 7 | 5 | 12 | 0 | 0 | 2 | 7 |
| C. acetobutylicum | 3941 | 364 | 19 | 117 | 33 | 319 | 0 | 0 | 21 | 342 |
| C. perfringens | 3031 | 275 | 10 | 84 | 16 | 125 | 0 | 0 | 8 | 91 |
| D. radiodurans 1 | 2649 | 470 | 9 | 9 | 29 | 36 | 0 | 0 | 23 | 162 |
| D. radiodurans 2 | 412 | 71 | 1 | 1 | 4 | 55 | 0 | 0 | 4 | 25 |
| E. coli K12 | 4639 | 383 | 8 | 45 | 27 | 297 | 2 | 108 | 19 | 181 |
| E. coli O157:H7 | 5528 | 549 | 7 | 22 | 55 | 370 | 0 | 0 | 31 | 220 |
| F. nucleatum | 2174 | 351 | 6 | 4 | 9 | 15 | 0 | 0 | 26 | 280 |
| H. influenzae | 1830 | 210 | 18 | 249 | 26 | 71 | 0 | 0 | 10 | 45 |
| Halobacterium NRC-1 | 2014 | 423 | 12 | 12 | 13 | 39 | 0 | 0 | 21 | 99 |
| H. pylori | 1668 | 343 | 38 | 261 | 46 | 161 | 0 | 0 | 28 | 122 |
| L. lactis | 2366 | 384 | 3 | 5 | 16 | 125 | 0 | 0 | 27 | 172 |
| L. innocua | 3011 | 368 | 3 | 27 | 11 | 107 | 1 | 39 | 30 | 1383 |
| L. monocytogenes | 2945 | 354 | 2 | 45 | 12 | 1397 | 0 | 0 | 27 | 203 |
| M. acetivorans | 5751 | 1363 | 87 | 288 | 735 | 3600 | 2 | 698 | 165 | 1914 |
| M. jannaschii | 1665 | 244 | 4 | 13 | 13 | 13 | 6 | 836 | 21 | 380 |
| M. kandleri | 1695 | 152 | 2 | 4 | 3 | 3 | 0 | 0 | 8 | 244 |
| M. loti | 7036 | 1158 | 22 | 120 | 73 | 3906 | 0 | 0 | 67 | 691 |
| M. mazei | 4096 | 891 | 149 | 657 | 558 | 2044 | 2 | 1554 | 132 | 1073 |
| M. thermoautotrophicum | 1751 | 321 | 4 | 8 | 24 | 39 | 2 | 2290 | 18 | 113 |
| M. genitalium | 580 | 60 | 17 | 71 | 2 | 58 | 0 | 0 | 6 | 72 |
| M. leprae | 3268 | 159 | 27 | 102 | 98 | 129 | 0 | 0 | 1 | 2 |
| M. pneumoniae | 816 | 137 | 8 | 15 | 18 | 71 | 0 | 0 | 5 | 42 |
| M. pulmonis | 964 | 116 | 23 | 134 | 4 | 1505 | 0 | 0 | 10 | 43 |
| M. tuberculosis | 4412 | 605 | 32 | 375 | 80 | 292 | 2 | 391 | 122 | 3405 |
| N. meningitidis | 2184 | 717 | 37 | 683 | 29 | 1075 | 1 | 134 | 86 | 942 |
| Nostoc PCC7120 | 6414 | 814 | 51 | 554 | 679 | 2615 | 2 | 378 | 118 | 1846 |
| P. multocida | 2257 | 217 | 4 | 62 | 11 | 229 | 2 | 906 | 11 | 92 |
| P. aeruginosa | 6264 | 1076 | 8 | 48 | 42 | 408 | 0 | 0 | 58 | 461 |
| P. abyssi | 1765 | 143 | 5 | 9 | 1 | 2 | 2 | 505 | 5 | 52 |
| P. aerophilum | 2222 | 200 | 5 | 8 | 5 | 5 | 2 | 1186 | 10 | 264 |
| P. horikoshii | 1739 | 151 | 3 | 3 | 2 | 2 | 5 | 1558 | 7 | 45 |
| R. solanacearum | 3716 | 617 | 21 | 131 | 26 | 98 | 0 | 0 | 41 | 540 |
| R. conorii | 1269 | 114 | 4 | 89 | 8 | 95 | 0 | 0 | 1 | 5 |
| R. prowazekii | 1112 | 43 | 1 | 1 | 8 | 13 | 0 | 0 | 0 | 0 |
| S. typhimurium | 4857 | 379 | 16 | 123 | 19 | 220 | 2 | 691 | 23 | 244 |
| S. meliloti | 3654 | 476 | 19 | 144 | 28 | 186 | 0 | 0 | 29 | 153 |
| S. aureus | 2815 | 417 | 7 | 73 | 25 | 949 | 0 | 0 | 40 | 356 |
| S. coelicolor | 8668 | 1148 | 138 | 418 | 416 | 1098 | 0 | 0 | 103 | 772 |
| S. pneumoniae | 2161 | 300 | 7 | 8 | 38 | 3653 | 0 | 0 | 13 | 76 |
| S. pyogenes | 1852 | 177 | 4 | 6 | 13 | 40 | 0 | 0 | 12 | 93 |
| S. solfataricus | 2992 | 253 | 6 | 12 | 22 | 70 | 5 | 5619 | 6 | 38 |
| S. tokodaii | 2695 | 240 | 3 | 5 | 14 | 23 | 5 | 6232 | 15 | 78 |
| Synechocystis sp | 3573 | 472 | 10 | 61 | 19 | 475 | 0 | 0 | 25 | 183 |
| T. acidophilum | 1565 | 68 | 0 | 0 | 3 | 3 | 1 | 570 | 1 | 3 |
| T. maritime | 1861 | 162 | 3 | 479 | 3 | 2 | 4 | 387 | 7 | 58 |
| T. tengcongensis | 2689 | 248 | 6 | 88 | 10 | 10 | 3 | 4063 | 2 | 4 |
| T. volcanium | 1585 | 61 | 0 | 0 | 2 | 2 | 2 | 280 | 1 | 6 |
| T. pallidum | 1138 | 79 | 3 | 2 | 7 | 60 | 0 | 0 | 4 | 37 |
| U. urealyticum | 752 | 72 | 14 | 25 | 6 | 48 | 0 | 0 | 2 | 4 |
| V. cholerae 1 | 2961 | 193 | 10 | 42 | 18 | 248 | 0 | 0 | 8 | 58 |
| V. cholerae 2 | 1072 | 119 | 0 | 0 | 13 | 45 | 0 | 0 | 23 | 520 |
| X. axonopodis | 5176 | 864 | 41 | 91 | 162 | 573 | 1 | 165 | 49 | 337 |
| X. campestris | 5076 | 895 | 49 | 185 | 159 | 850 | 0 | 0 | 77 | 925 |
| X. fastidiosa | 2679 | 359 | 38 | 239 | 66 | 898 | 0 | 0 | 25 | 254 |
| Y. pestis | 4654 | 532 | 39 | 130 | 221 | 833 | 0 | 0 | 34 | 233 |
The table displays the length of the chromosome, and the results for CR, SSR, TR, SPIDR, and “Others” classes of repeats. For each class, the number of elements (E) and the number of the repeats those elements contain (R) are displayed. For CR, these two quantities are similar (by definition).
Table 2.
Pearson's Correlations Between the Abundances of the Different Elements and Chromosome Length (CL)
| CR | SSR | TR | Others | SPIDR | |
|---|---|---|---|---|---|
| CL | 0.87 | 0.58 | 0.59 | 0.70 | NS |
| CR | 0.69 | 0.68 | 0.87 | NS | |
| SSR | 0.82 | 0.79 | NS | ||
| TR | 0.82 | NS | |||
| Others | NS |
NS stands for correlations not significantly different from zero.
Comparative Analysis of Classes
Table 1 can be analyzed in two fundamentally different ways, by looking at either the number of elements or the number of repeats per element in each class (see also Fig. 1). The number of elements indicates the number of potential recombination hotspots in the genome. The number of repeats per element adds information concerning the number of couples capable of engaging in recombination inside each element. Thus, it provides a measure of the intensity of recombination. When analyzing data on CRs, the two quantities are equivalent, but for the other classes they differ widely, because a typical TR, SSR, or SPIDR element has a substantial amount of multiple repeats. The majority of elements belong to CR in all chromosomes. Although CR elements exhibit lower recombination intensities, the sum of their repeats is larger than that of any other class in 37 of the 74 chromosomes. TR is typically the second most abundant class of repeats in bacterial genomes, and the largest one concerning elements containing multiple repeats, well ahead of SSR (which is less frequent than TR in 64 of the 74 chromosomes). The class “others” is constituted by very degenerate TR or SSR repeats and some small SPIDR elements. The elements of this class resemble the cryptic simplicity of certain DNA regions that have been proposed to play an important role in sequence variation (Tautz et al. 1986).
Figure 1.

The output of reputer consists of two-copy repeats that are classed into elements after being clustered together regarding their position in the chromosome and their overlapping with other repeats. CRs are composed of shortly spaced nonoverlapping nontandem repeats. SPIDRs contain multiple nontandem occurrences of repeats, whose spacers show no significant similarity. TRs/SSRs are composed of tandem motifs, which in SSRs are smaller than 6 nt, whereas in TRs they are larger. The elements that cannot be unambiguously classified are grouped as “others.” The repeatability profiles measure the extent of repeatability of each region of the element elsewhere in the element (see Methods).
Spaced Interspersed Direct Repeats (SPIDRs)
SPIDR elements are rare, but they exhibit high levels of recombination intensity because each of them consists of large numbers of repeats. They contain large runs (>10) of repeats averaging 30 nt, separated by spacers averaging 36 nt (Fig. 2), and they include the majority of repeats in 13 chromosomes. SPIDRs were searched without any a priori condition regarding their sequence, periodicity, or pattern. Still, all of the 64 identified elements seem to belong to the SPIDR family recently reviewed (Jansen et al. 2002), and identified by sequence similarity and repeatability pattern. This strongly suggests that large multiple-copy close repeats separated by spacers dissimilar in sequence but similar in length are a characteristic of this sole family, whose origin is unlikely to rely on slippage processes.
Figure 2.

Typical profiles of TRs (top panel), SPIDRs (bottom), and “others” (middle). The curves indicate the pattern matching of a 10-nt oligonucleotide (accepting up to two mismatches) in the remaining sequence. The black and gray boxes indicate the highly similar and degenerate repeats, respectively.
Distribution of Repeats Relative to the Presence of Genes
The repeats are distributed heterogeneously in the genome regarding the position of coding sequences (Table 3). The O/E abundance of CRs in genes, taking into account the genome gene density, is 0.97. Although this is significantly smaller than 1 (P < 0.01, signed-rank test), the difference does not seem very important. For the remaining classes this difference is much more relevant. A clear preference for intergenic regions is revealed by O/E values varying from 0.48 (Others) to 0.03 (for SPIDRs). One should take the exact values of this analysis with care, because repeated regions suffer frequent recombination events, and are very prone to sequencing and annotation errors. However, the difference between CRs and the other classes seems sufficiently important to be taken as biologically relevant. As a consequence, recombination among CRs will more often than not affect coding sequences, whereas variation of the other repeats affects predominantly regulatory regions.
Table 3.
Comparative Analysis Between Different Classes
| Repeats | Median length | Genes (O/E) | Elements | Repeats | RRP | RRI |
|---|---|---|---|---|---|---|
| CR | 16 | 0.97 | 25207 | 25207 | 3279 | 0.13 |
| SSR | 23 | 0.41 | 878 | 6582 | 2415 | 2.75 |
| TR | 28 | 0.30 | 4461 | 30290 | 10092 | 2.26 |
| SPIDR | 29 | 0.03 | 64 | 32722 | 1142 | 17.8 |
| Others | 22 | 0.48 | 1849 | 21233 | 2373 | 1.28 |
Median length of repeats; O/E, number of repeats in coding sequences; number of elements and repeats; corrected relative recombination potential (RRP), and corrected relative recombination intensity (RRI = RRP/number of elements), taking into account spacer length.
Correcting the Potential of Recombination for Spacer Length
These results add to previous works indicating an important role for the recombination between CRs to generate genetic diversity (Levinson and Gutman 1987; Achaz et al. 2002; Rocha et al. 2002). However, the frequency of illegitimate recombination is strongly dependent on the distance between the copies of the repeats, which is much larger in CRs (436 nt), than in the other classes. Because this effect may cancel out the relatively larger amount of CR, I tried to take into account the distance between repeats to build a corrected index of recombination potential. This was done by modeling the frequency of recombination in function of spacer length, using available data (Chédin et al. 1994) and then by assigning to each repeat a value of relative recombination potential (RRP) varying from 1 (tandem repeats) to 0 (spacers > 1000 nt; see Methods and additional material). The frequency of illegitimate recombination is also dependent on the repeat length (Bi and Liu 1996), but equally detailed data for an equivalent modeling of this effect is unavailable. The average length of repeats in the different classes varies by a factor of less than 2 (Table 3), whereas spacer length varies from 1 nt up to 1000 nt. Thus, neglecting the repeat length factor will probably have a minor impact in the comparison between the different classes of repeats.
As expected, the classes where repeats are closer, such as TR and SSR, exhibit relatively larger recombination potentials per repeat (Table 3). In this analysis, CR becomes a less preeminent class, although still the most represented in 15 of 74 genomes. One can also consider a measure of corrected recombination intensity (RRI) defined as the average RRP per element. This clearly shows that SSRs, TRs, and especially SPI-DRs are elements with very high recombination potentials, relative to CR (Table 3). The class of “others” has an intermediary behavior in this respect, probably because these repeats are degenerate and scattered on large regions.
This analysis confirms the intuition of biologists. SSRs and TRs are highly recombinogenic loci, relatively rare in bacteria and associated with intergenic regions. On the other hand, CRs are mostly in genes, possess a smaller potential for recombination, but as a class they may affect a much larger number of genes.
Shortly Spaced Repeats and Bacterial Pathogenicity
To test the role of shortly spaced repeats on sequence variation strategies implicated in pathogenesis, I classed all bacteria according to their pathogenic character and examined this in relation to the abundance of repeats in the genomes. The relative abundance of TRs varies very significantly among Bacteria, and pathogenesis seems partly responsible for it. In E. coli, for example, the strain K12 has less than half the number of TRs of the strain O157:H7 (27 vs. 55 elements, respectively), and one might be tempted to correlate such variations with the virulence of the latter. Although the genomes with larger numbers of such repeats (S. coelicolor, M. acetivorans, M. mazei, and Nostoc PCC7120) are not pathogenic, and closely related pathogens are not known in these groups, pathogens have on average higher densities of TRs (medians of 44 and 31 TR/Mb, P < 0.01, Wilcoxon test). Although SSR are rarer in most bacteria, they are abundant in the genomes of pathogens such as Clostridia and Haemophilus (Table 1). SSRs have commonly been regarded as major elements in the dynamics of the adaptation of bacterial pathogens (Deitsch et al. 1997). Indeed, the density of SSRs in pathogenic bacteria is 50% larger than in the other genomes (P < 0.005, Wilcoxon test). SSRs are composed of repeats of motifs of length from 1 to 5 nt, among which trinucleotides are overrepresented (36%), and nucleotides and dinucleotides underrepresented (10% and 7%, respectively). The densities of CRs, SPIDRs, or “others” were found to be independent of the pathogenic character of the Bacteria harboring them. Although SSR and TR elements are widespread among Bacteria, these results tend to confirm their roles in sequence variation of fast-adapting bacterial pathogens. The abundance of SSRs and TRs in intergenic regions is also in agreement with their role in contingency loci, by changing regulatory regions leading to variations on the expression patterns of genes relevant for pathogenicity (Moxon et al. 1994).
Close Repeats and the Generation of Duplications
Given the large abundance of CRs, I tried to identify potential roles for CRs in bacterial evolution. Slippage between CRs can lead to the duplication of the elements and the intervening sequences, thus producing large tandem duplications. It has been suggested that such events, followed by chromosomal rearrangements, are at the origin of repeats and duplicated genes of Eukaryotes and Bacteria (Levinson and Gutman 1987; Achaz et al. 2001, 2002). Interestingly, not only absolute numbers of CRs, but also CR density (number of CR/Kb) is well correlated with genome length (Spearman's rho=0.26, P < 0.05).
To identify recent cases of tandem duplications, I searched the genomes for close repeats repeated three times in strictly identical copies (for which Karlin's P < 0.001). Then I selected the elements for which the two spacers were similar in sequence (>90%) and had similar length (±5%). Further, I eliminated the elements followed by a region similar to the spacer (sequence similarity >70%). Despite these conservative thresholds, and the instability of such large tandem repeats, I identified such elements in 16 genomes (data not shown). These elements are composed of two large tandem repeats, but attentive inspection reveals that they end by a copy of the repeat which is at their initial positions (Fig. 3). This corresponds to an amplicon, which may then proceed to further duplication, and eventually be stabilized either by selection for a new function or by chromosomal rearrangements resulting in the separation of the repeats (Romero and Palacios 1997).
Figure 3.

Examples of the generation of tandem repeats by recombination between close repeats. (A) Dot-plot of a triple close repeat (black boxes) of 19 nt, with duplicated spacers of 62 nt (gray boxes). This repeat corresponds to the positions 842830 to 843000 of the genome of M. pulmonis. Dots correspond to a minimum of 19 matches for an oligonucleotide of length 25. (B) A SPIDR element that has probably been engaged in illegitimate recombination. This element comes from regions 236560 to 238180 of the M. jannaschii genome.
Although SPIDR elements have probably not originated from slipped mispair, they may be targeted by illegitimate recombination once they have arisen, just like CRs. To test whether they give rise to tandem duplications or to deletions, I compared the elements found in E. coli and S. enterica. One SPIDR exists near the iap gene in the E. coli K12 genome (Nakata et al. 1989). This element is deleted in E. coli O157:H7, even though the flanking genes are maintained. Since this element also exists in both strains of S. enterica, it has probably been deleted in O157:H7 by illegitimate recombination. This element is smaller in S. enterica typhi than in S. enterica typhimurium. Whether this corresponds to a deletion in the former or a multiplication by whatever mechanism creates SPIDR in the latter is difficult to determine. Still, illegitimate recombination between SPIDR can also give rise to duplications of the intervening genetic material. I found several cases of this. In one particularly remarkable case in M. jannaschii, the element's length was 50% enlarged by recombination that resulted in a tandem repeat inside the SPIDR element (Fig. 3). Thus, even if the mechanism of creation of SPIDR elements remains elusive, once they exist they seem to be targeted by illegitimate recombination, as any other close repeat.
Close Repeats, Gene Disintegration, and Genome Reduction
The genomes of non free-living bacteria suffer a process of genome reduction that is characterized by a loss of a significant part of nonessential genes, which can be compensated by the interaction with the host (Ochman and Moran 2001). Such genes become pseudogenes by point mutations and then they suffer multistep single-gene deletion, in such a way that the gene seems to have been surgically removed. This has been termed “gene disintegration” (Andersson and Andersson 2001; Silva et al. 2001). A bias of deletion versus insertion has been proposed to explain genome reduction and the high coding density of bacterial genomes (Lawrence et al. 2001; Mira et al. 2001). Because evidence of the mechanism of such bias is still elusive and because recombination between close repeats may induce deletions of genetic material, I put forward an analysis aimed at testing the association between the existence of large amounts of CR and the propensity of genes to delete.
This analysis was done using two genomes of Buchnera aphidicola (Sg and Ap) currently available (Shigenobu et al. 2000; Tamas et al. 2002). These genomes diverged around 50 million years ago, but they retain the same gene order and very few differences in terms of gene content. This stability is probably due to the lack of elements capable of disrupting the chromosomal structure, such as insertion sequences, large repeats, and probably even efficient homologous recombination (they contain the RecBCD system but lack RecA). Also, they are not subject to horizontal transfer (Ochman and Moran 2001), and therefore differences in gene content are mostly attributable to deletions.
I took B. aphidicola Ap as a reference and identified all genes that lacked a clear ortholog in B. aphidicola Sg. This includes 44 genes that are either absent, or under the form of pseudogenes or partially deleted genes. Then I tested whether these genes have more close repeats (larger than 9nt) than the average genes of B. aphidicola Ap. Indeed, these genes contain significantly more close repeats (averages of 20 and 14 repeats/kb, respectively, P < 0.005, Wilcoxon test). The opposite analysis, that is, the analysis of the genes deleted in B. aphidicola Ap and present in B. aphidicola Sg, also reveals that deleted genes have more repeats than nondeleted genes in the other genome (average of 20 and 17 repeats/kb, respectively). Thus, it is likely that close repeats, by promoting small deletions, play an important role in gene disintegration in bacterial genomes.
Conclusion
Most experimental work in bacterial recombination has been done using E. coli, its phages, or B. subtilis, which constitutes a problem when it comes to comparing different genomes, because one is implicitly obliged to assume that the different bacterial groups behave in a similar way. This is probably reasonable, because the studies of the two very distantly related bacteria B. subtilis and E. coli have given equivalent results in what concerns the dependence of illegitimate recombination on spacer and repeat lengths (Chédin et al. 1994; Lovett et al. 1994). Furthermore, the general features of illegitimate recombination are well supported by the systematic failure of genetic screens to reveal its dependence on any given gene (Bzymek and Lovett 2001). Nevertheless, some genes influence illegitimate recombination rates, such as DNA Pol III (Saveson and Lovett 1997) and mismatch repair (Heale and Petes 1995). The composition of the latter is widely variable in genomes, suggesting that some bacteria might be more prone to illegitimate recombination than others. Several bacteria lacking the MutSHL system, such as Mycoplasmas (Rocha and Blanchard 2002) and H. pylori (Saunders et al. 1998), show many antigenic variation strategies involving tandem repeats. In E. coli, single-strand annealing is thought to be inefficient due to the efficient degradation by the RecBCD enzyme (Bzymek and Lovett 2001), but RecBCD is absent in some bacterial genomes (e.g., in Rickettsia). Finally, illegitimate recombination also depends on the repeat's composition (Eckert and Yan 2000) and chromosome location (Trinh and Sinden 1991). Further studies in bacteria other than B. subtilis and E. coli will be required to allow the introduction of such constraints in comparative studies.
Despite the deficiencies of any study aiming at comparing genomes for which little experimental information is available, it seems clear that all bacteria contain a significant potential for variation by illegitimate recombination. Somewhat unexpectedly, SSR is rarely the class containing the largest fraction of the illegitimate recombination potential in bacterial genomes, even among pathogenic bacteria. TRs, SSRs, and SPIDRs are mostly confined to intergenic sequences, thus potentially modifying gene expression by altering regulatory elements such as promoters. The positive correlation between TR and SSR density and the pathogenic character of bacteria is likely to be a consequence of this.
The CR class has the smallest recombination potential per repeat, but includes the majority of the elements in all genomes. Because CRs are mostly in coding sequences, their recombination may play an important role in the duplication of protein motifs. CRs can also be at the origin of large tandem repeats, which, if rearrangements occur in the genome, turn into distant repeats. In the latter case they become protected from illegitimate recombination. The comparison of the number of CRs found in this work with the previously published data on the abundance of large distant repeats (Achaz et al. 2002) shows a strong correlation between the two (Spearman's ρ = 0.50, P > 0.001). All these observations seem to reinforce early propositions that small close repeats may lead to the formation of many of the large repeats that are observed in the genomes of bacteria and eukarya (Levinson and Gutman 1987; Achaz et al. 2002).
The results obtained regarding the correlation of CR abundance and gene loss in Buchnera indicate that CRs can also play an important role in the deletion of small DNA sequences. A recent study showed that small deletions (<2.5 kb) are predominant in E. coli strains (Ochman and Jones 2000). If this hypothesis holds, one is tempted to speculate that gene deletion in bacteria is the result of two different constraints. First, the likeliness of losing a gene is inversely proportional to the positive selection to which its function is subject. Second, mutational events resulting in the inactivation of genes can take place by point substitution or small deletions, the latter being irreversible in bacteria lacking horizontal transfer. As a consequence, among nonessential genes, the ones containing more repeats are more likely to be deleted.
METHODS
Data
The data on 74 complete bacterial chromosomes (69genomes) were taken from GenBank Genomes (ftp://ftp.ncbi.nih.gov/genomes/Bacteria/). With the exception of E. coli (for which both K12 and O157:H7 strains were analyzed), only one strain was analyzed per species.
Identification of Repeats
I used reputer (Kurtz and Schleiermacher 1999) to search for small exact repeats, and then I filtered out those with copies more than 1000 nt apart. reputer outputs all exact repeats larger than a given length threshold on a sequence. This threshold was computed in sliding windows of 1000 nt (P < 0.001), using the extreme statistics of Karlin and Ost (1985), which ranged from a maximum of 15 nt for highly biased regions in some genomes to a minimum of 13 nt (data not shown). The output of reputer consists of couples of two-copy repeats. Therefore, a degenerate large tandem repeat will appear under the form of multiple copies of two-copy repeats, which must be clustered in order to reconstitute an elementary repeat (see below). These elements are then classed into biologically pertinent classes.
Classification of Close Repeats
Five classes of elements were considered: close repeats (CRs), simple sequence repeats (SSRs), tandem repeats (TRs), spaced interspersed direct repeats (SPIDRs), and “others.” The first step in the classification procedure is the identification of all repeats that are present in the form of only two nontandem and nonoverlapping close copies (which constitute CRs). The remaining elements are then further classed into four categories. The repeats of these elements overlap partly or completely with other occurrences of repeats, thereby constituting what we shall call an element of multiple repeats (Fig. 1). Elements of multiple repeats are defined so that they do not overlap with other elements.
Identification of Multiple Elements
The identification of multiple elements starts by the precise definition of the region occupied by an element. For each repeat I identified all other repeats that partially or totally overlap with its sequence in the chromosome. I then defined the element as the region incorporating the complete set of repeats that share relationships of overlapping with the corresponding spacers (see Fig. 1). Thus, elements are necessarily independent from other elements because their sequences cannot overlap in the chromosome. These regions fall into two very different classes: Either they are constituted by tandem repeats (TRs and SSRs) or they are constituted by repeats separated by a spacer that is not repeated (SPIDRs).
Repeatability Profiles
To classify the elements containing multiple repeats into biologically pertinent classes, I analyzed their inner repeatability. This process starts by taking sliding windows of N nucleotides and a step S on the element's sequence. Each of these windows is then regarded as a pattern, and a pattern search is performed in the entire element accepting at most M mismatches (naturally at least one hit will be found, the one of the window with itself). After sliding the window over the entire element, a graph can be drawn that indicates the number of times a region of the element is repeated elsewhere in the element (see Figs. 1,2).
Classification of Spaced Interspersed Direct Repeats (SPIDRs)
The classification of SPIDRs is straightforward when the repeats are highly conserved: In TRs and SSRs the elements are homogeneously repetitive, whereas in SPIDR the elements are highly repetitive in the repeated regions and not repetitive at all in spacers (Fig. 1). Thus, the procedure starts by computing repeatability profiles (see above). Those of SPIDR are very typical and can be easily identified by eye (see Figs. 1,2). However, because this is not practical when analyzing 74 complete chromosomes, I devised an automated way to classify these elements. Repeatability profiles of all non-CR elements were done using nonoverlapping windows of 5 nt and accepting no mismatches. This provides a list with the number of the element's regions sharing a repeat with each region of the element. Each point is then coded with “+” or “-” regarding whether it is smaller or higher than the average. In SPIDRs, this gives rise to stretches such as “+++---+++---...,” whereas in non-well conserved SSRs it gives rise to nearly random stretches such as “+-++-+--+-.” Thus, I used the serial rank test (Zar 1996) on these stretches to identify aggregative behavior. When aggregation is significant (P < 0.05), the element is inspected by dot-plots before being classed as SPIDR. The analysis by eye of all elements of S. solfataricus, S. tokodaii, and T. acidophilum showed that all SPIDRs were found by the automatic procedure in the three chromosomes. The remaining elements can then be classed as SSR, TR, or Others.
Classification of TR, SSR, and Others
The distinction between TR and SSR allows separating elements that are tandem repeats of motifs from 1 to 5 nucleotides (SSR, e.g., GATGATGAT) from the others (TR). After removing SPIDRs, all elements in which less than 80% of the positions of the element were covered by at least one repeat were removed (see Fig. 1). These elements are classed as “others,” and consist of the elements that cannot be unambiguously classed. Finally, SSRs are differentiated from TRs by identifying the minimal repetitive motif of the elements. If this motif is smaller than 6 nt, then the element is classed as SSR, otherwise it is classed as TR. This is done using TRFinder (Benson 1999).
Regression of the Effect of Spacer Length on the Frequency of Recombination
Several groups have studied the dependency of the spacer length on the frequency of recombination (Chédin et al. 1994; Lovett et al. 1994), and Chédin et al. analyzed the effect of the frequency of recombination between an 18-bp repeat when the spacer was increased from 15 bp to 2.3 kb. Because this falls nicely on the range of repeat length and spacer distances analyzed here (Table 3), I took their data and modeled the frequency of recombination in function of spacer length. In their original work, Chédin et al. suggested the existence of two linear correlations between spacer length and recombination frequencies. One value of correlation would hold for small distances (<338 bp) and another for larger distances (correlations of -0.89 and -0.79, respectively). However, this type of data is usually handled more correctly by a Log transformation (Zar 1996). When this was done, I obtained a single homogeneous correlation of -0.97 between the two variables (P < 0.001). This corresponds to a regression formula (see also Supplemental material):
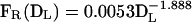 |
where FR is the frequency of recombination and DL is the length of the fragment (i.e., the sum of the length of the spacer and one copy of the repeat).
Definition of Relative Recombination Potential and Relative Corrected Intensity of Recombination Taking Into Account Spacer Length
One can use the above formula to compute FR for each couple of repeats. The division of this value by FR(DL = 18) provides a relative rate of recombination that is corrected in terms of the spacer length. Unfortunately, the range of fragments tested by Chédin et al. (1994) did not include a spacer of zero length (i.e., DL = 18). Because this regression formula does not allow safe extrapolations to the x = 0 region of the curve, I considered arbitrarily that for DL < 33 bp, the relative recombination potential (RRP) is 1. Thus, RRP for each repeat i is:
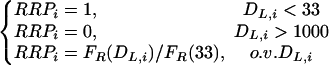 |
The RRP associated with each class of repeats is simply the sum of the RRP of each repeat included in the elements of the class. The relative recombination intensity (RRI) is the sum of the RRP of all repeats in a class of elements divided by the number of elements of that class (N).
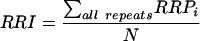 |
Acknowledgments
I thank the many people with whom I've discussed the questions associated with close repeats in bacteria, among them Guillaume Achaz, Alain Blanchard, Eric Coissac, Antoine Danchin, Erick Denamur, Isabelle Gonçalves, Pierre Netter, François Taddei, and Alain Viari. I also thank Guillaume Achaz and Isabelle Gonçalves for comments on the manuscript.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
E-MAIL erocha@abi.snv.jussieu.fr; FAX 33-1-44276312.
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.966203. Article published online before print in May 2003.
Footnotes
[Supplemental material is available online at www.genome.org and at http://www.abi.snv.jussieu.fr/~erocha/closerepeats.html.]
References
- Achaz, G., Netter, P., and Coissac, E. 2001. Study of intrachromosomal duplications among the eukaryote genomes. Mol. Biol. Evol. 18: 2280–2288. [DOI] [PubMed] [Google Scholar]
- Achaz, G., Rocha, E.P.C., Netter, P., and Coissac, E. 2002. Origin and fate of repeats in bacteria. Nucleic Acids Res. 30: 2987–2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albertini, A.M., Hofer, M., Calos, M.P., and Miller, J.H. 1982. On the formation of spontaneous deletions: The importance of short sequence homologies in the generation of large deletions. Cell 29: 319–328. [DOI] [PubMed] [Google Scholar]
- Andersson, J.O. and Andersson, S.G. 2001. Pseudogenes, junk DNA and the dynamics of Rickettsia genomes. Mol. Biol. Evol. 18: 829–839. [DOI] [PubMed] [Google Scholar]
- Benson, G. 1999. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 27: 573–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi, X. and Liu, L.F. 1996. A replication model for DNA recombination between direct repeats. J. Mol. Biol. 256: 849–858. [DOI] [PubMed] [Google Scholar]
- Bzymek, M. and Lovett, S.T. 2001. Instability of repetitive DNA sequences: The role of replication in multiple mechanisms. Proc. Natl. Acad. Sci. 98: 8319–8325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chédin, F., Dervyn, E., Ehrlich, S.D., and Noirot, P. 1994. Frequency of deletion formation decreases exponentially with distance between short direct repeats. Mol. Microbiol. 12: 561–569. [DOI] [PubMed] [Google Scholar]
- De Bolle, X., Bayliss, C.D., Field, D., van de Ven, T., Saunders, N.J., Hood, D.W., and Moxon, E.R. 2000. The length of a tetranucleotide repeat tract in Haemophilus influenzae determines the phase variation rate of a gene with homology to type III DNA methyltransferases. Mol. Microbiol. 35: 211–222. [DOI] [PubMed] [Google Scholar]
- Deitsch, K.W., Moxon, E.R., and Wellems, T.E. 1997. Shared themes of antigenic variation and virulence in bacterial, protozoal and fungal infections. Microbiol. Mol. Biol. Rev. 61: 281–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dsouza, M., Larsen, N., and Overbeek, R. 1997. Searching for patterns in genomic data. Trends Genet. 13: 497–498. [DOI] [PubMed] [Google Scholar]
- Eckert, K.A. and Yan, G. 2000. Mutational analyses of dinucleotide and tetranucleotide microsatellites in Escherichia coli: Influence of sequence on expansion mutagenesis. Nucleic Acids Res. 28: 2831–2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancock, J.M. and Armstrong, J.S. 1994. SIMPLE34: An improved and enhanced implementation for VAX and Sun computers of the SIMPLE algorithm for analysis of clustered repetitive motifs in nucleotide sequences. Comput. Appl. Biosci. 10: 67–70. [DOI] [PubMed] [Google Scholar]
- Heale, S.M. and Petes, T.D. 1995. The stabilization of repetitive tracts of DNA by variant repeats requires a functional DNA mismatch repair system. Cell 83: 539–545. [DOI] [PubMed] [Google Scholar]
- Hill, C.W. and Gray, J.A. 1988. Effects of chromosomal inversion on cell fitness in Escherichia coli K-12. Genetics 119: 771–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes, D. 1999. Impact of homologous recombination on genome organization and stability. In Organization of the prokaryotic genome (ed. R.L. Charlebois), pp. 109–128. ASM Press, Washington DC.
- Jansen, R., van Embden, J.D., Gaastra, W., and Schouls, L.M. 2002. Identification of a novel family of sequence repeats among prokaryotes. OMICS 6: 23–33. [DOI] [PubMed] [Google Scholar]
- Karlin, S. and Ost, F. 1985. Maximal segmental match length among random sequences from a finite alphabet. In Proceedings of the Berkeley Conference in honor of Jerzy Neyman and Jack Kiefer (eds L.M.L. Cam and R.A. Olshen), pp. 225–243. Wadsworth, Belmont, CA.
- Karlin, S., Morris, M., Ghandour, G., and Leung, M.Y. 1988. Efficient algorithms for molecular sequence analysis. Proc. Natl. Acad. Sci. 85: 841–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlin, S., Mrazek, J., and Campbell, A.M. 1997. Compositional biases of bacterial genomes and evolutionary implications. J. Bacteriol. 179: 3899–3913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King, S.R. and Richardson, J.P. 1986. Role of homology and pathway specificity for recombination between plasmids and bacteriophage λ. Mol. Gen. Genet. 204: 141–147. [DOI] [PubMed] [Google Scholar]
- Kurtz, S. and Schleiermacher, C. 1999. REPuter: Fast computation of maximal repeats in complete genomes. BioInformatics 15: 426–427. [DOI] [PubMed] [Google Scholar]
- Kuzminov, A. 2001. DNA replication meets genetic exchange: Chromosomal damage and its repair by homologous recombination. Proc. Natl. Acad. Sci. 98: 8461–8468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence, J.G., Hendrix, R.W., and Casjens, S. 2001. Where are the pseudogenes in bacterial genomes? Trends Microbiol. 9: 535–540. [DOI] [PubMed] [Google Scholar]
- Levinson, G. and Gutman, G.A. 1987. Slipped-strand mispairing: A major mechanism for DNA sequence evolution. Mol. Biol. Evol. 4: 203–221. [DOI] [PubMed] [Google Scholar]
- Lovett, S.T., Gluckman, T.J., Simon, P.J., Sutera, V.A., and Drapkin, P.T. 1994. Recombination between repeats in E. coli by a recA-independent, proximity-sensitive mechanism. Mol. Gen. Genet. 245: 294–300. [DOI] [PubMed] [Google Scholar]
- Michel, B. 1999. Illegitimate recombination in bacteria. In Organization of the prokaryotic genome (ed. R.L. Charlebois), pp. 129–150. ASM Press, Washington DC.
- Mira, A., Ochman, H., and Moran, N.A. 2001. Deletional bias and the evolution of bacterial genomes. Trends Genet. 17: 589–596. [DOI] [PubMed] [Google Scholar]
- Moxon, E.R., Rainey, P.B., Nowak, M.A., and Lenski, R.E. 1994. Adaptive evolution of highly mutable loci in pathogenic bacteria. Curr. Biol. 4: 24–33. [DOI] [PubMed] [Google Scholar]
- Nakata, A., Amemura, M., and Makino, K. 1989. Unusual nucleotide arrangement with repeated sequences in the Escherichia coli K-12 chromosome. J. Bacteriol. 171: 3553–3556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochman, H. and Jones, I.B. 2000. Evolutionary dynamics of full genome content in Escherichia coli. EMBO J. 19: 6637–6643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochman, H. and Moran, N.A. 2001. Genes lost and genes found: Evolution of bacterial pathogenesis and symbiosis. Science 292: 1096–1099. [DOI] [PubMed] [Google Scholar]
- Oliver, A., Baquero, F., and Blázquez, J. 2002. The Mismatch Repair System (mutS, mutL, and uvrD genes) in Pseudomonas aeruginosa: Molecular characterization of naturally occurring mutants. Mol. Microbiol. 43: 1641–1650. [DOI] [PubMed] [Google Scholar]
- Peeters, B.P., de Boer, J.H., Bron, S., and Venema, G. 1988. Structural plasmid instability in Bacillus subtilis: Effect of direct and inverted repeats. Mol. Gen. Genet. 212: 450–458. [DOI] [PubMed] [Google Scholar]
- Pericone, C.D., Bae, D., Shchepetov, M., McCool, T., and Weiser, J.N. 2002. Short-sequence tandem and nontandem DNA repeats and endogenous hydrogen peroxide production contribute to genetic instability of Streptococcus pneumoniae. J. Bacteriol. 184: 4392–4399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce, J.C., Kong, D., and Masker, W. 1991. The effect of the length of direct repeats and the presence of palindromes on deletion between directly repeated DNA sequences in bacteriophage T7. Nucleic Acids Res. 19: 3901–3905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rayssiguier, C., Thaler, D.S., and Radman, M. 1989. The barrier to recombination between E. coli and S. typhimurium is disrupted in mismatch-repair mutants. Nature 342: 396–401. [DOI] [PubMed] [Google Scholar]
- Rivals, E., Delgrange, O., Delahaye, J.P., Dauchet, M., Delorme, M.O., Henaut, A., and Ollivier, E. 1997. Detection of significant patterns by compression algorithms: The case of approximate tandem repeats in DNA sequences. Comput. Appl. Biosci. 13: 131–136. [DOI] [PubMed] [Google Scholar]
- Rocha, E.P.C. and Blanchard, A. 2002. Genomic repeats, genome plasticity and the dynamics of Mycoplasma evolution. Nucleic Acids Res. 30: 2031–2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocha, E.P.C., Danchin, A., and Viari, A. 1999. Analysis of long repeats in bacterial genomes reveals alternative evolutionary mechanisms in Bacillus subtilis and other competent prokaryotes. Mol. Biol. Evol. 16: 1219–1230. [DOI] [PubMed] [Google Scholar]
- Rocha, E.P.C., Matic, I., and Taddei, F. 2002. Over-representation of close repeats in stress response genes: A strategy to increase versatility under stressful conditions? Nucleic Acids Res. 30: 1886–1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero, D. and Palacios, R. 1997. Gene amplification and genomic plasticity in prokaryotes. Annu. Rev. Genet. 31: 91–111. [DOI] [PubMed] [Google Scholar]
- Sagot, M.F. and Myers, E.W. 1998. Identifying satellites and periodic repetitions in biological sequences. J. Comput. Biol. 5: 539–553. [DOI] [PubMed] [Google Scholar]
- Saunders, N.J., Peden, J.F., Hood, D.W., and Moxon, E.R. 1998. Simple sequence repeats in the Helicobacter pylori genome. Mol. Microbiol. 27: 1091–1098. [DOI] [PubMed] [Google Scholar]
- Saveson, C.J. and Lovett, S.T. 1997. Enhanced deletion formation by aberrant DNA replication in Escherichia coli. Genetics 146: 457–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigenobu, S., Watanabe, H., Hattori, M., Sakaki, Y., and Ishikawa, H. 2000. Genome sequence of the endocellular bacterial symbiont of aphids Buchnera sp. APS. Nature 407: 81–86. [DOI] [PubMed] [Google Scholar]
- Silva, F.J., Latorre, A., and Moya, A. 2001. Genome size reduction through multiple events of gene disintegration in Buchnera APS. Trends Genet. 17: 615–618. [DOI] [PubMed] [Google Scholar]
- Singer, B.S. and Westlye, J. 1988. Deletion formation in bacteriophage T4. J. Mol. Biol. 202: 233–243. [DOI] [PubMed] [Google Scholar]
- Smith, G.R. 1988. Homologous recombination in prokaryotes. Microbiol. Rev. 52: 1–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamas, I., Klasson, L., Canback, B., Naslund, A.K., Eriksson, A.S., Wernegreen, J.J., Sandstrom, J.P., Moran, N.A., and Andersson, S.G. 2002. 50 million years of genomic stasis in endosymbiotic bacteria. Science 296: 2376–2379. [DOI] [PubMed] [Google Scholar]
- Tautz, D., Trick, M., and Dover, G.A. 1986. Cryptic simplicity in DNA is a major source of genetic variation. Nature 322: 652–656. [DOI] [PubMed] [Google Scholar]
- Trinh, T.Q. and Sinden, R.R. 1991. Preferential DNA secondary structure mutagenesis in the lagging strand of replication in E. coli. Nature 352: 544–547. [DOI] [PubMed] [Google Scholar]
- van Belkum, A., Scherer, S., van Alphen, L., and Verbrugh. H. 1998. Short-sequence DNA repeats in prokaryotic genomes. Microbiol. Mol. Biol. Rev. 62: 275–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zar, J.H. 1996. Biostatistical analysis. Chapters 13 and 25. Prentice Hall, NJ.