

Background: β-Amyloid-induced neuron death and degeneration is considered to be central to the pathogenesis of Alzheimer disease.
Results: p53-up-regulated modulator of apoptosis (Puma), a protein of the B-cell lymphoma-2 family, is induced by transcription factor FoxO3a and participates in neuron death in response to β-amyloid.
Conclusion: β-Amyloid-induced neuron death requires induction of Puma.
Significance: Puma could be a potential target for disease therapeutics.
Keywords: Alzheimer Disease, Amyloid, Bcl-2 Family Proteins, Cell Death, Foxo, Neurodegeneration, Neurons, siRNA, Bim, Puma
Abstract
Neurodegeneration underlies the pathology of Alzheimer disease (AD). The molecules responsible for such neurodegeneration in AD brain are mostly unknown. Recent findings indicate that the BH3-only proteins of the Bcl-2 family play an essential role in various cell death paradigms, including neurodegeneration. Here we report that Puma (p53-up-regulated modulator of apoptosis), an important member of the BH3-only protein family, is up-regulated in neurons upon toxic β-amyloid 1–42 (Aβ(1–42)) exposure both in vitro and in vivo. Down-regulation of Puma by specific siRNA provides significant protection against neuron death induced by Aβ(1–42). We further demonstrate that the activation of p53 and inhibition of PI3K/Akt pathways induce Puma. The transcription factor FoxO3a, which is activated when PI3K/Akt signaling is inhibited, directly binds with the Puma gene and induces its expression upon exposure of neurons to oligomeric Aβ(1–42). Moreover, Puma cooperates with another BH3-only protein, Bim, which is already implicated in AD. Our results thus suggest that Puma is activated by both p53 and PI3K/Akt/FoxO3a pathways and cooperates with Bim to induce neuron death in response to Aβ(1–42).
Introduction
Alzheimer disease (AD)2 is an irreversible cognitive malfunctioning of brain that eventually dysregulates the integrity of the nervous system. Widespread synapse and neuron loss in selective areas of the brain are important characteristic features of AD along with other pathophysiological hallmarks, such as β-amyloid (Aβ) plaques and neurofibrillary tangles. The amyloid cascade hypothesis claims a central role of Aβ accumulation in the disease pathogenesis (1). It has also been shown that oligomeric Aβ is the main pathologic species that induces neurodegeneration in vivo and in vitro (2–4). However, downstream effectors of Aβ toxicity still remain elusive.
Accumulating evidence implicates a number of death-associated genes in AD-related neuron death (2, 4, 5). Proteins of the Bcl-2 (B-cell lymphoma 2) family regulate cell death in response to most if not all of the death insults. The Bcl-2 family comprises pro-survival (e.g. Bcl-2 and BclxL), multidomain proapoptotic (e.g. BAX and BAK), and BH3-only proapoptotic (e.g. Bim (Bcl-2-interacting mediator of cell death), Bid, and Puma) proteins (6). Activation of the proapoptotic members BAX and/or BAK is essential for cell death (7). The activity of BAX is controlled by the opposing action of prosurvival members and BH3-only proteins of the family. Recently, essential roles of Bim, Bid, and Puma have been shown in BAX activation and subsequent apoptosis during development (8). More importantly, it has been shown that inhibition of BAX protects neurons from Aβ toxicity in vitro and in vivo (2). Bim has also been shown to be an essential mediator of neuron death, yet knockdown of this molecule provides only transient protection in AD-relevant cell death models (5). This indicates that other death-associated molecules are also necessary in the molecular events of cell death in AD. Interestingly, recently, it has been shown that Puma cooperates with Bim in a critical apoptotic check point in autoreactive thymocytes (9) and in oncogene inactivation-induced apoptosis (10). Puma is induced by various apoptotic stimuli like DNA damage, endoplasmic reticulum stress, aberrant oncogene expression, serum or growth factor deprivation, and oxidative stress (11). However, its role in AD-related neurodegeneration has not yet been explored.
Puma is known to be a key transcriptional target of the tumor suppressor p53, and it carries out the cell death cascade in response to p53 activation (12–16). However, an increasing body of evidence shows that Puma is also regulated in a p53-independent manner (11, 17, 18). In healthy cells, Puma is kept in check by survival signals (19, 20). Inhibiting these signals through growth factor or cytokine withdrawal leads to p53-independent transcriptional activation of Puma. It has been shown that Forkhead transcription factor FoxO3a (Forkhead box, class O3a) is activated and up-regulates Puma in response to such survival factor withdrawal (21, 22). In surviving cells, FoxO3a is phosphorylated by Akt and remains in cytosol bound to 14-3-3 protein. However, in response to deprivation of growth factors or cytokines, it becomes dephosphorylated, translocates into the nucleus, and induces its target genes like Bim, Puma, etc. (22, 23). Recently, we have shown that FoxO3a is activated by multiple post-translational modifications, translocates to the nucleus, and mediates neuron death via Bim in response to Aβ (4). However, whether Puma is a target of activated FoxO3a in Aβ-treated neurons is yet to be discovered.
Because Aβ is widely considered to be the toxic species in AD, a number of drugs targeted to Aβ metabolism are in clinical trials to reduce the Aβ load. Although most of them have failed in clinical trials, targeting Aβ is still considered to be a very useful therapeutic strategy (24). In addition to that, it is believed that a complementary therapy may be necessary to block toxicity of the Aβ that cannot be removed completely. Therefore, it is essential to understand the molecular mechanism of Aβ-induced neuron death. In this study, we have investigated the role of Puma in Aβ-induced neuron death. Our findings suggest that Puma is induced transcriptionally by FoxO3a and cooperates with Bim to induce neuron death in response to Aβ toxicity.
EXPERIMENTAL PROCEDURES
Materials
Aβ(1–42) was purchased from American Peptide. Insulin, progesterone, putrescine, selenium, transferrin, NGF, and poly-d-lysine were purchased from Sigma. Anti-Puma and anti-FoxO3a antibodies were from Cell Signaling Technology. Protein A-agarose and HRP-conjugated secondary antibodies were from Santa Cruz Biotechnology, Inc. LY 294002 and pifithrin-α were from Calbiochem. Lipofectamine 2000, Alexa Fluor 488, Alexa Fluor 568, culture media, and serum were purchased from Invitrogen. Brain tissues of AβPPswe-PS1de9 mice and control littermates were kindly gifted by Dr. Anant B. Patel (Council of Scientific and Industrial Research-Centre for Cellular and Molecular Biology (CSIR-CCMB), Hyderabad, India).
Cell Culture
Cortical neurons from the neocortex of rat brain were cultured as described previously (25, 26). Briefly, neurons were isolated from the neocortex of day 18 embryos. The cells were plated on poly-d-lysine-coated culture plates and maintained in DMEM/F-12 medium supplemented with insulin (25 μg/ml), glucose (6 mg/ml), transferrin (100 μg/ml), progesterone (20 ng/ml), putrescine (60 μg/ml), and selenium (30 ng/ml). Cultured neurons were subjected to treatment after 6 days. Rat pheochromocytoma (PC12) cells were cultured as described previously (27) in RPMI medium supplemented with 10% heat-inactivated horse serum and 5% heat-inactivated fetal bovine serum. Neuronal differentiation was induced by NGF (100 ng/ml) in medium containing 1% horse serum for 6 days before the treatment, as described previously (28).
Preparation of Amyloid
Solution of oligomeric Aβ(1–42) from lyophilized, HPLC-purified Aβ(1–42) was prepared as described previously (29). First, 100% 1,1,1,3,3,3-hexafluoro-2-propanol was used to reconstitute Aβ(1–42) (1 mm), and then the 1,1,1,3,3,3-hexafluoro-2-propanol was removed by evaporation in a SpeedVac. The resultant pellet was then resuspended to 5 μm in anhydrous DMSO. This stock was diluted with PBS to a final concentration of 400 μm, and SDS was added to a final concentration of 0.2%. The resulting solution was then incubated at 37 °C for 18–24 h. The preparation was again incubated at 37 °C for 18–24 h after further dilution with PBS to a final concentration of 100 μm.
PCR
Total RNA of each sample is isolated from cultured cortical neurons by using TRI reagent (Sigma). The primers used for PCR amplification of rat Puma were 5′-GCGGAGACAAGAAGAGCAAC-3′ and 5′-CAAGGCTGGCAGTCCAGTAT-3′. The primers for α-tubulin were 5′-ATGAGGCCATCTATGACATC-3′ and 5′-TCCACAAACTGGATGGTAC-3′, and those for 18S were 5′-GCTTAATTTGACTCAACACGGGA-3′ and 5′-AGCTATCAATCTGTCAATCCTGTC-3′. Equal amounts of cDNA template were used for each PCR analysis of Puma or α-tubulin/18S. Primers were used at 0.2 μm concentration. For semiquantitative PCR, products were analyzed on a 1.5% agarose gel and visualized by staining with ethidium bromide. Quantitative PCR was performed using One Step SYBR Ex Taq qRT-Takara by using an Applied Biosystems 7500 Fast Real Time PCR System following the manufacturer's specifications.
Western Blotting
Cortical neurons were lysed, and proteins were analyzed by Western blotting as described previously (30). For each condition, 50 μg of protein were resolved in 12% SDS-PAGE and then transferred to PVDF membrane (Hybond, GE Healthcare). HRP-conjugated secondary antibodies against the primary antibodies were used. Detection was done by Amersham Biosciences ECL Western blotting detection reagent, according to the manufacturer's protocol. Bands were detected on an x-ray film (Eastman Kodak Co.) or Geldoc (4000 Pro, Carestream).
Immunocytochemical Staining
Cortical neurons were immunostained as described previously (28, 31). Briefly, the cells were fixed with 4% paraformaldehyde for 10 min and then were washed with PBS three times for 5 min each. The cells were then blocked in 3% goat serum in PBS containing 0.1% Triton X-100 for 2 h at room temperature. The cells were incubated with anti-Puma antibody in a blocking solution overnight at 4 °C. Alexa Fluor 546 was used as secondary antibody, and the nuclei were stained with Hoechst.
The intensities of staining for control or treated cells were quantified separately by ImageJ software (National Institutes of Health, Bethesda, MD). The corrected total cell fluorescence (CTCF) was determined by considering the integrated density of staining, area of the cell, and the background fluorescence for the different experimental conditions with the equation, CTCF = integrated density − (area of selected cell × mean fluorescence of background readings).
Chromatin Immunoprecipitation
Chromatin immunoprecipitation (ChIP) assays were done by using a ChIP assay kit from Millipore (Billerica, MA), following the manufacturer's protocol with a few exceptions. 5–8 × 106 cortical neurons were used after treatment with or without Aβ. Rabbit polyclonal anti-FoxO3a antibody was used to immunoprecipitate the protein-DNA complexes. The primers used for PCR amplification of the rat Puma promoter were 5′-AACTTGCATTCTCGCAGCTT-3′and 5′-GCTGCTCCCCAGTCTCACT-3′. PCR products were analyzed on a 1.5% agarose gel and visualized by staining with ethidium bromide. Association of FoxO3a with the Puma gene was also quantified by quantitative RT-PCR.
Transfection
The plasmid maxikit (Qiagen) was used to isolate DNA. For the survival assay, cortical neurons were transfected with 0.5 μg of either pSIREN-Puma-shRNA-zsgreen (shPuma) or pSIREN-Rand-shRNA-zsgreen (shRand). For the reporter assay, cells were cotransfected with 0.3 μg of Puma-luc reporter, 0.1 μg of Renilla vector, and 0.3 μg of either shPuma or shRand. Transfections were done in 500 μl of serum-free medium/well of a 24-well plate using Lipofectamine 2000. Six hours later, Lipofectamine containing medium was replaced by a fresh complete medium. Transfection was performed on the third day of culture. For endogenous FoxO3a down-regulation, naive PC12 cells were transfected with 1 μg of either shFoxO3a or shRand. Transfections were done in 1 ml of serum-free medium/well of a 12-well plate using Lipofectamine 2000. 24 h post-transfection, cells were differentiated in the presence of NGF. After 5 days of priming, cells were treated with either Aβ (5 μm) or left as untreated control.
Luciferase Assay
Cortical neurons were transfected with the constructs as mentioned above. Forty-eight hours after transfection, the cells were treated with or without Aβ(1–42) for 24 h. The cells were lysed in passive lysis buffer (Promega). The dual luciferase assay was done according to the manufacturer's protocol using a luminometer (PerkinElmer Life Sciences). Relative luciferase activities were obtained by normalizing the firefly luciferase activity against Renilla luciferase activity.
Site-directed Mutagenesis
The mutated Puma promoter reporter was generated by incorporating mutations into the consensus sequence for the FoxO binding site by PCR-based site directed mutagenesis using Pfu Turbo DNA polymerase (QuikChange site-directed mutagenesis kit, Stratagene) according to the manufacturer's protocol and was verified by sequencing. The primers used for generating mutations were 5′-GGCGGGTTTGTTTACAGGGAATGGGGTTCGGC-3′ and 5′-GCCCGAACCCCATTCCCTGTAAACAAACCCGCC-3′.
Sholl Analysis
Neural networks of cortical neurons were analyzed by Sholl analysis using ImageJ software as reported previously by Cuesto et al. (32), excepting few modifications as mentioned below. In brief, transfected cortical neurons either with shPuma or shRand were imaged at low magnification under a fluorescence microscope. Sholl analysis was performed on single neurons imaged at 0 and 72 h of Aβ(1–42) treatment using the ImageJ software (Sholl analysis plugin). Several concentric circles were drawn from a point of the cell body with gradually increasing radius of 40 μm in length. The number of branches that intersect the successive concentric circle was counted.
Survival Assay
Primary cortical neurons (5 days in vitro) were transfected with shRand or shPuma as mentioned above. After 48 h of transfection, the neurons were exposed to Aβ(1–42), and the number of transfected neurons (green) was counted (0 h). The number of surviving transfected neurons was also counted after 24, 48, and 72 h of treatment. Control and Aβ-treated transfected neurons were imaged under a fluorescence microscope (Leica, Wetzlar, Germany).
The cell viability was also checked by the intact nuclear counting method. This assay was performed as described previously (33). In brief, a detergent containing the buffer was added to the cells that dissolve only the cell membrane, leaving the nuclear membrane intact. The intact nuclei were then counted on a hemocytometer. The number of live cells was expressed as a percentage of the total cell population.
Oligomeric Aβ Infusion in Animals
Male Sprague-Dawley rats (300–380 g) were infused with oligomeric Aβ as described previously (4). Briefly, rats were anaesthetized by injecting a mixture of xylazine-ketamine and placed on a stereotaxic frame, and then a volume of 5 μl of 100 μm Aβ in PBS was infused in the right cerebral cortex at stereotaxic coordinates from bregma (AP, −4.1 mm; L, 2.5 mm; DV, 1.3 mm) according to the rat brain atlas. Control animals were injected with an equal volume of PBS. After 21 days of injection, animals were sacrificed. Following cardiac perfusion, the brains were dissected out and fixed in 4% paraformaldehyde for 24 h. The brains were further incubated in a 30% sucrose solution for 24 h, and then cryosectioning was done by using a cryotome (Thermo, West Palm Beach, FL).
Immunohistochemistry of Brain Slices
Cryosections of the brains from Aβ-infused or PBS-infused rats and wild-type or transgenic mice were immunostained as described previously (4). In brief, sections were blocked with 5% goat serum in PBS containing 0.3% Triton X-100 for 1 h at room temperature, incubated in primary antibody in a blocking solution overnight at 4 °C, washed with PBS, and then incubated with a fluorescence-tagged secondary antibody for 2 h at room temperature. After PBS wash and Hoechst staining for the nucleus, the sections were mounted and observed under fluorescence microscope.
Statistics
All experimental results are reported as mean ± S.E. Student's t test was performed as unpaired, two-tailed sets of arrays to evaluate the significance of difference between the means and presented as p values.
RESULTS
Puma Is Induced in Neurons in Response to Aβ in Cultures and in Vivo
Recent findings suggest that Aβ oligomers play an important role in the development of AD (34–36). Cortical neurons are severely affected in AD brains and undergo death after exposure to oligomeric Aβ in vitro (5, 26, 28, 29, 37–39). We have recently shown that the oligomeric Aβ(1–42) at a concentration of 1.5 μm induced significant neuron death after 24 h. Neuron death first becomes apparent by 12–16 h of Aβ exposure, and more than 40% of neurons die within 24 h (4). A time course reveals that Puma is up-regulated by oligomeric Aβ(1–42) in cultured neurons. Puma transcript is increased by 2-fold within about 2–4 h of Aβ treatment, as detected by semiquantitative PCR (Fig. 1A) as well as by real-time PCR (Fig. 1B). Protein level is increased by more than 2-fold within 8 h of Aβ exposure (Fig. 1, C and D). Thus, Puma induction by Aβ precedes overt signs of neuron death. Next, we checked whether Puma level is also elevated in vivo in response to Aβ. We and others have shown that adult rats infused with oligomeric Aβ(1–42) in brain resulted in Aβ deposition, caspase-3 activation, and loss of neuronal cells in the vicinity of the infusion site of Aβ(1–42) (4, 40). In this study, the right hemispheres of the brains of adult rats were infused with either Aβ(1–42) or PBS, and after 21 days, the animals were sacrificed, and brains were fixed, cryosectioned. The brain sections were co-immunostained with Puma and neuronal marker NeuN antibodies. Hoechst dye was used to stain nuclei. Results showed that Puma levels were greatly enhanced in neurons in Aβ(1–42)-infused but not in PBS-infused rat brains (Fig. 2). The presence of Aβ deposition in adjacent sections of Aβ(1–42) infused rat brains was verified by staining with Congo red (data not shown).
FIGURE 1.
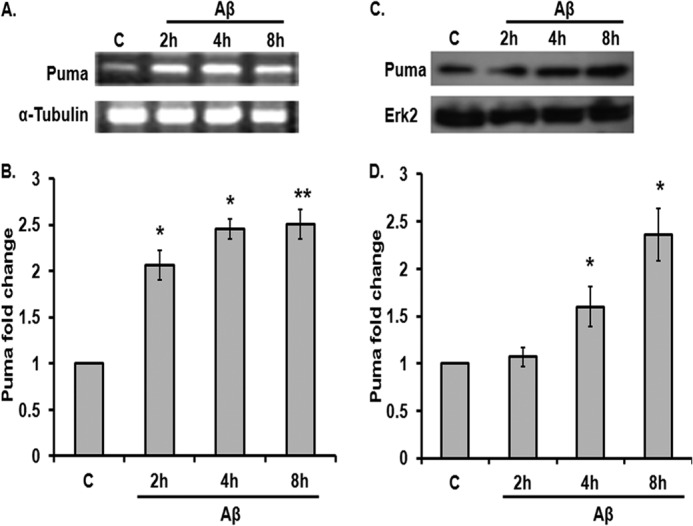
Puma is induced by Aβ in cortical neurons. A, rat cortical neurons (7 days in vitro) were subjected to oligomeric Aβ (1.5 μm) for the indicated times, and total RNA was isolated, reverse-transcribed, and analyzed by semiquantitative PCR for Puma transcripts. α-Tubulin was used as a loading control. B, graphical representation of changes in Puma transcript level upon Aβ (1.5 μm) treatment on primary cultured rat cortical neurons at the indicated times by real-time PCR. 18 S was used as a loading control. Data are presented as -fold increase relative to untreated control (C) and represent mean ± S.E. of three independent experiments. *, p < 0.05; **, p < 0.01. C, cortical neurons were subjected to Aβ (1.5 μm) for the indicated times, and total tissue lysates were analyzed by Western blot for Puma level. A representative immunoblot shows Puma protein level at the indicated time points. ERK2 was used as a loading control. D, graphical representation of -fold increase of Puma protein level after Aβ treatment at different time points expressed relative to untreated control. Data represent mean ± S.E. (error bars) of three experiments. *, statistically significant differences from 0 h control; p < 0.05.
FIGURE 2.
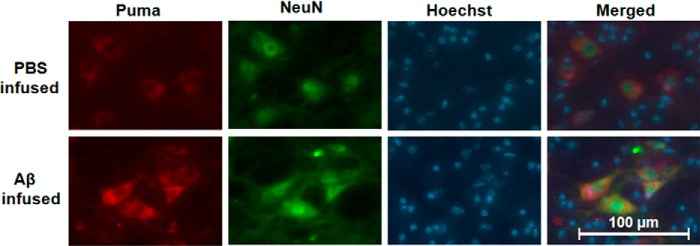
Puma is induced following Aβ(1–42) infusion in vivo. Right hemispheres of brains of adult rats were infused with either Aβ(1–42) or PBS, and after 21 days, the animals were sacrificed, and brains were taken out following cardiac perfusion. The brains were cryosectioned and co-immunostained with Puma and NeuN antibodies; nuclei were stained with Hoechst dye. Representative images of five sections from three animals of each group with similar results are shown here. Scale bar, 100 μm. Images were taken for each case by using an inverted fluorescence microscope and camera set to the same exposure.
The use of synthetic Aβ may result in different effects compared with naturally secreted Aβ. Therefore, we also examined the brain sections of AβPPswe-PS1de9 (Swedish mutation in APP and PS1 mutation) transgenic mice for Puma expression. Brains of transgenic or control littermate mice were cryosectioned and stained with Congo red to confirm deposition of Aβ plaques (Fig. 3A). Then the brain sections were co-immunostained with Puma and NeuN antibodies. Nuclei were stained with Hoechst dye. Our results showed that the levels of Puma expression were much higher in transgenic mice compared with control littermates (Fig. 3B). Collectively, these findings clearly establish that Puma is induced in neurons in response to Aβ(1–42) in vitro and in vivo.
FIGURE 3.

Puma is elevated in AD transgenic mice brain. The brain sections of AβPPswe-PS1de9 transgenic mice and wild-type mice were analyzed for level of Puma expression. A, transgenic and wild-type brain slices were stained with Congo red to see the deposition of Aβ plaques. These plaques (arrows) were seen in transgenic brains, whereas they were absent in wild-type brain tissue. B, the brain sections of transgenic mice were co-immunostained with Puma and NeuN antibodies. Hoechst was used to stain nuclei. Representative images from six sections from three animals of each group with similar results are shown here. Scale bar, 100 μm. Inset, magnified version of the same section in each case. Images were taken for each case using an inverted fluorescence microscope and a camera set to the same exposure.
Puma Knockdown Prevents Cortical Neuron Death upon Aβ Treatment
Next, we employed an shRNA-mediated knockdown strategy to assess whether Puma plays any role in Aβ(1–42)-mediated neuron death. Cultured cortical neurons were transfected with a previously described shRNA construct against the Puma gene (shPuma) (12) along with a control shRNA construct (shRand), maintained for 48 h, and then subjected to Aβ treatment (1.5 μm) for 72 h. The number of surviving cells (green) was counted under a fluorescence microscope. Down-regulation of Puma by shRNA provided significant protection of cortical neurons from death evoked by Aβ(1–42). A significant number of shPuma-expressing neurons survived with intact neurites even after 72 h of Aβ exposure compared with shRand-expressing neurons (Fig. 4, A and B).
FIGURE 4.

Down-regulation of Puma by shRNA protects cortical neurons from death. A, primary cultured rat cortical neurons (5 days in vitro) were transfected with pSIREN-shPuma-zsgreen or control pSIREN-shRand-zsgreen (scrambled shRNA) and maintained for 48 h and then subjected to Aβ (1.5 μm) treatment for 72 h. Representative pictures of transfected neurons that were maintained in the presence or absence of Aβ for the indicated time periods are shown. Images were taken under a ×20 objective. B, graphical representation of the percentage of viable green cells after each time point. The numbers of surviving transfected (green) cells were counted under a fluorescence microscope just before Aβ treatment (C) and after 24, 48, and 72 h of the same treatment. Data are from three independent experiments, each with comparable results, and are shown as mean ± S.E., performed in triplicates. The asterisks denote statistically significant differences from control (shRand) at corresponding time points: *, p < 0.05; **, p < 0.001. C, Puma knockdown prevents neuronal degeneration. Sholl analysis of single imaged neurons by using ImageJ was done as described under “Experimental Procedures.” Data represent the mean ± S.E. (error bars) of six different neurons from three independent cultures for each class. *, statistically significant differences from shRand (control); p < 0.001.
We have also quantitatively measured the preservation of neural networks by shPuma in Aβ treated cells by Sholl analysis (Fig. 4C). A single cortical neuron transfected with shPuma or shRand was analyzed by ImageJ as described under “Experimental Procedures.” The analyses showed that shPuma-transfected cells retain most of the neurites even after 72 h of Aβ treatment compared with shRand (control)-transfected cells, where diminution of neural networks is evident. Taken together, these results suggest that Puma is required for neuron death and degeneration evoked by Aβ toxicity.
FoxO3a Regulates Puma Expression upon Aβ Toxicity
It is known that Puma is regulated in both a p53-dependent and -independent manner (11, 17, 18). We have also found that basal expression of Puma was down-regulated when p53 was inhibited by pifithrin-α (Fig. 5, A and B). However, inhibition of p53 did not completely block the Aβ-mediated up-regulation of Puma. Fig. 5, A and B, shows that the increase in the level of Puma by Aβ was only partially suppressed by pifithrin α, suggesting that other regulatory pathways independent of p53 are operating in the induction of Puma by Aβ (see below). Previous reports showed that the detrimental effect of Aβ can be abolished by pifithrin α to a considerable extent (41). Under stress stimuli, p53 gets activated upon phosphorylation at serine 15 (42, 43). The rapid increase of p53 phosphorylation at serine 15 evoked by Aβ is evident in our study and can be inhibited by pifithrin α in the presence of Aβ (Fig. 5, C and D). We also checked whether pifithrin-α has any effect on cell viability in this model. We found that this compound significantly protected neurons from death induced by Aβ toxicity (Fig. 5E). These results suggest that p53 is induced and is required for neuron death in response to Aβ. It is known that both Aβ and p53 induce BAX (2, 44, 45), which acts as a downstream effector of PUMA in the cell death cascade (46, 47). Therefore, we checked the level of Bax and found that it was elevated in response to Aβ (Fig. 5, F and G).
FIGURE 5.

Inhibition of PI3K signaling induces Puma in cortical neurons. A, primary cultures of rat cortical neurons were treated with LY294002 (50 μm) or pifithrin-α (50 μm) for 8 h with or without Aβ (1.5 μm), and the proteins were analyzed by Western immunoblotting using enhanced chemiluminescence for the expression of Puma and actin (loading control). B, graphical representation of densitometric analysis of -fold change of Puma level in the indicated conditions. Data represent ± S.E. (error bars) of three experiments. *, significant differences from control; p < 0.03. C, cultured cortical neurons were treated with pifithrin-α with or without Aβ for 8 h, and the expression of phospho-p53Ser-15 and total p53 was assessed by Western blot. D, graphical representation of densitometric analysis of fold change of phospho-p53Ser-15 level in the indicated conditions. Data represent means ± S.E. of three experiments. *, p < 0.05. E, cultured cortical neurons were treated with or without Aβ in the presence and absence of pifithrin-α. Data are represented as mean ± S.E. of three independent experiments. *, p < 0.05. F, cortical neurons were subjected to Aβ (1.5 μm) for the indicated times, and total tissue lysates were analyzed by Western blot for BAX. Actin was used as loading control. G, graphical representation of -fold increase of BAX protein level after Aβ treatment at different time points expressed relative to untreated control. Data represent mean ± S.E. of three experiments with three replicate cultures. *, significant differences from control; p < 0.03.
Besides p53, it has been shown that dysregulated PI3K/Akt signaling in response to growth factor withdrawal activates transcription factor FoxO3a, which in turn up-regulates Puma (10, 21, 22). It has also been reported that the PI3K/Akt signaling pathway is inhibited by Aβ toxicity (48). Therefore, we investigated whether Puma is regulated by the PI3K/Akt/FoxO3a pathway in Aβ-treated neurons. First we checked whether inhibition of PI3K signaling is capable of inducing Puma. We found that PI3K inhibition by a specific PI3K inhibitor, LY294002, led to the elevation of Puma about 2-fold compared with control (Fig. 5, A and B). The extent of the increase was comparable with the Puma level induced by Aβ treatment. Interestingly, treatment with Aβ in presence of LY294002 did not enhance the Puma level further (Fig. 5, A and B). This result implies that Puma induction in Aβ-treated neurons also occurs through inhibition of PI3K pathway. Because the inhibition of PI3K signaling is known to activate FoxO3a, we investigated the role of FoxO3a in the induction of Puma by Aβ. Cultured cortical neurons were transfected with a previously described shRNA construct against FoxO3a (4) and then immunostained with Puma antibody. Results revealed that shFoxO3a-expressing cells had less intense staining of Puma compared with shRand-expressing cells upon Aβ treatment (Fig. 6A). A quantitative analysis showed that about 80% of shFoxO3a-transfected neurons and 20% of shRand-expressing neurons had low staining of Puma after 8 h of Aβ exposure (Fig. 6B). We further quantified the total cell fluorescence intensity of Puma in shFoxO3a- and shRand-expressing cells by ImageJ and found significantly less intense Puma staining in FoxO3a-expressing cells compared with shRand-expressing cells upon Aβ treatment (Fig. 6C). We also validated the down-regulation of Puma by shFoxO3a by Western blot. PC12 cells were transfected with shFoxO3a or shRand and then primed with NGF, and we checked the level of FoxO3a and Puma. In this condition, more than 60% cells were transfected, and FoxO3a was markedly down-regulated by shFoxO3a (Fig. 6, D and E). We found that the Puma level was significantly reduced by shFoxO3a- compared with shRand-transfected cells (Fig. 6, F and G). Collectively, these results suggest that Puma is a target of FoxO3a in Aβ-treated neurons.
FIGURE 6.

Knockdown of FoxO3a by shRNA represses up-regulation of endogenous Puma in neuronal cells subjected to Aβ treatment. A, cortical neurons were transfected with shFoxO3a or shRand and maintained for 48 h and then treated with Aβ (1.5 μm) for 8 h, after which they were immunostained with antibodies against Puma (red). Images were taken under a ×63 objective. B, percentage of stained cells pertains to the proportions of transfected cells (green) that show Puma staining either greater than that of non-treated control neurons (High) or equal to or less than that of non-treated neurons (Low). Data represent the mean ± S.E. (error bars) of three experiments. The number of cells evaluated per culture was ∼50. *, p < 0.01. C, graphical representation of corrected total cell fluorescence of Puma in neurons transfected with shRand or shFoxO3a following Aβ exposure. Difference in intensity of Puma staining was quantified by ImageJ as described under “Experimental Procedures.” Data represent mean ± S.E. of 60 different cells from three independent experiments. *, p < 0.03. D, PC12 cells were transfected with shFoxO3a or shRand and primed as described under “Experimental Procedures,” and then the down-regulation of endogenous FoxO3a was analyzed by Western blotting with anti-FoxO3a antibody. E, graphical representation of densitometric analysis of -fold change of FoxO3a level upon transfection with shFoxO3a or shRand in the presence or absence of Aβ (5 μm). Data represent mean ± S.D. (error bars) of two independent experiments. *, p < 0.05. F, PC12 cells were transfected with shFoxO3a or shRand and primed, and then the down-regulation of endogenous Puma was analyzed by Western blotting with anti-Puma antibody. G, graphical representation of densitometric analysis of -fold change of Puma level upon transfection with shFoxO3a or shRand in the presence or absence of Aβ (5 μm). Data represent mean ± S.D. of two independent experiments; *, p < 0.05.
FoxO3a Directly Activates Puma Gene in Response to Aβ
A FoxO response element (FHRE) in intron 1 of the Puma gene, which is conserved between human and mouse, has been reported (22). We have also found a similar FHRE in intron 1 of the rat gene. Therefore, we were interested to see whether FoxO3a directly binds with the Puma gene and is required for its activation during Aβ-induced neuron death. First, we prepared a construct of luciferase reporter driven by a segment of the rat Puma gene that contains the FHRE (Fig. 7A). Cultured cortical neurons were co-transfected with the luciferase reporter and shRand or shFoxO3a and exposed to Aβ. Puma promoter-driven luciferase activity was increased severalfold upon Aβ(1–42) treatment for 24 h when neurons were co-transfected with shRand (Fig. 7B). However, this increase in luciferase activity in response to Aβ(1–42) is diminished when neurons were co-transfected with shFoxO3a (Fig. 7B).
FIGURE 7.
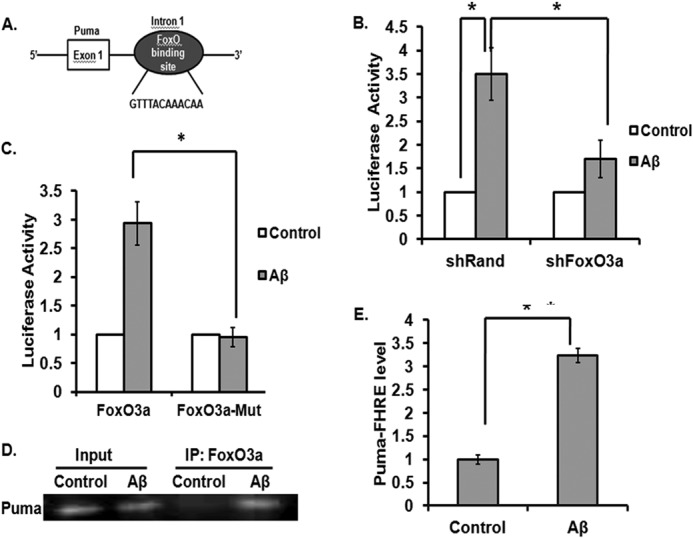
FoxO3a directly binds with intron 1 of the rat Puma gene and regulates its induction upon Aβ treatment. A, schematic representation of Puma-luc reporter consisting of intron-1 of the rat Puma gene. B, cortical neurons were co-transfected with 0.3 μg of Puma-luc reporter and 0.1 μg of Renilla luciferase expression construct pRL-CMV with 0.3 μg of either shRand (control) or shFoxO3a. The cultures were maintained for 48 h and then subjected to overnight Aβ treatment, after which luciferase activity was assayed and represented as -fold change of luciferase activity. Data represent mean ± S.E. (error bars) of four experiments. *, p < 0.05. C, cortical neurons were co-transfected with 0.4 μg of either wild type Puma-luc reporter or FoxO3a-mutated construct and 0.1 μg of Renilla luciferase expression construct pRL-CMV. The cultures were maintained for 48 h and then subjected to overnight Aβ treatment, after which luciferase activity was assayed and represented as -fold change of luciferase activity. Data represent mean ± S.E. of four experiments. *, p < 0.05. D, primary cultures of rat cortical neurons were treated with or without Aβ for 8 h. An equal number of cells were processed for ChIP assay using anti-FoxO3a antibody for immunoprecipitation. The immunoprecipitated materials were subjected to PCR using primers against the portion of the Puma promoter that flanks the FoxO3a-binding site. PCR products were verified by agarose gel electrophoresis. Templates were DNA from cells before ChIP (Input) or DNA from immunoprecipitated (IP) materials. PCR assays were conducted after ChIP, using samples from cells that were either left untreated (Control) or treated with Aβ. E, graphical representation of FoxO3a association with the Puma gene. Quantitative PCR was performed using material derived from cultured cortical neurons treated as in D. Association of FoxO3a with Puma Forkhead response element (Puma FHRE level) in the presence or absence of Aβ was determined by quantitative PCR after ChIP, using samples from cells that were either left untreated (Control) or treated with Aβ. Numbers on the y axis represent the levels of FoxO3a association with the Puma promoter region after normalizing to Ct values from input samples. Data shown are means ± S.E. *, p < 0.05.
To further validate the dependence of Puma induction on FoxO3a, we generated mutations in the consensus FoxO3a binding site in the Puma promoter reporter construct. The mutations abolished the Puma-luc reporter activity induced by Aβ (Fig. 7C). Thus, our result suggests that FoxO3a is necessary for activation of the Puma promoter in response to Aβ.
Next, we performed a ChIP assay to see whether FoxO3a directly binds with the Puma gene in response to Aβ. Results showed that a significant amount of FoxO3a was bound to the endogenous Puma gene after 8 h of Aβ(1–42) exposure, whereas there was negligible binding in control cells (Fig. 7D). As a negative control, an irrelevant antibody was used, which did not precipitate Puma DNA, and PCR of FoxO3a immunoprecipitate with primers specific for α-tubulin did not produce any product (data not shown). Quantitative PCR analyses of immunoprecipitated DNA also showed that there was a severalfold increase in binding of FoxO3a with the Puma promoter when cortical neurons were exposed to Aβ for 8 h compared with control conditions, where cells were not treated with Aβ (Fig. 7E). Taken together, our results indicate that Puma is induced in response to Aβ, at least in part by FoxO3a.
Puma Cooperates with Bim in Neuron Death Induced by Aβ
It has been reported that Bim and Puma cooperate in developmental apoptosis of lymphocytes (49) and autoreactive thymocytes (9) and tumor regression in response to tyrosine kinase inhibitors in breast and lung cancers (10). We therefore determined the effects of Bim and Puma knockdown, either alone or together, on Aβ-induced neuron death. We used a previously described shRNA construct that effectively silences rat Bim (31) and protects neurons from Aβ-induced death (5). Silencing both Bim and Puma led to a modest but significant increase in protection at 24 and 48 h of Aβ treatment (Fig. 8). These data indicate that Bim and Puma cooperate in neuron death evoked by Aβ and that Puma and Bim may activate separate apoptotic pathways.
FIGURE 8.
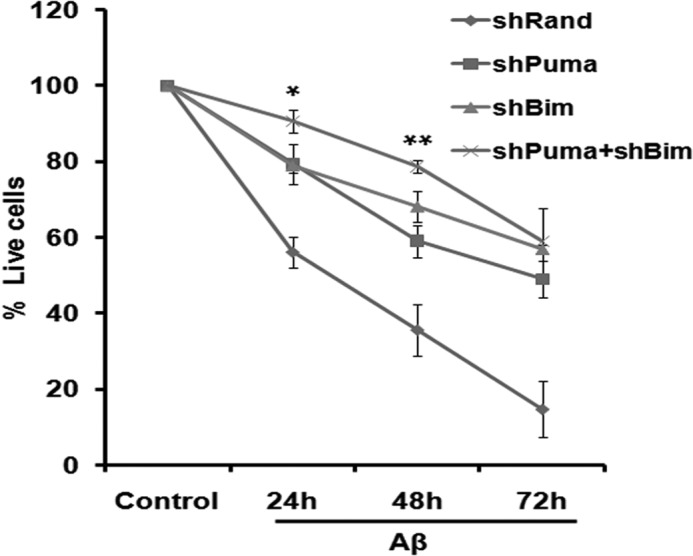
Knockdown of both Bim and Puma together provided better protection than knockdown of individual genes against Aβ-induced neuron death. Primary cultured rat cortical neurons (5 days in vitro) were transfected with pSIREN-shPuma-zsgreen or with pSIREN-shBim-zsgreen or co-transfected with both or with pSIREN-shRand-zsgreen (control); maintained for 48 h; and then subjected to Aβ (1.5 μm) treatment for the indicated times. Live cells were counted under a fluorescence microscope after each time point. Data represent three independent experiments, each with comparable results, and are shown as mean ± S.E. (error bars), performed in triplicates. The asterisks denote statistically significant differences from shPuma at corresponding time points: *, p < 0.05; **, p < 0.01.
DISCUSSION
Although recent research in multidisciplinary areas points out that interactions of multiple factors, including Aβ, Tau, apoE4, and aging result in development of AD, it does not rule out the original hypothesis that accumulation of Aβ due to altered metabolism and clearance is central to the disease pathogenesis (1, 24). Cultured neurons or animals exposed to aggregated Aβ and transgenic mice that produce more Aβ all manifest neurodegeneration and AD-like pathology. Recent radiological tools clearly demonstrate the accumulation of Aβ in the affected areas of patients' brain (50). However, reducing Aβ load by various means does not improve the conditions of patients in various clinical trials. These observations clearly indicate the need of alternative measures to counter the toxicity of Aβ and prevent the progressive loss of neurons by the residual Aβ that cannot be removed completely and safely by any Aβ-reducing drug.
The BH3-only proteins of the Bcl-2 family that act as sentinels of apoptotic stimuli and are required for mitochondrial pathway of apoptosis could be appropriate targets to reduce the toxicity of Aβ. In this report, we showed that the BH3-only protein Puma is induced by Aβ in vitro and in vivo and plays an essential role in AD-associated neurodegeneration. We have observed significant increase of Puma transcripts as well as its protein levels in cortical neurons upon Aβ treatment in a time-dependent manner. The expression of Puma is also enhanced in AD transgenic mice and Aβ-infused rat brains. We further observed that knockdown of Puma by the shRNA construct provides significant protection against neuron death and helps in retention of the neural network for longer time points.
Accumulating evidence also implicates Puma in several other neurodegenerative disorders. Puma is essential for neuron death evoked by 6-OHDA, a model of Parkinson disease (12). Deletion of Puma rescues motor neurons from apoptosis caused by endoplasmic reticulum stress in amyotrophic lateral sclerosis (51). It has also been reported that Puma is up-regulated in hippocampal CA1 neurons after transient global cerebral ischemia and thus could be a potential molecular target for therapy in stroke (52). Our study now implicates its involvement in AD-related neurodegeneration.
We have also investigated the regulatory pathways of Puma induction in response to Aβ treatments. Accumulating evidence suggests that Puma is regulated in both a p53-dependent and p53-independent manner in various systems (11, 18). Consistent with these reports, we found that inhibition of p53 is not sufficient to block the induction of Puma by Aβ, indicating the involvement of additional regulatory pathways. The survival pathway directed by PI3K-Akt is seen to be compromised by Aβ toxicity. This can be correlated with our recent findings that the activation and subsequent translocation of one of its downstream transcription factors, FoxO3a, to the nucleus induce proapoptotic protein Bim (4). The present findings indicate that FoxO3a also up-regulates another proapoptotic protein of the same family, Puma, in response to Aβ and causes consequent neuron death. Therefore, our results suggest that both p53 and FoxO3a regulate Puma expression.
In this context, we thought it would be interesting to know whether Bim and Puma act cooperatively in Aβ-induced neuron death. Cooperative function of Bim and Puma has been reported in various apoptotic paradigms. Bean et al. (10) have reported that Bim and Puma mediate oncogene inactivation-induced apoptsosis in vitro and in vivo. They found that full apoptotic response initiated by oncogene deletion required both Bim and Puma. Similarly, it has been found that the combined loss of Bim and Puma is required for accumulation of immature thymocytes (9). They found that Bim−/−/Puma−/− mice had more extensive splenomegaly and lymphadenopathy than Bim−/− mice. In agreement with these findings, we found that combined knockdown of Bim and Puma provided significantly increased protection against Aβ-induced death up to 48 h of treatment compared with that of single knockdown of either of the proteins. Thus, our results indicate that both Bim and Puma are required for Aβ-induced neuron death. However, it remains to be examined whether they activate separate apoptotic pathways.
In comparison with the prominent role of Puma in p53-dependent apoptosis, the function of it in p53-independent cell death remains to be fully addressed. It has already been shown that under stress conditions like serum deprivation or cytokine withdrawal, Puma gets activated by FoxO3a in a p53-independent fashion (19, 22, 53). Mast cells deficient in FoxO3a were markedly resistant to cytokine withdrawal compared with wild-type cells (54). Puma knock-out mice were resistant to apoptosis induced by the withdrawal of interleukin IL-3 and IL-6 (18, 55). In p53-deficient cancer cells, Puma is induced in response to serum starvation, and cells are resistant to apoptosis after knockdown of Puma (56). Puma is also known to be induced by transcription factors other than FoxO3a like CHOP, E2F1, or even Trb3 (20, 57, 58). We are currently in search of other probable signaling pathways that may regulate Puma in cooperation with p53 and FoxO3a in response to Aβ.
Considering our recent report (4) and the findings of this study, we propose a model for Aβ-induced neuron death (Fig. 9). Under Aβ-treated conditions, p53 is activated, which partly induces Puma. FoxO3a is also activated as the PI3K/Akt pathway is inhibited in Aβ-treated neurons. Activated FoxO3a translocates to the nucleus and binds directly to Bim and Puma genes, leading to their enhanced expression. Puma in cooperation with Bim activates the intrinsic apoptotic pathway that results in neuron death. In view of the fact that both Bim and Puma can be regulated by various transcription factors in other systems (17, 59), there is a possibility that multiple transcription factors may be required for activation of both Bim and Puma in response to Aβ. Further studies are required to validate this assumption and evaluate if those factors work independently or synergistically to activate Bim and Puma in Aβ-induced neuron death.
FIGURE 9.
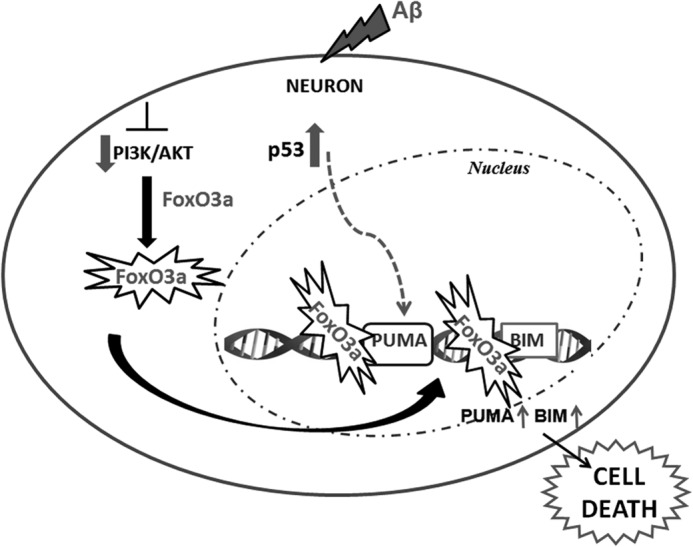
Schematic representation of Puma activation by FoxO3a and of neuron death in response to Aβ.
Acknowledgments
We thank Dr. K. P. Mohanakumar for allowing use of the stereotaxic facility and Raghavendra Singh for help in developing the animal model. We also thank Dr. A. B. Patel for providing tissues of transgenic animals of the Alzheimer model and Vivek Tiwari for help in processing the transgenic brains. We also thank Dr. P. K. Sarkar and Dr. Sumantra Das for critical reading of the manuscript and helpful discussions.
This work is supported in part by the Department of Biotechnology, Government of India, Project BT/PR14383/MED/12/475/2010 and CSIR-Supra Institutional Project BenD BSC0206.
- Aβ
- β-amyloid
- AD
- Alzheimer disease
- BH
- Bcl-2 homology domain.
REFERENCES
- 1. Hardy J., Selkoe D. J. (2002) The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297, 353–356 [DOI] [PubMed] [Google Scholar]
- 2. Kudo W., Lee H. P., Smith M. A., Zhu X., Matsuyama S., Lee H. G. (2012) Inhibition of Bax protects neuronal cells from oligomeric Aβ neurotoxicity. Cell Death Dis. 3, e309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kudo W., Lee H. P., Zou W. Q., Wang X., Perry G., Zhu X., Smith M. A., Petersen R. B., Lee H. G. (2012) Cellular prion protein is essential for oligomeric amyloid-β-induced neuronal cell death. Hum. Mol. Genet. 21, 1138–1144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sanphui P., Biswas S. C. (2013) FoxO3a is activated and executes neuron death via Bim in response to β-amyloid. Cell Death Dis. 4, e625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Biswas S. C., Shi Y., Vonsattel J. P., Leung C. L., Troy C. M., Greene L. A. (2007) Bim is elevated in Alzheimer's disease neurons and is required for β-amyloid-induced neuronal apoptosis. J. Neurosci. 27, 893–900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Youle R. J., Strasser A. (2008) The BCL-2 protein family: opposing activities that mediate cell death. Nat. Rev. Mol. Cell Biol. 9, 47–59 [DOI] [PubMed] [Google Scholar]
- 7. Wei M. C., Zong W. X., Cheng E. H., Lindsten T., Panoutsakopoulou V., Ross A. J., Roth K. A., MacGregor G. R., Thompson C. B., Korsmeyer S. J. (2001) Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science 292, 727–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ren D., Tu H. C., Kim H., Wang G. X., Bean G. R., Takeuchi O., Jeffers J. R., Zambetti G. P., Hsieh J. J., Cheng E. H. (2010) BID, BIM, and PUMA are essential for activation of the BAX- and BAK-dependent cell death program. Science 330, 1390–1393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gray D. H., Kupresanin F., Berzins S. P., Herold M. J., O'Reilly L. A., Bouillet P., Strasser A. (2012) The BH3-only proteins Bim and Puma cooperate to impose deletional tolerance of organ-specific antigens. Immunity 37, 451–462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bean G. R., Ganesan Y. T., Dong Y., Takeda S., Liu H., Chan P. M., Huang Y., Chodosh L. A., Zambetti G. P., Hsieh J. J., Cheng E. H. (2013) PUMA and BIM are required for oncogene inactivation-induced apoptosis. Sci. Signal. 6, ra20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yu J., Zhang L. (2008) PUMA, a potent killer with or without p53. Oncogene 27, S71–S83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Biswas S. C., Ryu E., Park C., Malagelada C., Greene L. A. (2005) Puma and p53 play required roles in death evoked in a cellular model of Parkinson disease. Neurochem. Res. 30, 839–845 [DOI] [PubMed] [Google Scholar]
- 13. Bock F. J., Villunger A. (2011) GSK3 TIPping off p53 to unleash PUMA. Mol. Cell 42, 555–556 [DOI] [PubMed] [Google Scholar]
- 14. Follis A. V., Chipuk J. E., Fisher J. C., Yun M. K., Grace C. R., Nourse A., Baran K., Ou L., Min L., White S. W., Green D. R., Kriwacki R. W. (2013) PUMA binding induces partial unfolding within BCL-xL to disrupt p53 binding and promote apoptosis. Nat. Chem. Biol. 9, 163–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hemann M. T., Zilfou J. T., Zhao Z., Burgess D. J., Hannon G. J., Lowe S. W. (2004) Suppression of tumorigenesis by the p53 target PUMA. Proc. Natl. Acad. Sci. U.S.A. 101, 9333–9338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nakano K., Vousden K. H. (2001) PUMA, a novel proapoptotic gene, is induced by p53. Mol. Cell 7, 683–694 [DOI] [PubMed] [Google Scholar]
- 17. Galehdar Z., Swan P., Fuerth B., Callaghan S. M., Park D. S., Cregan S. P. (2010) Neuronal apoptosis induced by endoplasmic reticulum stress is regulated by ATF4-CHOP-mediated induction of the Bcl-2 homology 3-only member PUMA. J. Neurosci. 30, 16938–16948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jeffers J. R., Parganas E., Lee Y., Yang C., Wang J., Brennan J., MacLean K. H., Han J., Chittenden T., Ihle J. N., McKinnon P. J., Cleveland J. L., Zambetti G. P. (2003) Puma is an essential mediator of p53-dependent and -independent apoptotic pathways. Cancer Cell 4, 321–328 [DOI] [PubMed] [Google Scholar]
- 19. Han J., Flemington C., Houghton A. B., Gu Z., Zambetti G. P., Lutz R. J., Zhu L., Chittenden T. (2001) Expression of bbc3, a pro-apoptotic BH3-only gene, is regulated by diverse cell death and survival signals. Proc. Natl. Acad. Sci. U.S.A. 98, 11318–11323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zou C. G., Cao X. Z., Zhao Y. S., Gao S. Y., Li S. D., Liu X. Y., Zhang Y., Zhang K. Q. (2009) The molecular mechanism of endoplasmic reticulum stress-induced apoptosis in PC-12 neuronal cells: the protective effect of insulin-like growth factor I. Endocrinology 150, 277–285 [DOI] [PubMed] [Google Scholar]
- 21. Amente S., Zhang J., Lavadera M. L., Lania L., Avvedimento E. V., Majello B. (2011) Myc and PI3K/AKT signaling cooperatively repress FOXO3a-dependent PUMA and GADD45a gene expression. Nucleic Acids Res. 39, 9498–9507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. You H., Pellegrini M., Tsuchihara K., Yamamoto K., Hacker G., Erlacher M., Villunger A., Mak T. W. (2006) FOXO3a-dependent regulation of Puma in response to cytokine/growth factor withdrawal. J. Exp. Med. 203, 1657–1663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gilley J., Coffer P. J., Ham J. (2003) FOXO transcription factors directly activate bim gene expression and promote apoptosis in sympathetic neurons. J. Cell Biol. 162, 613–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Huang Y., Mucke L. (2012) Alzheimer mechanisms and therapeutic strategies. Cell 148, 1204–1222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Park D. S., Morris E. J., Padmanabhan J., Shelanski M. L., Geller H. M., Greene L. A. (1998) Cyclin-dependent kinases participate in death of neurons evoked by DNA-damaging agents. J. Cell Biol. 143, 457–467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Troy C. M., Rabacchi S. A., Friedman W. J., Frappier T. F., Brown K., Shelanski M. L. (2000) Caspase-2 mediates neuronal cell death induced by β-amyloid. J. Neurosci. 20, 1386–1392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Greene L. A., Tischler A. S. (1976) Establishment of a noradrenergic clonal line of rat adrenal pheochromocytoma cells which respond to nerve growth factor. Proc. Natl. Acad. Sci. U.S.A. 73, 2424–2428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Biswas S. C., Shi Y., Sproul A., Greene L. A. (2007) Pro-apoptotic Bim induction in response to nerve growth factor deprivation requires simultaneous activation of three different death signaling pathways. J. Biol. Chem. 282, 29368–29374 [DOI] [PubMed] [Google Scholar]
- 29. Barghorn S., Nimmrich V., Striebinger A., Krantz C., Keller P., Janson B., Bahr M., Schmidt M., Bitner R. S., Harlan J., Barlow E., Ebert U., Hillen H. (2005) Globular amyloid β-peptide oligomer: a homogenous and stable neuropathological protein in Alzheimer's disease. J. Neurochem. 95, 834–847 [DOI] [PubMed] [Google Scholar]
- 30. Biswas S. C., Greene L. A. (2002) Nerve growth factor (NGF) down-regulates the Bcl-2 homology 3 (BH3) domain-only protein Bim and suppresses its proapoptotic activity by phosphorylation. J. Biol. Chem. 277, 49511–49516 [DOI] [PubMed] [Google Scholar]
- 31. Biswas S. C., Liu D. X., Greene L. A. (2005) Bim is a direct target of a neuronal E2F-dependent apoptotic pathway. J. Neurosci. 25, 8349–8358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cuesto G., Enriquez-Barreto L., Caramés C., Cantarero M., Gasull X., Sandi C., Ferrús A., Acebes Á., Morales M. (2011) Phosphoinositide-3-kinase activation controls synaptogenesis and spinogenesis in hippocampal neurons. J. Neurosci. 31, 2721–2733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rideout H. J., Wang Q., Park D. S., Stefanis L. (2003) Cyclin-dependent kinase activity is required for apoptotic death but not inclusion formation in cortical neurons after proteasomal inhibition. J. Neurosci. 23, 1237–1245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gilbert B. J. (2013) The role of amyloid β in the pathogenesis of Alzheimer's disease. J. Clin. Pathol. 66, 362–366 [DOI] [PubMed] [Google Scholar]
- 35. Lesné S. E., Sherman M. A., Grant M., Kuskowski M., Schneider J. A., Bennett D. A., Ashe K. H. (2013) Brain amyloid-β oligomers in ageing and Alzheimer's disease. Brain 136, 1383–1398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yoshiike Y., Chui D. H., Akagi T., Tanaka N., Takashima A. (2003) Specific compositions of amyloid-β peptides as the determinant of toxic β-aggregation. J. Biol. Chem. 278, 23648–23655 [DOI] [PubMed] [Google Scholar]
- 37. Biswas S. C., Zhang Y., Iyirhiaro G., Willett R. T., Rodriguez Gonzalez Y., Cregan S. P., Slack R. S., Park D. S., Greene L. A. (2010) Sertad1 plays an essential role in developmental and pathological neuron death. J. Neurosci. 30, 3973–3982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Estus S., Tucker H. M., van Rooyen C., Wright S., Brigham E. F., Wogulis M., Rydel R. E. (1997) Aggregated amyloid-β protein induces cortical neuronal apoptosis and concomitant “apoptotic” pattern of gene induction. J. Neurosci. 17, 7736–7745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pike C. J., Walencewicz A. J., Glabe C. G., Cotman C. W. (1991) In vitro aging of β-amyloid protein causes peptide aggregation and neurotoxicity. Brain Res. 563, 311–314 [DOI] [PubMed] [Google Scholar]
- 40. Frautschy S. A., Baird A., Cole G. M. (1991) Effects of injected Alzheimer β-amyloid cores in rat brain. Proc. Natl. Acad. Sci. U.S.A. 88, 8362–8366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fogarty M. P., McCormack R. M., Noonan J., Murphy D., Gowran A., Campbell V. A. (2010) A role for p53 in the β-amyloid-mediated regulation of the lysosomal system. Neurobiol. Aging 31, 1774–1786 [DOI] [PubMed] [Google Scholar]
- 42. She Q. B., Chen N., Dong Z. (2000) ERKs and p38 kinase phosphorylate p53 protein at serine 15 in response to UV radiation. J. Biol. Chem. 275, 20444–20449 [DOI] [PubMed] [Google Scholar]
- 43. Fogarty M. P., Downer E. J., Campbell V. (2003) A role for c-Jun N-terminal kinase 1 (JNK1), but not JNK2, in the β-amyloid-mediated stabilization of protein p53 and induction of the apoptotic cascade in cultured cortical neurons. Biochem. J. 371, 789–798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Schuler M., Green D. R. (2001) Mechanisms of p53-dependent apoptosis. Biochem. Soc. Trans. 29, 684–688 [DOI] [PubMed] [Google Scholar]
- 45. Selznick L. A., Zheng T. S., Flavell R. A., Rakic P., Roth K. A. (2000) Amyloid β-induced neuronal death is bax-dependent but caspase-independent. J. Neuropathol. Exp. Neurol. 59, 271–279 [DOI] [PubMed] [Google Scholar]
- 46. Braun F., Bertin-Ciftci J., Gallouet A. S., Millour J., Juin P. (2011) Serum-nutrient starvation induces cell death mediated by Bax and Puma that is counteracted by p21 and unmasked by Bcl-xL inhibition. PLoS One 6, e23577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Gallenne T., Gautier F., Oliver L., Hervouet E., Noël B., Hickman J. A., Geneste O., Cartron P. F., Vallette F. M., Manon S., Juin P. (2009) Bax activation by the BH3-only protein Puma promotes cell dependence on antiapoptotic Bcl-2 family members. J. Cell Biol. 185, 279–290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lee H. K., Kumar P., Fu Q., Rosen K. M., Querfurth H. W. (2009) The insulin/Akt signaling pathway is targeted by intracellular β-amyloid. Mol. Biol. Cell 20, 1533–1544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Erlacher M., Labi V., Manzl C., Böck G., Tzankov A., Häcker G., Michalak E., Strasser A., Villunger A. (2006) Puma cooperates with Bim, the rate-limiting BH3-only protein in cell death during lymphocyte development, in apoptosis induction. J. Exp. Med. 203, 2939–2951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Selkoe D. J. (2012) Preventing Alzheimer's disease. Science 337, 1488–1492 [DOI] [PubMed] [Google Scholar]
- 51. Kieran D., Woods I., Villunger A., Strasser A., Prehn J. H. (2007) Deletion of the BH3-only protein puma protects motoneurons from ER stress-induced apoptosis and delays motoneuron loss in ALS mice. Proc. Natl. Acad. Sci. U.S.A. 104, 20606–20611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Niizuma K., Endo H., Nito C., Myer D. J., Chan P. H. (2009) Potential role of PUMA in delayed death of hippocampal CA1 neurons after transient global cerebral ischemia. Stroke 40, 618–625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. You H., Yamamoto K., Mak T. W. (2006) Regulation of transactivation-independent proapoptotic activity of p53 by FOXO3a. Proc. Natl. Acad. Sci. U.S.A. 103, 9051–9056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ekoff M., Kaufmann T., Engström M., Motoyama N., Villunger A., Jönsson J. I., Strasser A., Nilsson G. (2007) The BH3-only protein Puma plays an essential role in cytokine deprivation induced apoptosis of mast cells. Blood 110, 3209–3217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ekert P. G., Jabbour A. M., Manoharan A., Heraud J. E., Yu J., Pakusch M., Michalak E. M., Kelly P. N., Callus B., Kiefer T., Verhagen A., Silke J., Strasser A., Borner C., Vaux D. L. (2006) Cell death provoked by loss of interleukin-3 signaling is independent of Bad, Bim, and PI3 kinase, but depends in part on Puma. Blood 108, 1461–1468 [DOI] [PubMed] [Google Scholar]
- 56. Ming L., Sakaida T., Yue W., Jha A., Zhang L., Yu J. (2008) Sp1 and p73 activate PUMA following serum starvation. Carcinogenesis 29, 1878–1884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Futami T., Miyagishi M., Taira K. (2005) Identification of a network involved in thapsigargin-induced apoptosis using a library of small interfering RNA expression vectors. J. Biol. Chem. 280, 826–831 [DOI] [PubMed] [Google Scholar]
- 58. Li J., Lee B., Lee A. S. (2006) Endoplasmic reticulum stress-induced apoptosis: multiple pathways and activation of p53-up-regulated modulator of apoptosis (PUMA) and NOXA by p53. J. Biol. Chem. 281, 7260–7270 [DOI] [PubMed] [Google Scholar]
- 59. Liu D. X., Biswas S. C., Greene L. A. (2004) B-myb and C-myb play required roles in neuronal apoptosis evoked by nerve growth factor deprivation and DNA damage. J. Neurosci. 24, 8720–8725 [DOI] [PMC free article] [PubMed] [Google Scholar]