

Background: Lithium exerts a mood-stabilizing effect and inhibits myo-inositol monophosphatase (IMPase).
Results: IMPase mutant mice had impaired jaw formation and mimicked lithium-induced behaviors.
Conclusion: Craniofacial development and brain function require intracellular inositol production.
Significance: This mouse model reveals molecular mechanisms relevant to understanding lithium's efficacy and inositol-mediated developmental processes.
Keywords: Bone, Brain, Circadian Rhythms, Craniofacial Development, Inositol Phosphates, Mouse Genetics, Mutagenesis, Neurodevelopment, Phosphatase
Abstract
myo-Inositol is an essential biomolecule that is synthesized by myo-inositol monophosphatase (IMPase) from inositol monophosphate species. The enzymatic activity of IMPase is inhibited by lithium, a drug used for the treatment of mood swings seen in bipolar disorder. Therefore, myo-inositol is thought to have an important role in the mechanism of bipolar disorder, although the details remain elusive. We screened an ethyl nitrosourea mutant mouse library for IMPase gene (Impa) mutations and identified an Impa1 T95K missense mutation. The mutant protein possessed undetectable enzymatic activity. Homozygotes died perinatally, and E18.5 embryos exhibited striking developmental defects, including hypoplasia of the mandible and asymmetric fusion of ribs to the sternum. Perinatal lethality and morphological defects in homozygotes were rescued by dietary myo-inositol. Rescued homozygotes raised on normal drinking water after weaning exhibited a hyper-locomotive trait and prolonged circadian periods, as reported in rodents treated with lithium. Our mice should be advantageous, compared with those generated by the conventional gene knock-out strategy, because they carry minimal genomic damage, e.g. a point mutation. In conclusion, our results reveal critical roles for intracellular myo-inositol synthesis in craniofacial development and the maintenance of proper brain function. Furthermore, this mouse model for cellular inositol depletion could be beneficial for understanding the molecular mechanisms underlying the clinical effect of lithium and myo-inositol-mediated skeletal development.
Introduction
Lithium salts are used as a first-line drug to treat psychiatric illnesses, particularly bipolar (manic depressive) disorder. Evidence indicates that the mood-stabilizing action of lithium is mediated by inhibiting myo-inositol monophosphatase (IMPase,2 EC 3.1.3.25) activity, thereby inducing intracellular inositol depletion (1–3). IMPase generates myo-inositol, a substrate for the membrane phospholipid phosphatidylinositol, from inositol monophosphate species, which are produced in cells by the multistep dephosphorylation of higher inositol phosphates (“recycling” of inositol) or by the isomerization of d-glucose 6-phosphate (“de novo synthesis” of inositol). Mammalian cells express IMPase 1 and IMPase 2, which are encoded by Impa1/IMPA (4, 5) and Impa2/IMPA2 (6, 7), respectively. Their primary structures are closely related to each other, whereas their three-dimensional structures and enzymatic characteristics vary slightly but significantly (8–10). Importantly, IMPase 1 is more sensitive to lithium inhibition than IMPase 2 in our in vitro assay. This finding strengthens the importance of IMPase 1 as a bona fide target for lithium therapy. However, genetic variations in IMPA2, but not in IMPA1, have been implicated in multiple neuropsychiatric diseases, including schizophrenia (11), bipolar disorder (12, 13), and febrile seizures (14), suggesting a role for the IMPA2 gene in the genetic risk for these illnesses. Although these lines of evidence support critical roles for IMPase and myo-inositol in maintaining normal brain function, it still remains unclear whether and how inositol depletion mediates the therapeutic efficacy of lithium or how intracellular synthesis of myo-inositol impacts the normal development of the brain and other organs. To clarify this point, establishing reliable animal models in which the biological effects of inositol depletion are easily detectable as phenotypes is essential.
The potent mutagen, N-ethyl-N-nitrosourea (ENU) primarily causes single base substitutions. We generated a library of ENU-mutated mice and employed a high throughput screening system to detect mutations. We refer to these techniques collectively as RIKEN ENU-based gene-driven mutagenesis system (RGDMS) (15–18). Using RGDMS to generate and analyze mice with ENU-induced mutations, we uncovered unexpected functions for Impa1 in the development of the skull, as well as in mouse behavior.
EXPERIMENTAL PROCEDURES
Mice
All protocols using animals were approved by the Animal Experiment Committee of RIKEN. Mice were housed in groups under constant temperature and humidity with a 12-h light/dark cycle (lights on at 08:00 h). They had ad libitum access to standard lab chow and water. In the rescue experiment, 2% myo-inositol in drinking water was provided to dams until weaning.
Screening of the ENU Mutant Mouse Library
The basic concept of our mutant mouse screening system (RGDMS) is shown in Fig. 1A. The library for Impa mutant mice was screened by PCR using the primer sets, followed by temperature gradient capillary electrophoresis. The information for used primers is available upon request. The mutations identified by temperature gradient capillary electrophoresis were confirmed by Sanger sequencing. Ova from normal mice were fertilized in vitro with sperm stocks harboring one of the identified missense mutations, and the zygotes were implanted in the uteri of female mice to create heterozygous (G2) mice. Founder mice were crossed at least six times with inbred C57BL/6N females (Japan SLC, Shizuoka, Japan) to dilute the original genetic background and irrelevant mutations. Heterozygous males and females were mated to generate homozygotes. The mutant strains are available from the RIKEN BioResource Center under the RBRC numbers shown in Table 1.
FIGURE 1.

ENU-induced mutations in Impa1. A, schematic representation of the gene-driven ENU mutagenesis system. The library was screened for mutations in the two IMPase genes. B, sequence data for genomic DNA from three Impa1 mutants. Note that each mutant strain (G0) is a heterozygote for the corresponding mutation. Arrows indicate the position of the mutation. C, amino acid sequence alignment of mouse and human IMPases. Identical and conserved amino acids are highlighted in black and gray, respectively. Positions of missense mutations are indicated by colored dots as follows: blue, Impa1 Phe-81; red, Impa1 Thr-95; yellow, Impa1 Thr-96; and green, Impa2 Ile-282.
TABLE 1.
Mutations identified in Impa1 and Impa2 genes
| Gene | Mutant allele | Stock no.a | Position | Exon/intron | Amino acid change | PolyPhen-2 |
|---|---|---|---|---|---|---|
| Impa1 | Rgsc01422 | RBRC-GD000153 | c.85G>A | Intron 1 | ||
| Rgsc01496 | RBRC-GD000155 | c.44T>A | Intron 1 | |||
| Rgsc01494 | RBRC-GD000154 | c.63 + 46G>A | Intron 2 | |||
| Rgsc01411b | RBRC-GD000162 | c.197 + 55C>T | Intron 3 | |||
| Rgsc00210 | RBRC-GD000158 | c.241T>C | Exon 4 | F81L | Benign | |
| Rgsc01846 | RBRC-GD000159 | c.284C>A | Exon 4 | T95K | Probably damaging | |
| Rgsc01827 | RBRC-GD000160 | c.286A>G | Exon 4 | T96A | Possibly damaging | |
| Rgsc01639 | RBRC-GD000157 | c.322T>C | Exon 4 | Synonymous | ||
| Rgsc01835 | RBRC-GD000161 | c.302 + 94G>A | Intron 4 | |||
| Rgsc01418 | RBRC-GD000151 | c.303–4T>A | Intron 4 | |||
| Rgsc01495 | RBRC-GD000156 | c.458–101A>G | Intron 6 | |||
| Rgsc01423 | RBRC-GD000152 | c.566 + 43C>T | Intron 7 | |||
| Impa2 | Rgsc01365 | RBRC-GD000146 | c.103–62C>T | Intron 1 | ||
| Rgsc01384 | RBRC-GD000149 | c.342–29A>G | Intron 3 | |||
| Rgsc01373 | RBRC-GD000150 | c.387 + 21T>C | Intron 4 | |||
| Rgsc01363 | RBRC-GD000147 | c.496 + 85C>T | Intron 5 | |||
| Rgsc01362 | RBRC-GD000148 | c.845T>C | Exon 8 | I282T | Possibly damaging | |
a Mutant strains are available from RIKEN BioResource Center.
b This mutation was found in other G1 mice derived from the same G0 male, indicating that this mutation existed in the G0 male and was transmitted to G1 mice.
Genotyping of Mutant Mice
Genotyping of the Impa1 missense mutants (F81L, T95K, and T96A) was performed as follows: genomic DNA purified from mouse tails was amplified using the primer set forward primer 5′-CTTCATCGTGTTTATTATTATCATCCTC-3′ and reverse primer: 5′-TTGGTCCCTTGCTCCACAGCTTAGA-3′. Amplicons were sequenced directly, using the forward primer and the BigDye Terminator Version 3.1 cycle Sequencing kit (Invitrogen).
In Vitro Enzyme Assays of Recombinant Proteins
IMPase assays were performed as described (8). In brief, a DNA fragment spanning the open reading frame of mouse Impa1 was amplified from mouse brain Marathon-Ready cDNA (TaKaRa Bio, Ohtsu, Japan), using the primer set mIM1-Fw1, 5′-GTGCGCTCGCGCGAGATAATGGCAGAC-3′, and mIM1-Rv1, CCCAGGGACAGCAAGGATGACACTGGA-3′, followed by a second PCR assay with the primer set mIM1cds-Fw-EcoRV, 5′-AGTGAGATATCAATGGCAGACCCTTGGCAGGAG-3′, and mIM1cds-Rv-XhoI, AGTGACTCGAGCTAGCTTTCGTCGTCTCTTTG-3′ (underline sequences denote restriction enzyme recognition sites) to introduce restriction enzyme recognition sites into the PCR product. After digestion with EcoRV and XhoI, the resultant fragment was cloned into the EcoRV/XhoI site of SR-HA, a mammalian expression vector (19) that expresses proteins with N-terminal HA tags, generating SR-HA-mImpa1 WT. Site-directed mutagenesis following the standard DpnI method was performed to produce the three Impa1 mutant constructs as follows: SR-HA-mImpa1 F81L, SR-HA-mImpa1 T95K, and SR-HA-mImpa1 T96A. The nucleotide sequence of each construct was verified. Human kidney-derived HEK293T cells were transfected with one these three constructs or SR-HA (empty vector), using the calcium phosphate method, and then cultured for 2 days. The HA-tagged recombinant proteins were purified from lysates to near-homogeneity using HA antibody affinity beads. Protein preparations were analyzed using SDS-PAGE, followed by silver staining and Western blotting with an anti-HA antibody.
Bone and Neurofilament Staining
T95K heterozygous male and female mice were intercrossed, and pregnant females were euthanized by cervical dislocation at E10.5, E14.5, or E18.5. Fetuses were removed quickly from uteri, and the tissue samples were harvested. E18.5 and E14.5 fetuses were stained with Alcian blue and alizarin red (20) to visualize bones. E10.5 embryos were subjected to whole-mount immunohistochemistry using an anti-neurofilament antibody (clone 2H3, Developmental Studies Hybridoma Bank, Iowa City, IA) and standard procedures to visualize neural fiber organization. Immune complexes were detected using a combination of anti-mouse IgG labeled with horseradish peroxidase and 3,3′-diaminobenzidine. Western blotting of the tissue extract was performed as described (8). Hematoxylin and eosin staining of brain paraffin sections was performed according to a standard procedure.
Behavioral Tests
Mice were 3–5 months old when tested. Behavioral tests were conducted as described (21, 22), except for circadian rhythm, which was evaluated using a wheel-running apparatus (23) with minor modifications. In brief, individual mice were housed in cages (28 cm wide × 12 cm deep × 15 cm high) equipped with a steel wheel (5.5 cm wide × 15 cm in diameter). For the assessment of circadian rhythm, wheel-running activity was monitored using a computer (O'Hara & Co., Tokyo, Japan) during regular light-dark cycles. Light intensity was set to 150 lux. The ClockLab software (Actimetrics, Wilmette, IL) was used to determine the circadian period.
Statistical Analysis
We used Student's t test to compare the two groups subjected to behavioral examinations. When data show a biased distribution, the nonparametric (Mann-Whitney U) test was used. When necessary, data were analyzed using two-way repeated measures analysis of variance followed by post hoc Fisher's protected least significant difference test. The segregation ratio of pup genotypes was tested for significance using the χ2 test. A p value <0.05 was defined as significant.
RESULTS
Screening of the ENU mouse library for mutations in coding exons and flanking intron sequences of Impa1 and Impa2 (Fig. 1A) led to the identification of 17 mutations (12 in Impa1 and 5 in Impa2) (Table 1), of which four were missense (nonsynonymous) mutations (Impa1: F81L, T95K, and T96A; Impa2: I282T) (Fig. 1, B and C, and Table 1). The Ile-282 residue is conserved between human and mouse IMPase homologs (Fig. 1C, green dot), raising the possibility that it impacts the biological function of IMPase 2. However, because Impa2 knock-out (KO) mice lack a detectable phenotype (21), we focused on the three Impa1 mutations. In Fig. 2A, the positions of the mutant amino acid residues are mapped on the crystal structure of the mouse IMPase 1 homodimer (Protein Data Bank 4AS5) (24). The Thr-95 and Thr-96 residues (Fig. 1C, red and yellow dots, respectively) are close to the catalytic site, with Thr-95 being conserved between human and mouse IMPase homologs. The T95K mutation introduces a positive charge, potentially affecting conformation and enzymatic activity. In contrast, the substitution of Leu for Phe-81 may not have a deleterious effect as it is distant from the catalytic site and close to the surface (Fig. 2A). Moreover, Leu occupies a position corresponding to the mouse Impa1 Phe-81 residue in human IMPase 1 and IMPase 2 and in mouse IMPase 2 (Fig. 1C, blue dot). The PolyPhen-2 software tool (25) employs a three-step grading system to predict the impact of given mutations on the biological function of a protein as follows: benign, possibly damaging, and probably damaging. PolyPhen-2 predicted that the three missense mutations, F81L, T95K, and T96A, were benign, probably damaging, and possibly damaging, respectively (Table 1). These analyses strongly support the conclusion that the Lys-95 mutation may exert the strongest effect on biological function.
FIGURE 2.

T95K mutation inactivates the intrinsic activity of IMPase 1. A, three-dimensional structure of the mouse IMPase 1 homodimer (Protein Data Bank code 4AS5) (chains A and B), showing the mutant amino acid residues. The CueMol software was used for the three-dimensional model construction. The Thr-95 and Thr-96 residues are located proximal to the enzyme catalytic site. B, recombinant HA-tagged IMPase 1 proteins purified from cDNA-transfected HEK293 cells were analyzed using SDS-PAGE, followed by silver staining. Western blot (WB) data using an anti-HA antibody is shown in the lower panel. Each sample contained equal loadings of recombinant protein. C, kinetics of phosphatase activity. Enzymatic activity of the T95K mutant (red line) was undetectable. D, lateral views of wild-type control (Wild), heterozygote (Het), and homozygote (Homo) mice. Homozygous T95K mice rarely survived into adulthood.
To test this possibility, wild-type and mutant HA-tagged IMPase 1 recombinant proteins were affinity-purified from cDNA-transfected HEK293T cells (Fig. 2B) and then tested for activity using a published method (8). Consistent with in silico results, the Lys-95 (T95K) mutant lacked detectable activity (Fig. 2C). In contrast, there was no significant difference between the activity of wild-type and Ala-96 (T96A) proteins. Interestingly, the Leu-81 (F81L) mutant produced higher activity compared with the wild-type protein.
Next, we investigated the phenotypes of mice harboring these three mutations. Heterozygous mice (G (generation) 2 mice shown in Fig. 1A) were generated by in vitro fertilization. We detected no abnormalities in the heterozygotes (data not shown). Therefore, they were backcrossed to the inbred C57BL/6N strain for six generations to dilute unrelated mutations on a homogeneous genetic background (Fig. 1A). Heterozygous males and females from the three strains were mated, and the genotypes of the offspring were determined at weaning. T95K heterozygotes grew normally (Fig. 2D), but only one homozygote (1/84 (1.2%)) was identified (Table 2), implying an essential role for IMPase 1 activity in survival. The lone homozygote, which was undersized and weak, died shortly after weaning (data not shown). In contrast, homozygous and F81L and T96A mice were present at the expected segregation ratio without any visible phenotypic changes (Fig. 2D and data not shown). We therefore focused on the Impa1 T95K (Lys-95) strain.
TABLE 2.
Distribution of offspring's genotypes by mating between Impa1 Lys-95 heterozygotes
The abbreviations used are as follows: Wild, wild-type control; Het, heterozygote; and Homo, homozygote mice.
| Stage | Wild | Het | Homo | Sum | χ2 test (df = 1) | |
|---|---|---|---|---|---|---|
| Standard diet during pregnancy and lactation | ||||||
| E10.5 | Observed | 12 | 23 | 11 | 46 | χ2 = 0.0435, p = 0.83 |
| Expected | 11.5 | 23 | 11.5 | |||
| E14.5 | Observed | 9 | 20 | 7a | 36 | χ2 = 0.0667, p = 0.41 |
| Expected | 9 | 18 | 9 | |||
| E18.5 | Observed | 68 | 136 | 30a,b | 204 | χ2 = 18.5, p = 1.69 × 10−5 |
| Expected | 58.5 | 117 | 58.5 | |||
| Weaning | Observed | 30 | 53 | 1 | 84 | χ2 = 25.8, p = 3.81 × 10−7 |
| Expected | 21 | 42 | 21 | |||
| Inositol supplementation during pregnancy and lactation | ||||||
| E18.5 | Observed | 8 | 26 | 12 | 46 | χ2 = 1.48, p = 0.22 |
| Expected | 11.5 | 23 | 11.5 | |||
| Weaning | Observed | 146 | 325 | 127 | 598 | χ2 = 5.73, p = 0.017 |
| Expected | 149.5 | 299 | 149.5 | |||
a All fetuses/embryos show mandible hypoplasia.
b Data include one dead fetus with a normal body size and mandible hypoplasia.
To determine when Lys-95 homozygous pups die, we again intercrossed heterozygous T95K mice and determined the segregation ratios of offspring genotypes at E18.5. There were significantly fewer homozygous embryos at E18.5 than expected (30/204 (14.7%), χ2, p = 1.69 × 10−5) (Table 2). The body sizes of the surviving homozygotes (Fig. 3A, denoted as Homo) ranged from being slightly to significantly smaller than those of wild-type (denoted as Wild) and heterozygous (denoted as Het) littermates. The head to body ratio of homozygotes appeared smaller than wild-type controls (Fig. 3A, and data not shown). Surprisingly, the lower jaws of homozygous fetuses were abnormally shorter (mandibular micrognathia) than those of wild-type controls (Fig. 3, A and B, and data not shown), indicating aberrant craniofacial development. Moreover, homozygotes had poorly developed tongues (Fig. 3, A and B) and mandible hypoplasia, revealed by Alcian blue and alizarin red staining (Fig. 3C). These cranial malformations showed 100% penetrance. The severity of mandible hypoplasia did not correlate with body size (Fig. 3A, and data not shown), supporting the conclusion that the mandible phenotype was not a result of delayed growth. Notably, we found no clear defects in the upper jaw or palate closure of homozygotes (Fig. 3, A and B). In addition to lower jaw malformation, ∼30% of homozygotes exhibited asymmetric sternum-rib fusion (Fig. 3D). Two homozygous pups exhibited exencephaly (data not shown), and one exhibited a cleft palate (data not shown). However, because the number of these phenotypic abnormalities was not statistically significant, we could not eliminate the possibility that the mutations were not causal. The homozygotes that survived until E18.5 stopped moving within a few minutes after Caesarean section delivery, whereas wild-type and heterozygous littermates continued to move and were resuscitated. Because abnormal peripheral neuronal development is a major cause of paralysis after delivery, we examined neural fiber formation in E10.5 embryos by whole-mount immunohistochemistry, with an anti-neurofilament antibody. We found no detectable differences in neural organization between wild-type and homozygous embryos (Fig. 4A). At E10.5, embryo genotypes from heterozygote intercrossing were present at the expected segregation ratios (Table 2), and there were no visible morphological abnormalities in homozygous embryos (Fig. 4A). These data show that the retardation in mandible formation appears slowly after E10.5 in homozygotes. To investigate this point, we examined heterozygote intercrosses at E14.5. Even at this stage, we were clearly able to discriminate homozygotes from wild-type and heterozygous littermates, based on hypoplasia of the lower jaw (Fig. 4B, yellow arrowheads) and the Meckel's cartilage (Fig. 4B, red arrowheads), which is laid down prior to mandible formation in the developmental process. We detected no evidence of biased genotype distribution in offspring at E14.5 (Table 2). Importantly, with the exception of the mandible, the gross architecture of the skull, including the maxilla, appeared to be normal (Fig. 4B). Collectively, these results indicate that the cause of mandible hypoplasia is most likely due to abnormal differentiation of the mandibular process from the first pharyngeal arch.
FIGURE 3.
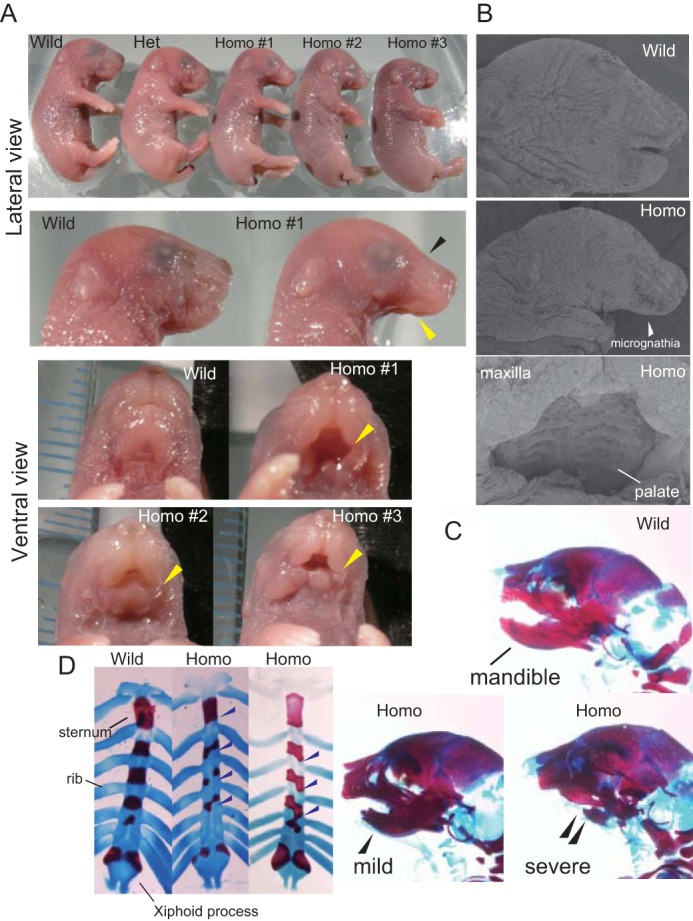
Lys-95 homozygotes exhibit characteristic developmental defects at E18.5. A, E18.5 fetuses from a single litter mating of heterozygotes are shown (top panel). This litter contained one wild-type (Wild), one heterozygote (Het), and three (Homo #1–3) homozygote (Homo) mice. The tips of mouse tails were removed for genotyping. Magnified lateral views of wild-type (Wild) and Homo #1 fetal heads are shown in the 2nd row. Homozygotes show incompletely developed mandibles (yellow arrowheads) and malformed heads (black arrowheads). Ventral views of wild-type, Homo #1 (severe), Homo #2 (mild), and Homo #3 (moderate) mice are shown in the bottom four panels. Hypoplasia of the mandible (yellow arrowheads, micrognathia) is evident in homozygotes. B, scanning electron microscopic images of mouse heads. Homozygous fetuses lack most of the lower jaw and tongue (middle). The head of a wild-type littermate is shown at the top. The palate is formed in homozygotes (bottom). C, skulls of wild-type and homozygote fetuses were visualized using Alcian blue and alizarin red staining. Images show typical wild-type (Wild) and homozygote (Homo) mice, demonstrating mild and severe mandible hypoplasia. Hypoplasia is indicated by arrowheads. D, Alcian blue and alizarin red staining of rib cages. Images show typical wild-type (Wild) and homozygote (Homo) mice. Fusion of ribs to the sternum is asymmetric in E18.5 homozygotes. The bifida sternum is evident in the homozygotes. Blue arrowheads indicate the sites of asymmetric connection of ribs to the sternum.
FIGURE 4.
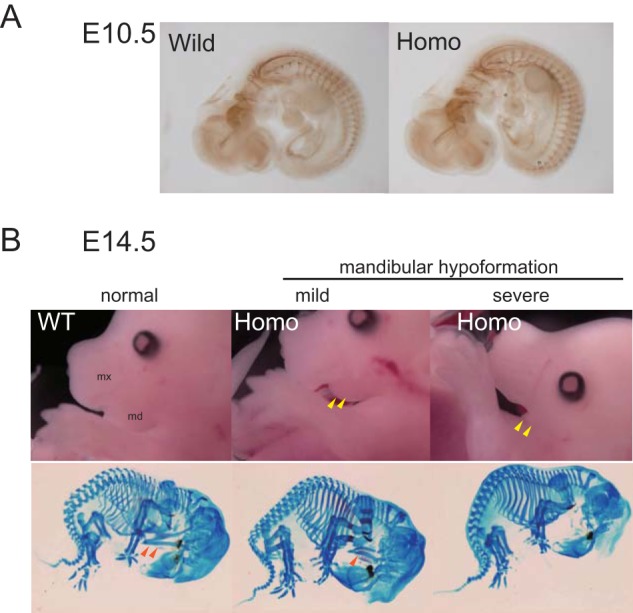
Hypoformation of Meckel's cartilage in Lys-95 homozygotes. A, neural fiber organization of an E10.5 embryo was analyzed in whole mounts by immunochemical staining (2H3, anti-neurofilament antibody). There was no apparent difference between wild-type (Wild) and homozygous T95K (Homo) littermates. B, homozygous embryos show hypoformation of Meckel's cartilage. Typical pictures are shown for wild-type (Wild) and homozygous (Homo) E14.5 mouse embryos. Yellow arrowheads indicate hypoformation of the lower jaw. Bone preparations stained by Alcian blue and alizarin red are also shown. Red arrowheads (Meckel's cartilage) indicate hypoformation of the lower jaw.
To test whether the Lys-95/Lys-95 phenotypes were caused by a reduction in cellular myo-inositol levels, we supplemented the drinking water of heterozygous females with 2% myo-inositol before mating. myo-Inositol supplementation, which continued until weaning (Fig. 5A), significantly enhanced the survival of homozygotes (127/598, 21.2%) (Table 2). The survivors showed no visible abnormalities in craniofacial development (Fig. 5, B and C) or gross brain structure (Fig. 5D). Moreover, we found no homozygous fetuses with malformation of the rib cage (Fig. 5B). Based on these lines of evidence, we postulate a direct relationship between developmental defects in homozygotes and a relative reduction of cellular myo-inositol. The levels of Impa1 mRNA did not differ in various tissues among the three genotype cohorts fed water, instead of myo-inositol after weaning (data not shown), indicating that the mutation had no effect on the transcription or stability of Impa1 mRNA. Homozygotes and wild-type controls consistently expressed similar levels of IMPase 1 protein (Fig. 5E). Neither Impa2 mRNA nor protein levels were elevated to compensate for deficits in IMPase 1 activity (Fig. 5E and data not shown). The rescued homozygotes died from unknown causes before the wild-type controls (Fig. 5F).
FIGURE 5.
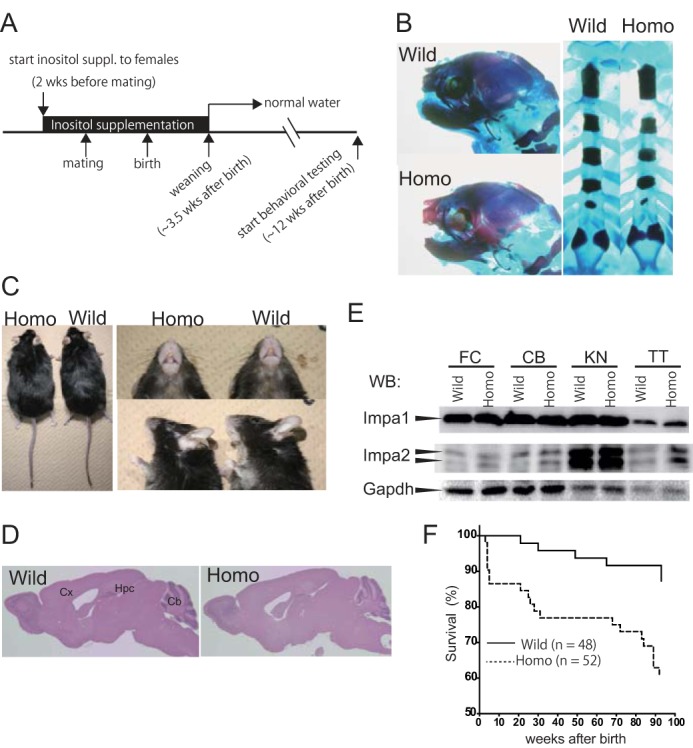
myo-Inositol supplementation during pregnancy rescues the lethality and craniofacial defects of Lys-95 homozygotes. A, schematic of the protocol for myo-inositol supplementation. B, Alcian blue and alizarin red staining of rib cages at E18.5. Typical pictures show wild-type (Wild) and homozygote (Homo) mice. Note that malformation of the jaw or rib cage is not visible in homozygotes. C, there was no gross difference between adult wild-type and homozygote mice (left panel). Abnormal lower jaw morphology was rescued by dietary supplementation with inositol during pregnancy (right panels). Top right, ventral view; bottom right, lateral view. D, there were no gross structural abnormalities in the brains of homozygotes stained with hematoxylin and eosin. Cx, cerebral cortex; Hpc, hippocampus; Cb, cerebellum. E, expression level of the Impa1 protein in homozygotes was comparable with that of wild-type controls. Lysates (40 μg of protein) prepared from K95/K95 and control mice were analyzed by Western blotting (WB) with anti-Impa1, anti-Impa2, or anti-Gapdh antibodies. FC, frontal cortex; CB, cerebellum; KN, kidney, and TT, testis. F, Kaplan-Meier survival analysis of mice rescued with myo-inositol. Homozygotes (Homo) showed normal morphology, but their life spans were shorter than those of wild-type (Wild) controls. Note that after weaning, mice drank water without myo-inositol.
As stated earlier, IMPase 1 is the likely molecular target for lithium when used as a mood stabilizer. Therefore, we explored whether loss of IMPase 1 activity could mimic behavioral changes seen in animals given lithium, using inositol-rescued adult mice (Fig. 5A). We examined mice of both sexes, aged 12 weeks and older, using a comprehensive battery of behavioral tests (Table 3) covering multiple domains of the brain functionality, such as motor function, affective traits, and cognitive and sensorimotor gating functions (26–31). Many researchers have detected antidepressant-like effects for lithium when administered to rodents in various experimental paradigms, including the forced swim and tail suspension tests. Both of these are basic tests to evaluate manic or depressive moods in rodents, where the antidepressant-like effect of lithium is detected as decreased immobility time. Impa1 T95K homozygotes of both sexes exhibited hyperactivity, collectively determined based on the following observations: 1) increased total distances traveled in the dark- and light-box test (Fig. 6A); 2) decreased immobility time in the forced swim test (Fig. 6B); 3) increased entry number in the Y-maze test (Fig. 6C), and 4) reduced freezing behavior on the conditioning day of the fear-conditioning test (Table 3). We also observed a significant increase in the total distance in the elevated plus-maze test and in daytime locomotor activity in the home cage for females, as well as an increased tendency for both measures in males. In addition, we detected increased locomotor activity in the open field test for males (Fig. 6D), whereas there were no significant differences between homozygotes and wild-type controls during the first 10 min of the open field test (Table 3 and Fig. 6D).
TABLE 3.
Summary of behavioral test battery of homozygotes
All the analyses were done using 8–24 mice/genotype. NS means not significant in male and female; ASR means acoustic startle response; CS means conditioned stimuli; US means unconditioned stimuli; PP means prepulse; P means pulse.
| Test | Measure | Difference from wild-type control |
|---|---|---|
| Open field (10 min) | Total distance | NSa |
| Center time (%) | Decreaseda,b | |
| Home cage activity | Total activity (/24 h) | NSa |
| Day time (/12 h) | Increaseda,c | |
| Night time (/12 h) | NSa | |
| Elevated plus maze | Open stay (%) | Increaseda,b |
| Closed stay (%) | Decreaseda,b | |
| Center (%) | NSd | |
| Total distance (cm) | Increaseda,c | |
| Light/dark transition | Light distance (cm) | NSa |
| Dark distance (cm) | Increaseda,e | |
| Total distance (cm) | Increaseda,e | |
| Light time (s) | NSa | |
| Dark time (s) | NSa | |
| Entry numbers | NSe | |
| Latency to dark box (s) | Decreasedb,d | |
| Tail suspension | Immobility (%) | NSa |
| Forced swim | Immobility time (5 min) | Decreaseda,e |
| Y maze | Alteration (%) | NSd |
| Entry numbers | Increasedd,e | |
| Fear conditioning (freezing (%)) | Conditioning (pre-+post-US/CS) | Decreasedd,e |
| Contextual | NSa | |
| Cued (pre-CS) | NSd | |
| Cued (post-CS) | NSd | |
| Prepulse inhibition | 74/120 (PP/P db) (%) | NSf |
| 80/120 (PP/P db) (%) | ||
| 86/120 (PP/P db) (%) | ||
| ASR | at 120 db | NSa |
a Student's t test.
b Difference was detected only in males.
c Difference was detected only in females.
d Mann-Whitney's U test.
e Significant difference was detected in both sexes.
f Repeated measures two-way analysis of variance (genotype effect).
FIGURE 6.

T95K homozygous mutant mice show hyperactivity. A–C, male and female homozygotes were tested in the light-dark (LD) box transition (A), forced swim (B), and Y-maze (C) tests. Data are shown as the mean ± S.E. (n = 10 for each genotype). D, male homozygotes (Homo) were tested in the longer version (60 min) of the open field test. Data are shown as the mean ± S.E. (n = 10 for each genotype).
Because Impa1 is highly expressed in mouse cerebellar Purkinje cells (8), we examined whether Impa1 Lys-95 homozygotes suffered from a motor coordination deficiency. We found that performance in the rotarod test was unaffected (data not shown). Interestingly, ∼5% of older homozygotes (>1.5 years) exhibited epileptic responses when removed from their home cages and placed on the floor (supplemental Movie 1). Typically they recovered within 10 min of being returned to their home cages. Taken together with the behavioral abnormalities of the Impa1 Lys-95 homozygotes, we conclude that Impa1-dependent myo-inositol turnover plays crucial roles in the generation and maintenance of brain function integrity.
A large body of evidence demonstrates a correlation between affective disorders (including bipolar disorder and major depressive disorder) and circadian dysregulation (32, 33), represented by high comorbidity of affective disorders and sleep disturbances. To identify possible effects for the Impa1 T95K mutation on circadian functions, we examined the properties of rest-activity rhythms of homozygotes under entrained and free-running conditions. As lithium prolongs the circadian period (τ) when administered to rodents, this period in homozygotes may be prolonged if lithium targets IMPase 1 in vivo. Our homozygotes showed a normal activity rhythm under regular 12-h light:12-h dark conditions (Fig. 7A). Interestingly, we observed that both the onset and offset (Fig. 7B, red and blue arrowheads, respectively) of the free-running activity rhythms of homozygotes were gradually delayed compared with that of the wild types under constant darkness (DD) (Fig. 7B). This trend was clearly visible between DD cycles 11 and 13 (Fig. 7C). We analyzed this same data set and determined the free-running circadian period, and we found that mutants showed significantly lengthened circadian periods (Fig. 7D). It is highly plausible that the delayed timing of activity rhythms induced by this mutation is the result of prolonged circadian periods. Importantly, a similar prolonging circadian period effect has been reported repeatedly in animals, including rodents treated with lithium, as we will discuss later.
FIGURE 7.

Disturbance of circadian rhythm control in T95K mutants. A, wheel-running activity in 12-h light:12-h dark (LD) cycles was monitored for 2 weeks. Data are shown as the mean without error bars (n = 10 for each genotype). B, wheel-running activity in DD cycles was monitored for 13 days. Data are shown as the mean without error bars. Homozygotes show delayed activity (blue and red arrowheads) at later stages of the test. Red and blue arrowheads indicate delayed onsets and offsets, respectively. C, wheel-running activity between DD cycles 11 and 13. Data are shown as the mean ± S.E. (n = 10 for each genotype). Blue asterisks show significant differences between the two genotypes (p < 0.05, post hoc Fisher's PLSD test following two-way repeated analysis of variance). Time points with p < 0.1 are indicated by green asterisks. D, circadian period length of homozygotes. The circadian period (tau) is determined. Data are shown as the mean ± S.E. *, p < 0.05 (Student's t test, n = 10 for each genotype).
In addition to the beneficial effects on mood, lithium therapy frequently causes multiple side effects, including poisoning, making it essential to monitor and control lithium serum concentrations. Some undesirable effects of lithium can potentially be mediated by inhibiting IMPase. The typical side effects of lithium therapy, including tremor and polyuria, were not exhibited by the homozygote mice (data not shown), and there were no detectable abnormalities in blood cell counts or blood biochemistry (data not shown). Because rescued homozygotes had a shorter life span compared with wild-type controls (Fig. 5F), we cannot exclude the possibility that the premature deaths were related to side effects of lithium therapy.
DISCUSSION
In this study, using the powerful and robust RGDMS system, we demonstrated in mice that IMPase 1 activity is required for normal development of the mandible and rib cage, as well as normal brain function. Homozygous Impa1 mutants (Lys-95) rarely survived until weaning, and none survived longer than 7 min after Caesarean delivery at E18.5. It is highly likely that they would still die shortly after birth following a normal gestation period. Of note, Smit1 (sodium myo-inositol transporter 1) KO mice die of congenital central apnea, caused by abnormal respiratory rhythmogenesis (34, 35). Considering the phenotype of Smit1 KO mice, the lack of IMPase 1 activity in Lys-95 mutants could cause central apnea due to abnormal neuronal development, resulting from reduced intracellular myo-inositol concentrations.
We have already reported the spatial expression patterns of Impa1 and Impa2 in embryonic stages (E9.5 and E10.5) (21), as well as brain expression during early post-natal stages (P7 and P21) (8). Those results showed Impa1 is expressed in various areas of the mouse, including the first brachial arch that generates the lower and upper jaws. Although the maxilla (upper jaw) and palate do not appear to be severely affected, the process of mandibular formation is specifically altered in homozygotes. In fact, Meckel's cartilage, an essential structure for mandibular formation, is poorly developed in homozygotes at E14.5 (Fig. 4B). In conjunction with the observation that Smit1 knock-out mice also display a deficiency in osteogenesis (36), these data support a crucial role for free myo-inositol in bone formation. Intriguingly, we observed no abnormalities in limb formation (Figs. 3A and 4), although the expression of Impa1 was relatively high in limb buds during embryogenesis (21). A possible explanation could be that expression of inositol transporters such as SMIT and HMIT masks the deleterious effect of Impa1 deficiency during limb development. Supporting this idea is the observation that Smit1 knock-out mice have shorter limbs (36). Some genetically engineered mice (e.g. Dlx5/6, Zic2, Chd, and Nog KOs) also show abnormal jaw formation (37–39), and some knock-out animals (Dppa4, Ephrin B1, and EphB2/EphB3) exhibit asymmetric fusion of ribs to the sternum (40, 41). Whether and how these morphogenic genes are involved in inositol or inositol phosphate metabolism remain to be determined.
Here, we show that Impa1 Lys-95 homozygotes were more active in various behavioral tests, compared with their wild-type littermates, demonstrating that IMPase 1 activity is required for normal brain function. In keeping with our results, Cryns et al. (42). reported that Impa1 KO mice are hyperactive in the open field test and exhibit shorter immobility times in the forced swim test compared with control mice. The hyperactivity and altered circadian control seen in our homozygotes were not restored by dietary supplementation with 4% myo-inositol during adulthood (Fig. 8, A–C). Although the simplest explanation may be that loss of cellular inositol in the adult brain does not precipitate behavioral deficits, we cannot exclude the possibility that myo-inositol ingested by these adult animals did not sufficiently penetrate the blood-brain barrier, possibly due to its hydrophilic nature. It should be noted that Cryns et al. (42) did not detect a significant reduction in myo-inositol content within the brains of adult Impa1 knock-out mice supplemented with myo-inositol, up to the stage of weaning.
FIGURE 8.

Inositol supplementation does not alleviate behavioral alterations in homozygote mice. A, schematic presentation of experiment schedules. Inositol was supplied for 6 weeks during the weaning period to rescue mutant mice. After this, rescued mice were raised on normal water for 6 weeks and then maintained with or without 4% myo-inositol. B, open field test. Note that inositol supplementation produced no apparent effect on open field activity in either genotype. C, wheel-running activity between DD cycles 11 and 13 under inositol supplementation. Wheel-running activity is presented as in Fig. 7, C and D.
Lithium salts are a first-line drug therapy for bipolar patients, preventing extreme mood swings. In addition, they are often used to treat refractory depression. When administered to normal mice, lithium promotes antidepressant-like effects in multiple behavioral tests, such as the forced swim and tail suspension tests. As stated earlier, Impa1 Lys-95 homozygotes and Impa1 KO mice (42) also exhibit hyper-locomotion. These results support the idea that inhibition of IMPase 1 activity exerts an antidepressant-like effect, similar to that of lithium in rodents and humans. Although mammals express IMPase 2, its activity was only slightly inhibited by lithium in our in vitro assay system (8), and Impa2 KO mice do not exhibit significant behavioral changes (21, 43). Pretreating mice with lithium lowers the threshold for the convulsant effect of pilocarpine, and Impa1 KO mice show greater sensitivity to pilocarpine (42). The Impa1 Lys-95 homozygotes, despite low penetrance, develop an epileptic phenotype without pretreatment with pilocarpine and lithium. In totality, these results support the conclusion that IMPase 1 may serve as a molecular target for lithium, in the lithium-pilocarpine model.
Molecular genetics studies suggest pathophysiological associations between mood disorders and circadian dysregulation, including abnormal period and phase of rest-activity rhythms (44–49). This is the first study showing a potential role for the free inositol-producing system in the regulation of activity rhythm. We discovered that shutdown of the IMPase 1 pathway led to a prolonged circadian period. Although lithium is known to lengthen the circadian period in various experimental systems, until now, this effect was thought to be a result of lithium's inhibition of GSK3α/β (50–52). Our results put forward the novel possibility that IMPase inhibition, especially that of IMPase 1, may at least in part, through the effects of lithium, alter the circadian rhythm and thus be related to the efficacy of this drug. Although it is still unknown how inositol depletion mediates its biological consequences, especially on brain function, research groups have set about trying to answer this question. First, Andreassi et al. (53) found that Impa1 mRNA is the most abundant transcript in the axons of rat sympathetic neurons and that axon-specific down-regulation of Impa1 mRNA induces axon degeneration. Second, IMPase plays an essential role in maintaining neuronal polarity as demonstrated by studies on the Caenorhabditis elegans gene, ttx-7, which is the only nematode gene to encode an IMPase. Mutants of ttx-7 are defective in thermotaxis due to disruption of membrane polarity in the RIA neuron (54, 55) and can be rescued by enforced expression of human IMPA1 or IMPA2 (21), suggesting that mammalian IMPases and nematode Ttx-7 play similar roles in neurons.
The Impa1 T95K mutant may therefore serve as a useful tool to dissect the mechanistic action of lithium as a mood stabilizer. Moreover, defining the mechanisms underlying its morphological phenotypes may provide new insights into bone development. Finally, this study illustrates the power of the RGDMS platform to decipher gene function based on the introduction of point mutations, compared with more invasive KO techniques.
Acknowledgments
We thank Drs. Shiroishi, Sakuraba, Uchiyama, Sezutsu, Kaneda, and Furuse for their technical support in identifying mutants. We also thank members of the Research Resources Center of RIKEN BSI for their technical support in DNA sequencing, animal care, blood biochemistry, and cell counting; Y. Ishizuka for animal maintenance, and Dr. Akagi (RRC, RIKEN BSI) and D. Inoue (Materials Characterization Support Unit, RIKEN) for their technical support with electron microscopy observations. We are also grateful to Drs. Abe and Fujisawa for useful discussions.
This work was supported by in part by grant-in-aid from the Ministry of Education, Culture, Sports, Science and Technology of Japan (to T. O. and T. Y.), RIKEN BSI funds, grants from the Mitsubishi Pharma Research Foundation (to T. Y.), Astra Zeneca (to T. O.), Takeda Science Foundation (to T. O.), JSPS KAKENHI Grants 15200032 (to Y. G.), 21240043 (to Y. G. and T. M.), and 25241016 (to Y. G.), and grant-in-aid for scientific research on innovative areas (Unraveling the Micro-endophenotypes of Psychiatric Disorders at the Molecular, Cellular and Circuit Levels) from the Ministry of Education, Culture, Sports, Science and Technology (to T. Y.). A portion of this study is the result of the “Development of Biomarker Candidates for Social Behavior” Project carried out under the Strategic Research Program for Brain Sciences from Ministry of Education, Culture, Sports, Science and Technology (to T. Y.).

This article contains supplemental Movie 1.
- IMPase
- myo-inositol monophosphatase
- ENU
- N-ethyl-N-nitrosourea
- RGDMS
- RIKEN ENU-based gene-driven mutagenesis system
- DD
- constant darkness.
REFERENCES
- 1. Berridge M. J., Downes C. P., Hanley M. R. (1989) Neural and developmental actions of lithium: a unifying hypothesis. Cell 59, 411–419 [DOI] [PubMed] [Google Scholar]
- 2. Williams R. S., Cheng L., Mudge A. W., Harwood A. J. (2002) A common mechanism of action for three mood-stabilizing drugs. Nature 417, 292–295 [DOI] [PubMed] [Google Scholar]
- 3. Harwood A. J. (2005) Lithium and bipolar mood disorder: The inositol-depletion hypothesis revisited. Mol. Psychiatry 10, 117–126 [DOI] [PubMed] [Google Scholar]
- 4. Diehl R. E., Whiting P., Potter J., Gee N., Ragan C. I., Linemeyer D., Schoepfer R., Bennett C., Dixon R. A. (1990) Cloning and expression of bovine brain inositol monophosphatase. J. Biol. Chem. 265, 5946–5949 [PubMed] [Google Scholar]
- 5. McAllister G., Whiting P., Hammond E. A., Knowles M. R., Atack J. R., Bailey F. J., Maigetter R., Ragan C. I. (1992) cDNA cloning of human and rat brain myo-inositol monophosphatase. Expression and characterization of the human recombinant enzyme. Biochem. J. 284, 749–754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sjøholt G., Molven A., Løvlie R., Wilcox A., Sikela J. M., Steen V. M. (1997) Genomic structure and chromosomal localization of a human myo-inositol monophosphatase gene (IMPA). Genomics 45, 113–122 [DOI] [PubMed] [Google Scholar]
- 7. Yoshikawa T., Turner G., Esterling L. E., Sanders A. R., Detera-Wadleigh S. D. (1997) A novel human myo-inositol monophosphatase gene, IMP. 18p maps to a susceptibility region for bipolar disorder. Mol. Psychiatry 2, 393–397 [DOI] [PubMed] [Google Scholar]
- 8. Ohnishi T., Ohba H., Seo K. C., Im J., Sato Y., Iwayama Y., Furuichi T., Chung S. K., Yoshikawa T. (2007) Spatial expression patterns and biochemical properties distinguish a second myo-inositol monophosphatase IMPA2 from IMPA1. J. Biol. Chem. 282, 637–646 [DOI] [PubMed] [Google Scholar]
- 9. Arai R., Ito K., Ohnishi T., Ohba H., Akasaka R., Bessho Y., Hanawa-Suetsugu K., Yoshikawa T., Shirouzu M., Yokoyama S. (2007) Crystal structure of human myo-inositol monophosphatase 2, the product of the putative susceptibility gene for bipolar disorder, schizophrenia, and febrile seizures. Proteins 67, 732–742 [DOI] [PubMed] [Google Scholar]
- 10. Fujita S., Ohnishi T., Okuda S., Kobayashi R., Fukuno S., Furuta D., Kikuchi T., Yoshikawa T., Fujita N. (2011) In silico study on the substrate binding manner in human myo-inositol monophosphatase 2. J. Mol. Model. 17, 2559–2567 [DOI] [PubMed] [Google Scholar]
- 11. Yoshikawa T., Kikuchi M., Saito K., Watanabe A., Yamada K., Shibuya H., Nankai M., Kurumaji A., Hattori E., Ishiguro H., Shimizu H., Okubo Y., Toru M., Detera-Wadleigh S. D. (2001) Evidence for association of the myo-inositol monophosphatase 2 (IMPA2) gene with schizophrenia in Japanese samples. Mol. Psychiatry 6, 202–210 [DOI] [PubMed] [Google Scholar]
- 12. Sjøholt G., Ebstein R. P., Lie R. T., Berle J. Ø., Mallet J., Deleuze J. F., Levinson D. F., Laurent C., Mujahed M., Bannoura I., Murad I., Molven A., Steen V. M. (2004) Examination of IMPA1 and IMPA2 genes in manic-depressive patients: association between IMPA2 promoter polymorphisms and bipolar disorder. Mol. Psychiatry 9, 621–629 [DOI] [PubMed] [Google Scholar]
- 13. Ohnishi T., Yamada K., Ohba H., Iwayama Y., Toyota T., Hattori E., Inada T., Kunugi H., Tatsumi M., Ozaki N., Iwata N., Sakamoto K., Iijima Y., Iwata Y., Tsuchiya K. J., Sugihara G., Nanko S., Osumi N., Detera-Wadleigh S. D., Kato T., Yoshikawa T. (2007) A promoter haplotype of the inositol monophosphatase 2 gene (IMPA2) at 18p11.2 confers a possible risk for bipolar disorder by enhancing transcription. Neuropsychopharmacology 32, 1727–1737 [DOI] [PubMed] [Google Scholar]
- 14. Nakayama J., Yamamoto N., Hamano K., Iwasaki N., Ohta M., Nakahara S., Matsui A., Noguchi E., Arinami T. (2004) Linkage and association of febrile seizures to the IMPA2 gene on human chromosome 18. Neurology 63, 1803–1807 [DOI] [PubMed] [Google Scholar]
- 15. Gondo Y., Fukumura R. (2009) ENU-induced mutant mice for a next-generation gene-targeting system. Prog. Brain Res. 179, 29–34 [DOI] [PubMed] [Google Scholar]
- 16. Gondo Y., Fukumura R., Murata T., Makino S. (2009) Next-generation gene targeting in the mouse for functional genomics. BMB Rep. 42, 315–323 [DOI] [PubMed] [Google Scholar]
- 17. Gondo Y., Fukumura R., Murata T., Makino S. (2010) ENU-based gene-driven mutagenesis in the mouse: A next-generation gene-targeting system. Exp. Anim. 59, 537–548 [DOI] [PubMed] [Google Scholar]
- 18. Sakuraba Y., Sezutsu H., Takahasi K. R., Tsuchihashi K., Ichikawa R., Fujimoto N., Kaneko S., Nakai Y., Uchiyama M., Goda N., Motoi R., Ikeda A., Karashima Y., Inoue M., Kaneda H., Masuya H., Minowa O., Noguchi H., Toyoda A., Sakaki Y., Wakana S., Noda T., Shiroishi T., Gondo Y. (2005) Molecular characterization of ENU mouse mutagenesis and archives. Biochem. Biophys. Res. Commun. 336, 609–616 [DOI] [PubMed] [Google Scholar]
- 19. Yamashita A., Ohnishi T., Kashima I., Taya Y., Ohno S. (2001) Human SMG-1, a novel phosphatidylinositol 3-kinase-related protein kinase, associates with components of the mRNA surveillance complex and is involved in the regulation of nonsense-mediated mRNA decay. Genes Dev. 15, 2215–2228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. McLeod M. J. (1980) Differential staining of cartilage and bone in whole mouse fetuses by Alcian blue and alizarin red S. Teratology 22, 299–301 [DOI] [PubMed] [Google Scholar]
- 21. Ohnishi T., Tanizawa Y., Watanabe A., Nakamura T., Ohba H., Hirata H., Kaneda C., Iwayama Y., Arimoto T., Watanabe K., Mori I., Yoshikawa T. (2013) Human myo-inositol monophosphatase 2 rescues the nematode thermotaxis mutant ttx-7 more efficiently than IMPA1: Functional and evolutionary considerations of the two mammalian myo-inositol monophosphatase genes. J. Neurochem. 124, 685–694 [DOI] [PubMed] [Google Scholar]
- 22. Ohnishi T., Watanabe A., Ohba H., Iwayama Y., Maekawa M., Yoshikawa T. (2010) Behavioral analyses of transgenic mice harboring bipolar disorder candidate genes, IMPA1 and IMPA2. Neurosci. Res. 67, 86–94 [DOI] [PubMed] [Google Scholar]
- 23. Kasahara T., Kubota M., Miyauchi T., Noda Y., Mouri A., Nabeshima T., Kato T. (2006) Mice with neuron-specific accumulation of mitochondrial DNA mutations show mood disorder-like phenotypes. Mol. Psychiatry 11, 577–593 [DOI] [PubMed] [Google Scholar]
- 24. Singh N., Halliday A. C., Knight M., Lack N. A., Lowe E., Churchill G. C. (2012) Cloning, expression, purification, crystallization, and x-ray analysis of inositol monophosphatase from Mus musculus and Homo sapiens. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 68, 1149–1152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Adzhubei I. A., Schmidt S., Peshkin L., Ramensky V. E., Gerasimova A., Bork P., Kondrashov A. S., Sunyaev S. R. (2010) A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Arguello P. A., Gogos J. A. (2006) Modeling madness in mice: One piece at a time. Neuron 52, 179–196 [DOI] [PubMed] [Google Scholar]
- 27. Baker M. (2011) Animal models: inside the minds of mice and men. Nature 475, 123–128 [DOI] [PubMed] [Google Scholar]
- 28. Beaulieu J. M., Zhang X., Rodriguiz R. M., Sotnikova T. D., Cools M. J., Wetsel W. C., Gainetdinov R. R., Caron M. G. (2008) Role of GSK3β in behavioral abnormalities induced by serotonin deficiency. Proc. Natl. Acad. Sci. U.S.A. 105, 1333–1338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Clapcote S. J., Lipina T. V., Millar J. K., Mackie S., Christie S., Ogawa F., Lerch J. P., Trimble K., Uchiyama M., Sakuraba Y., Kaneda H., Shiroishi T., Houslay M. D., Henkelman R. M., Sled J. G., Gondo Y., Porteous D. J., Roder J. C. (2007) Behavioral phenotypes of Disc1 missense mutations in mice. Neuron 54, 387–402 [DOI] [PubMed] [Google Scholar]
- 30. Del Pino I., García-Frigola C., Dehorter N., Brotons-Mas J. R., Alvarez-Salvado E., Martínez de Lagrán M., Ciceri G., Gabaldón M. V., Moratal D., Dierssen M., Canals S., Marín O., Rico B. (2013) Erbb4 deletion from fast-spiking interneurons causes schizophrenia-like phenotypes. Neuron 79, 1152–1168 [DOI] [PubMed] [Google Scholar]
- 31. Powell C. M., Miyakawa T. (2006) Schizophrenia-relevant behavioral testing in rodent models: A uniquely human disorder? Biol. Psychiatry 59, 1198–1207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. McClung C. A. (2011) Circadian rhythms and mood regulation: insights from pre-clinical models. Eur. Neuropsychopharmacol. 21, S683–S693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. McClung C. A. (2013) How might circadian rhythms control mood? Let me count the ways. Biol. Psychiatry 74, 242–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Berry G. T., Wu S., Buccafusca R., Ren J., Gonzales L. W., Ballard P. L., Golden J. A., Stevens M. J., Greer J. J. (2003) Loss of murine Na+/myo-inositol cotransporter leads to brain myo-inositol depletion and central apnea. J. Biol. Chem. 278, 18297–18302 [DOI] [PubMed] [Google Scholar]
- 35. Chau J. F., Lee M. K., Law J. W., Chung S. K., Chung S. S. (2005) Sodium/myo-inositol cotransporter-1 is essential for the development and function of the peripheral nerves. FASEB J. 19, 1887–1889 [DOI] [PubMed] [Google Scholar]
- 36. Dai Z., Chung S. K., Miao D., Lau K. S., Chan A. W., Kung A. W. (2011) Sodium/myo-inositol cotransporter 1 and myo-inositol are essential for osteogenesis and bone formation. J. Bone Miner. Res. 26, 582–590 [DOI] [PubMed] [Google Scholar]
- 37. Inoue T., Hatayama M., Tohmonda T., Itohara S., Aruga J., Mikoshiba K. (2004) Mouse Zic5 deficiency results in neural tube defects and hypoplasia of cephalic neural crest derivatives. Dev. Biol. 270, 146–162 [DOI] [PubMed] [Google Scholar]
- 38. Stottmann R. W., Anderson R. M., Klingensmith J. (2001) The BMP antagonists Chordin and Noggin have essential but redundant roles in mouse mandibular outgrowth. Dev. Biol. 240, 457–473 [DOI] [PubMed] [Google Scholar]
- 39. Depew M. J., Lufkin T., Rubenstein J. L. (2002) Specification of jaw subdivisions by Dlx genes. Science 298, 381–385 [DOI] [PubMed] [Google Scholar]
- 40. Madan B., Madan V., Weber O., Tropel P., Blum C., Kieffer E., Viville S., Fehling H. J. (2009) The pluripotency-associated gene Dppa4 is dispensable for embryonic stem cell identity and germ cell development but essential for embryogenesis. Mol. Cell. Biol. 29, 3186–3203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Compagni A., Logan M., Klein R., Adams R. H. (2003) Control of skeletal patterning by ephrinB1-EphB interactions. Dev. Cell 5, 217–230 [DOI] [PubMed] [Google Scholar]
- 42. Cryns K., Shamir A., Van Acker N., Levi I., Daneels G., Goris I., Bouwknecht J. A., Andries L., Kass S., Agam G., Belmaker H., Bersudsky Y., Steckler T., Moechars D. (2008) IMPA1 is essential for embryonic development and lithium-like pilocarpine sensitivity. Neuropsychopharmacology 33, 674–684 [DOI] [PubMed] [Google Scholar]
- 43. Cryns K., Shamir A., Shapiro J., Daneels G., Goris I., Van Craenendonck H., Straetemans R., Belmaker R. H., Agam G., Moechars D., Steckler T. (2007) Lack of lithium-like behavioral and molecular effects in IMPA2 knockout mice. Neuropsychopharmacology 32, 881–891 [DOI] [PubMed] [Google Scholar]
- 44. Artioli P., Lorenzi C., Pirovano A., Serretti A., Benedetti F., Catalano M., Smeraldi E. (2007) How do genes exert their role? Period 3 gene variants and possible influences on mood disorder phenotypes. Eur. Neuropsychopharmacol. 17, 587–594 [DOI] [PubMed] [Google Scholar]
- 45. Lamont E. W., Legault-Coutu D., Cermakian N., Boivin D. B. (2007) The role of circadian clock genes in mental disorders. Dialogues Clin. Neurosci. 9, 333–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. McClung C. A. (2007) Circadian genes, rhythms and the biology of mood disorders. Pharmacol. Ther. 114, 222–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Barnard A. R., Nolan P. M. (2008) When clocks go bad: neurobehavioural consequences of disrupted circadian timing. PLoS Genet. 4, e1000040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kripke D. F., Nievergelt C. M., Joo E., Shekhtman T., Kelsoe J. R. (2009) Circadian polymorphisms associated with affective disorders. J. Circadian Rhythms 7, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Mendlewicz J. (2009) Disruption of the circadian timing systems: Molecular mechanisms in mood disorders. CNS Drugs 23, 15–26 [DOI] [PubMed] [Google Scholar]
- 50. Dokucu M. E., Yu L., Taghert P. H. (2005) Lithium- and valproate-induced alterations in circadian locomotor behavior in Drosophila. Neuropsychopharmacology 30, 2216–2224 [DOI] [PubMed] [Google Scholar]
- 51. Lavoie J., Hébert M., Beaulieu J. M. (2013) Glycogen synthase kinase-3β haploinsufficiency lengthens the circadian locomotor activity period in mice. Behav. Brain Res. 253, 262–265 [DOI] [PubMed] [Google Scholar]
- 52. Yin L., Wang J., Klein P. S., Lazar M. A. (2006) Nuclear receptor Rev-erbα is a critical lithium-sensitive component of the circadian clock. Science 311, 1002–1005 [DOI] [PubMed] [Google Scholar]
- 53. Andreassi C., Zimmermann C., Mitter R., Fusco S., De Vita S., Devita S., Saiardi A., Riccio A. (2010) An NGF-responsive element targets myo-inositol monophosphatase-1 mRNA to sympathetic neuron axons. Nat. Neurosci. 13, 291–301 [DOI] [PubMed] [Google Scholar]
- 54. Kimata T., Tanizawa Y., Can Y., Ikeda S., Kuhara A., Mori I. (2012) Synaptic polarity depends on phosphatidylinositol signaling regulated by myo-inositol monophosphatase in Caenorhabditis elegans. Genetics 191, 509–521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Tanizawa Y., Kuhara A., Inada H., Kodama E., Mizuno T., Mori I. (2006) Inositol monophosphatase regulates localization of synaptic components and behavior in the mature nervous system of C. elegans. Genes Dev. 20, 3296–3310 [DOI] [PMC free article] [PubMed] [Google Scholar]