

Abstract
Iron is an essential nutrient that facilitates cell proliferation and growth. However, iron also has the capacity to engage in redox cycling and free radical formation. Therefore, iron can contribute to both tumour initiation and tumour growth; recent work has also shown that iron has a role in the tumour microenvironment and in metastasis. Pathways of iron acquisition, efflux, storage and regulation are all perturbed in cancer, suggesting that reprogramming of iron metabolism is a central aspect of tumour cell survival. Signalling through hypoxia-inducible factor (HIF) and WNT pathways may contribute to altered iron metabolism in cancer. Targeting iron metabolic pathways may provide new tools for cancer prognosis and therapy.
Elemental iron has crucial functions in mammalian cells. Iron enables the function of vital iron- and haem-containing enzymes, including mitochondrial enzymes that are involved in respiratory complexes, enzymes involved in DNA synthesis and the cell cycle, detoxifying enzymes such as peroxidase and catalase, and many more1. Therefore, iron is essential for cell replication, metabolism and growth. However, the ability to gain and lose electrons — the very attribute that makes iron useful enzymatically — also enables iron to participate in potentially deleterious free radical-generating reactions. Among these is the Fenton reaction, in which ferrous iron donates an electron in a reaction with hydrogen peroxide to produce the hydroxyl radical, a reactive oxygen species (ROS). This reaction not only damages lipids and proteins, but also causes oxidative damage to DNA, including DNA base modifications and DNA strand breaks2,3, which can be mutagenic4. Therefore, iron is both essential and potentially toxic.
Both the beneficial and deleterious effects of iron have a role in cancer. For example, iron may accelerate tumour initiation by enhancing the formation of free radicals, as well as function as a nutrient that fosters tumour cell proliferation. The extent to which and the mechanisms by which iron has such roles have been debated for decades. As early as 1940, exposure to iron oxide dust was shown to triple the incidence of pulmonary tumours in mice5; in the 1950s, intramuscular injection of iron–dextran was shown to induce sarcoma in rats6. In the 1990s, it was demonstrated that the growth rate of tumour xenografts could be influenced by levels of dietary iron7,8. Many years and experiments later, a clearer picture linking excess iron and altered iron metabolism to cancer is emerging, based on evidence ranging from epidemiological to molecular (TABLE 1).
Table 1.
Some cancers in which iron has been implicated
| Type of cancer | Type of evidence | Refs |
|---|---|---|
| Non-small-cell lung cancer | Cell culture, animal models and epidemiological | 5,12,54,79,80,166,172,173 |
| Breast cancer | Cell culture, animal models, human tissue studies and epidemiological |
7,8,20,27,43,47,48,69–71, 126,138,165 |
| Renal cell carcinoma | Cell culture and animal models | 55,139,141–145,174 |
| Hepatocellular cancer | Cell culture, animal models and epidemiological | 7,22,24–26,36,94,175–177 |
| Oesophageal, stomach, aerodigestive and gastric cancer |
Human tissue studies, animal models and epidemiological |
17,26,178,179 |
| Colorectal cancer | Cell culture, human tissue studies, animal models and epidemiological |
7,12,27,45,46,50,95,107, 134–137,180 |
| Prostate cancer | Cell culture and epidemiological | 19,128 |
| Haematological cancers (leukaemias, lymphomas and myeloma) |
Cell culture, animal models, epidemiological and clinical case study |
75,76,137,148,154,181 |
| Melanoma | Cell culture and animal model | 151 |
| Pancreatic cancer | Cell culture, animal models and clinical trial | 37,111 |
| Bladder cancer | Cell culture | 182 |
Unravelling the complex relationship between iron and cancer has been facilitated by the recent discovery of new proteins that participate in and control iron metabolism. For example, newly identified iron efflux pumps, systemic iron regulators, oxidases and reductases that maintain iron in the appropriate valence state, as well as siderophore-binding proteins, are providing resolution to the picture of how tumour cells reprogramme iron metabolism. Studying the role of iron and cancer has also revealed that proteins involved in iron metabolism may be multifunctional and can contribute to malignancy in ways that are independent of their primary role in iron metabolism. Recent studies not only provide insights into cellular and systemic iron metabolism that explain and redefine the relationships between iron and cancer, but may also provide new tools for cancer therapy and for determining prognosis.
Clinical and population-based studies
Population-based studies have taken four general approaches to examine the relationship between iron and cancer risk. Although the results are not always consistent, these studies collectively support a model in which increased levels of iron in the body are associated with increased cancer risk. An overview of mechanisms that regulate the content of body iron in humans and its metabolism in cells is described below (FIGS 1,2). Following uptake from the diet, iron is loaded onto transferrin (TF), which can bind two atoms of ferric (Fe3+) iron. TF-bound iron circulates in the bloodstream and delivers iron to peripheral tissues by binding to transferrin receptor 1 (TFR1), which is a broadly expressed cell surface receptor. The diferric iron–TF–TFR1 complex is endocytosed; in the acidic environment of the endosome, and with the assistance of STEAP reductases, ferric iron is reduced to ferrous iron (Fe2+). Divalent metal transporter 1 (DMT1; also known as NRAMP2) then facilitates the egress of ferrous iron from the endosome into a pool of loosely bound iron, which is termed the labile iron pool. From this pool, iron is delivered to multiple intracellular destinations. It is incorporated into the active site of proteins such as ribonucleotide reductase, where it participates in the catalytic conversion of ribonucleotides to deoxyribonucleotides; iron is also used in the synthesis of haem and iron–sulphur clusters, which are in turn incorporated into proteins that carry out the citric acid cycle, oxidative phosphorylation and many other essential functions. Excess iron, which exceeds the levels required for the synthesis of these proteins, is stored in the iron storage protein ferritin. Iron can also be exported from cells through ferroportin (also known as SLC40A1), which is an iron efflux pump. This process involves oxidases such as ceruloplasmin or hephaestin, which oxidize iron back to the ferric form before loading onto TF. Levels of ferroportin are partly regulated by the circulating peptide hormone hepcidin, which binds to ferroportin and triggers its degradation.
Figure 1. Key features of systemic iron homeostasis in humans.

Dietary iron (predominantly in the form of ferric iron (Fe3+)) is absorbed in the duodenum through the concerted action of a reductase, such as duodenal cytochrome b (DCYTB), which produces ferrous iron (Fe2+), and divalent metal transporter 1 (DMT1). Iron exits the basolateral surface of the enterocyte through the iron efflux pump ferroportin, which functions together with the oxidase hephaestin to oxidize ferrous iron to form ferric iron, which is loaded onto transferrin (TF). The diferric iron transferrin complex (TF–[Fe3+]2) circulates through the bloodstream to deliver iron to sites of utilization. Principal among these sites is the bone marrow, where iron is used in the synthesis of haemoglobin and red blood cells (RBCs). RBCs circulate for approximately 90 days before they are catabolized by macrophages of the reticuloendothelial (RE) system. Iron is released from catabolized haem and effluxed out of the macrophage through the action of ferroportin, where it is loaded onto TF in the bloodstream, in a process termed iron recycling. TF–[Fe3+]2 is also delivered to peripheral tissues and the liver, which is the primary organ for the storage of excess iron. Although small amounts of iron are lost through desquamation, there is no excretory pathway for iron, so levels of iron in the body are primarily regulated at the absorption step. Excess iron induces the synthesis of the peptide hormone hepcidin (HP), which serves as a master regulator of systemic iron homeostasis. HP binds to ferroportin and triggers its degradation, inhibiting both delivery of dietary iron through the enterocyte and iron recycling through the macrophage. HP is also induced in response to inflammatory cytokines and thus contributes to the anaemia of cancer.
Figure 2. Key steps in mammalian cellular iron metabolism.

Iron circulates throughout the body bound to transferrin (TF), which can bind two atoms of ferric iron (Fe3+). TF-bound iron binds to transferrin receptor 1 (TFR1) on the plasma membrane of most cells, and the TF– [Fe3+]2–TFR1 complex is endocytosed. In the acidic environment of the endosome, ferric iron is released from TF and is reduced to ferrous iron (Fe2+) through the ferrireductase activity of STEAP3. The apotransferrin–TFR1 complex then recycles back to the cell surface, where apotransferrin participates in further rounds of iron uptake. In the meantime, ferrous iron is transported out of the endosome into the cytosol by divalent metal transporter 1 (DMT1), and enters the metabolically active pool of iron (the labile iron pool). Iron then traffics to multiple destinations. It is inserted into cytosolic enzymes that are required for DNA synthesis, such as ribonucleotide reductase, and is also used in haem synthesis and the biogenesis of iron–sulphur clusters, processes that occur partly in the mitochondria and partly in the cytosol. Excess iron is stored in ferritin, an iron storage protein. Iron leaves the cell through the activity of ferroportin, an iron efflux pump, and an oxidase such as ceruloplasmin or hephaestin, which can re-oxidize iron to ferric iron to enable the loading onto TF.
In an early study examining the association between biochemical markers of iron stores and cancer, Stevens and co-workers analysed more than 14,000 participants in the first US National Health and Nutrition Examination Survey (NHANES)9,10. They demonstrated that saturation of the iron-binding transporter TF at study enrolment was significantly higher in men who subsequently developed cancer than in those who did not develop cancer. Subsequent studies revealed similar trends11–13. However, analysis did not include other measures that are thought to more closely mirror levels of iron in the body, such as serum levels of ferritin, which is a limitation of these studies.
A second analytical approach has explored the association between dietary iron intake and cancer risk. A meta-analysis of 33 studies assessing iron intake and colorectal cancer revealed that approximately three-quarters of these studies associated higher iron intake with an increased risk of colorectal cancer14. A less consistent association between iron consumed either in the diet or as a supplement and an increased risk of developing other types of cancer has also been observed (for example, see REFS 15,16). Variable results can be partly attributed to inherent difficulties in conducting these studies, such as inferential assessments of iron intake, variability in iron absorption post-intake, assessment of iron intake at a single point in time as a surrogate for long-term exposure and sample size. Recent studies suggest that the dietary source of iron (haem versus non-haem) and genetic polymorphisms in antioxidant enzymes may further complicate these studies17–20.
A third approach has been to use genetically induced accumulation of excess iron (known as iron overload) to infer the consequences on cancer. Hereditary haemochromatosis is a genetic disorder that is characterized by iron overload, particularly in the parenchymal cells of the liver, heart and endocrine organs (reviewed in REF. 21). Liver cirrhosis with hepatocellular carcinoma accounts for approximately 20–30% of deaths in untreated or poorly treated patients with hereditary haemochromatosis22,23, which is a 20–200-fold increased risk for these patients22,24,25. Subjects that carry mutations in haemochromatosis (HFE), one of the mutated genes that underlies hereditary haemochromatosis, may also be at an increased risk of extrahepatic cancer, including breast, colorectal and other cancers26,27.
The effect of a clinically driven reduction in body iron stores on cancer risk is the fourth epidemio logical link between iron and cancer. Thus, repeated phlebotomy over approximately 4.5 years in elderly men with peripheral artery diseases reduced the overall cancer risk (hazard ratio (HR) 0.65; P = 0.036) and cancer-specific mortality (HR 0.49; P = 0.009). Although the authors interpreted their results cautiously, this is consistent with other observations of a decreased risk of several cancers (such as liver, lung, colon, stomach and oesophageal cancer) in individuals who frequently donate blood28.
Intracellular iron regulation is modified in cancer
The past decade, which has been marked by the discovery of new iron-associated proteins and pathways, has been described as a ‘golden age’ of iron metabolism29 (FIG. 2). This decade has reaped the rewards of these discoveries by applying them to deciphering molecular relationships between iron and cancer. Many proteins that were originally studied for their roles in normal iron metabolism have now been shown to contribute to malignant growth. It seems that cancer cells retain most elements of the general iron metabolism pathway, although for many cancer types this has not been rigorously or explicitly studied. However, cancer cells differ from their non-malignant counterparts in the levels or activity of many of the proteins that are involved in iron metabolism. In many cases, the net result of these cancer-specific alterations is an increase in intra cellular iron levels that fuels the activity of iron-dependent proteins and enables enhanced proliferation. In some cases, the downstream consequences of changes in iron-regulated proteins remain unclear.
The regulation of iron uptake in cancer
FIGURE 3 depicts some crucial changes in iron uptake and efflux that have been identified in malignant cells. TFR1 is highly expressed in many cancers, including breast cancer, leukaemia, lymphoma, bladder cancer, lung cancer, glioma and others30. Consequently, TFR1 antibodies have been used to inhibit tumour growth31,32; toxic moieties conjugated to its ligand (TF) have also been widely used for the tumour-selective delivery of anticancer agents30. Some members of the STEAP family of metalloreductases (which participate in iron uptake by reducing endosomal ferric iron to ferrous iron)33 are also over-expressed in cancer, including STEAP1, STEAP2 and STEAP3. However, because STEAP1 does not possess reductase activity34, not all STEAP family members may be associated with cancer through their effects on iron metabolism.
Figure 3. Iron uptake and efflux in malignant and non-malignant cells.

a | Normal epithelial cells express low levels of transferrin (TF) receptor 1 (TFR1) and hepcidin and high levels of ferroportin, which collectively lead to a small pool of labile iron. In breast cells, lipocalin 2 (LCN2), in a complex with a siderophore (SD), may further reduce levels of intracellular iron by capturing and effluxing SD-bound iron from these cells, although this is currently hypothetical. b | Cancer cells show increased expression of TFR1 and hepcidin and low levels of ferroportin, which lead to an increased labile iron pool. In breast cancer cells, LCN2, in a complex with SD-bound iron, may serve as a further source of iron. LCN2R, LCN2 receptor.
Lipocalin 2 (LCN2; also known as NGAL and 24p3) is a less well-studied protein that is involved in an alternative pathway of iron uptake that is also upregulated in some cancers, including breast35, liver36 and pancreatic cancer37. LCN2 binds siderophores, which are low molecular mass iron-binding ligands that are best known for their roles in iron acquisition by bacteria and fungi38. Mammalian cells synthesize catechol, which is an iron-binding molecule with the properties of a siderophore, and iron circulates in the blood bound to an LCN2–catechol complex39. This complex binds to the cell surface receptor 24p3R (also known as SLC22A17) and can serve as a mechanism of iron delivery. By contrast, internalization of unligated LCN2 can serve as a mechanism of iron efflux and can lead to cell death40. Overexpression of LCN2 in MCF7 breast cancer cells increases proliferation41 and increases angiogenesis in a corneal pocket angiogenesis assay42. Conversely, inhibition of LCN2 inhibits breast tumorigenesis in two different mouse models of cancer35,43, although no correlation between LCN2 expression and breast tumour aggressiveness was observed in a different genetic background44. In mouse models of colorectal cancer, LCN2 expression was associated with reduced metastasis45 although, paradoxically, it was also associated with the decreased overall survival of patients with colorectal cancer46. Nevertheless, recent results suggest that LCN2 expression is associated with shorter disease-free survival47 and is an independent prognostic factor for decreased disease-free survival in primary human breast cancer48; LCN2 is also associated with a poor prognosis in hepatocellular carcinoma36.
Regulation of iron storage in cancer
Most cells, including cancer cells, store excess intracellular iron in ferritin, where it can be safely sequestered from participation in radical-generating reactions (FIG. 2). Ferritin is a 24-subunit protein that can store up to 4,500 iron atoms in a ferrihydrite mineral core. It is composed of two subunit types, termed ferritin heavy chain (FTH) and ferritin light chain (FTL) subunits. Ferritin is regulated by iron-regulatory protein 1 (IRP1; also known as ACO1) and IRP2 (also known as IREB2), which post-transcriptionally repress ferritin expression and increase TFR1 expression (discussed further below) (FIG. 4). Expression of the proto-oncogene MYC in B cells induces IRP2 expression and represses ferritin expression49. It has been suggested that the consequent reduction in iron storage and increase in TFR1 (a downstream effect of IRP2 activation) could increase the intracellular availability of iron for metabolic and proliferative purposes. A direct effect of MYC on TFR1 and DMT1 expression was subsequently demonstrated in colon cancer50. The E1a oncogene found in adenovirus similarly represses ferritin51. Studies on cells expressing the HRAS oncogene revealed that the downregulation of ferritin increases the labile iron pool and stimulates proliferation52,53, demonstrating that oncogene-induced changes in the iron storage protein ferritin are sufficient to increase the labile iron pool and increase proliferation. The tumour suppressor p53 may exert the opposite effect: p53 induces ferritin by inactivating IRPs54,55, which was suggested to contribute to p53-mediated growth arrest by restricting the availability of iron54. Downregulation of ferritin increases the sensitivity of breast cancer cells to the chemotherapeutic agents doxorubicin56 and carmustine57, presumably by increasing intracellular oxidative stress.
Figure 4. Control of cellular iron metabolism by the IRE–IRP regulatory axis.

Iron-regulatory protein 1 (IRP1) and IRP2 are crucial proteins in the maintenance of cellular iron homeostasis. These proteins bind to iron-response elements (IREs) present in either the 5′ or the 3′ untranslated region (UTR) of mRNAs. IREs are found in the 5′ UTR of mRNAs encoding the ferritin heavy chain (FTH) and ferritin light chain (FTL) subunits, ferroportin and hypoxia-inducible factor 2α (HIF2α), and in the 3′ UTR of mRNAs encoding transferrin receptor 1 (TFR1) and IRE-containing isoforms of divalent metal transporter 1 (DMT1). Binding of IRPs to 5′ IREs inhibits translation, whereas binding to 3′ IREs stabilizes mRNA. IRPs bind to IREs under conditions of low iron levels; under conditions of high iron levels, IRP1 loses its IRE-binding activity and acquires enzymatic activity as a cytosolic aconitase, whereas IRP2 is degraded. Thus, under conditions of low iron levels, the IRE–IRP system functions to increase iron uptake (by stabilizing mRNAs that encode TFR1 and presumably IRE-containing isoforms of DMT1) and decreases iron storage and efflux (by inhibiting the translation of ferritin and ferroportin). Binding of IRPs to mRNAs encoding HIF2α may function as a feedback loop to inhibit erythropoiesis when iron levels are low171.
Ferritin is also intimately connected to nuclear factor-κB (NF-κB) signalling. NF-κB is a widely expressed transcription factor that is involved in many processes, including inflammation and cancer58. FTH1 is transcriptionally induced by tumour necrosis factor (TNF)59, an effect that is mediated through NF-κB in fibroblasts60. An increase in the ferritin protein (through the induction of FTH1 expression) has a key role in NF-κB-mediated survival signalling: by sequestering iron, ferritin thereby prevents oxidative stress and proapoptotic JUN N-terminal kinase (JNK) signalling, which is activated by oxidative stress61. Functioning upstream of the pathway, extracellular ferritin may also induce NF-κB signalling in hepatic stellate cells62. It is intriguing that the connection between NF-κB and ferritin has not generally been made in cancer cells themselves, but has been made in fibroblasts and inflammatory cells that may model the tumour microenvironment. Thus, both the downregulation and the upregulation of ferritin63 may contribute to tumour survival in the appropriate cellular context: ferritin upregulation may support the survival of inflammatory and stromal cells in the microenvironment, whereas ferritin down-regulation may increase the metabolic availability of iron in the cancer cells themselves, albeit at the cost of persistent oxidative stress64.
The regulation of iron efflux in cancer
Cancer cells increase metabolically available iron not only by increasing iron uptake and decreasing iron storage, but also by decreasing iron efflux. One of the most important recent discoveries in iron biology has been the identification of the ferroportin–hepcidin regulatory axis65. These two proteins, which substantially contribute to the regulation of systemic iron levels, also have a key role in cancer. Ferroportin is the only known iron efflux pump in vertebrates. Its expression on the cell surface of enterocytes is regulated by the circulating peptide hormone hepcidin. When intracellular storage and circulating levels of iron are high, hepcidin is induced in hepatocytes via a bone morphogenetic protein (BMP)-mediated pathway and is secreted into the circulation66. Hepcidin binds to ferroportin on the basolateral side of enterocytes and triggers ferroportin internalization into clathrin-coated pits and its subsequent lysosomal degradation67, thus blocking the delivery of iron from the digestive tract to the blood. Simultaneously, hepcidin binds to ferroportin on macrophages and blocks iron recycling, which further limits iron availability (FIG. 1). Mutations in the ferroportin–hepcidin axis that disrupt this pathway lead to inappropriate iron accumulation and hereditary haemochromatosis21.
Ferroportin is expressed not only in tissues that are important for the regulation of systemic iron homeostasis, but also in breast tissue68,69. Somewhat surprisingly, hepcidin is also expressed in breast epithelial cells69, thus creating the potential for a local autocrine and/or paracrine iron-regulatory loop. Ferroportin is down-regulated in breast cancer cell lines70 and in human breast cancer samples69. Decreased ferroportin levels were associated with increased levels of the labile iron pool in cultured breast cancer cells and with the increased growth of breast tumour xenografts69. Decreased ferroportin expression was significantly associated with a poor prognosis in four separate cohorts comprising approximately 800 patients with breast cancer69. Further, when all patients were combined, in women whose tumours expressed high levels of ferroportin, concomitant expression of high levels of hepcidin reduced distant metastasis-free survival. Interrogation of gene expression in a homogeneously treated subpopulation of 276 oestrogen receptor-positive (ER+) tamoxifen-treated patients demonstrated that the combination of high ferroportin and low hepcidin was associated with a favourable prognosis, even in women with lymph node metastases. This suggests that the measurement of ferroportin and hepcidin levels in breast tumours could be used in breast cancer prognosis. Furthermore, because the end point of these analyses was the presence of metastasis, these studies indirectly suggest that ferroportin levels are important not only in the growth of primary tumours, but also in metastatic spread.
Subsequent work revealed the association of additional ‘iron genes’ with breast cancer prognosis71. Sixty-one genes with functions related to iron metabolism were selected and analysed for their association with the survival of patients with breast cancer71. The expression of 49% of these genes was significantly associated with distant metastasis-free survival, a much larger proportion than would be expected by chance (P <0.02). Most of the prognostic information contained in these genes could be captured using 16 of the 61 genes; this 16 gene set was termed the iron-regulatory gene signature (IRGS), and could be successfully used to discriminate among patients with breast cancer who had a high, a medium or a low risk of distant metastasis-free survival. This included lymph node-positive tamoxifen-treated patients, who are generally considered a high-risk group; assessment of the IRGS in these patients may spare some of these women unneeded chemotherapy.
Several gene signatures are likely to be embedded in the IRGS71. For example, the expression of two separate gene dyads was particularly important in determining patient outcome: TFR1 (also known as TFRC) and HFE, which control iron import; and ferroportin and hepcidin, which control iron export71. These two dyads seem to represent alternative pathways that lead to the same outcome: tumours with a low iron uptake profile (TFR1lowHFEhi) did not frequently overlap with those that expressed a high iron efflux profile (ferroportinhihepcidinlow), but both conferred a favourable prognosis71. These results suggest that the expression of multiple iron pathways affects the prognosis of patients with breast cancer.
In addition to the effects of hepcidin that is synthesized by tumours, systemic hepcidin that is synthesized by the liver contributes to cancer in other ways. Many cancer patients suffer from anaemia of chronic disease, a syndrome that is characterized by iron deregulation and anaemia72. It has been argued that the systemic lack of iron availability that is characteristic of this syndrome, which is sometimes referred to as the iron withholding response, is a host response that combats cancer by depriving tumours of iron73,74. Whatever its teleological explanation, anaemia of chronic disease is linked to the upregulation of hepcidin — which is induced by BMP2 in myeloma75 and by interleukin-6 (IL-6) in Hodgkin’s lymphoma76. Targeting this pathway may be of substantial clinical benefit in ameliorating this syndrome, which is estimated to affect 40–70% of cancer patients72. However, the potential of the ensuing increase in iron absorption to promote tumour growth will have to be considered when designing such a strategy.
Iron-regulatory proteins
The conceptual link between cancer and proteins that regulate iron uptake or efflux is straightforward, because levels of these proteins directly affect intracellular iron content. How other proteins that are associated with iron regulation influence cancer is less certain; for example, IRP1 and IRP2. These proteins are master regulators of intracellular iron homeostasis that function to increase iron uptake when intracellular iron levels are low and decrease cellular iron uptake when intracellular iron levels are high (FIG. 4). This is accomplished by IRP-mediated translational regulation of the proteins that are associated with iron import, storage and efflux. When iron is scarce, IRPs bind to iron-response elements (IREs) in the 5′ untranslated region (UTR) of ferritin and ferroportin mRNAs, which blocks their translation and consequently inhibits iron storage and efflux. At the same time, IRPs also bind to IREs in the 3′ UTR of TFR1 mRNA, which stabilizes the mRNA and increases iron import. A 3′ IRE is also present and contributes to iron-dependent regulation of IRE-containing isoforms of the iron transporter DMT1, presumably through a mechanism similar to that of TFR1 (REFS 77,78). Despite the apparently similar roles of IRP1 and IRP2, overexpression of IRP1 reduces tumour growth in vivo, whereas overexpression of IRP2 promotes tumour growth in vivo79,80. The reason for these opposing phenotypes has not been explained, although it has been suggested that IRP2 may have functions that are unrelated to its role in iron metabolism, such as the induction of MYC and MAPK signalling79, which may account for its tumour-promoting effects.
Iron and the tumour microenvironment
Tumours exist in a rich microenvironment that includes endothelial cells and macrophages, among other cell types81. M2-polarized macrophages, which resemble tumour-associated macrophages, were recently shown to exhibit a gene expression profile that is consistent with iron efflux: an increase in ferroportin and a decrease in ferritin82. Conditioned media from M2 macrophages promoted tumour cell proliferation, an effect that was inhibited by iron chelation, suggesting that macrophages may foster tumour growth partly by providing tumour cells with iron82,83. Ferritin, which is best known for its role in intracellular iron storage, is also secreted by macrophages84. Independently of its role as a potential additional iron source85,86, secreted ferritin may promote tumour angiogenesis by binding cleaved high molecular weight kininogen (HMWK; also known as kininogen 1, the cleaved form of HMWK is termed HKa), which is an endogenous angiogenesis inhibitor87,88.
The iron ‘metabolic switch’ and hypoxia
Frataxin is a mitochondrial protein that is implicated as a cause of the neurodegenerative disorder Friedreich’s ataxia. Although cancer is not a hallmark of the disease, there are several reports of cancer in young patients with Friedreich’s ataxia89,90. Frataxin has a crucial role in the initial steps of iron–sulphur cluster biogenesis, possibly by serving as an iron donor or as an allosteric regulator (reviewed in REF. 91). Frataxin dysfunction leads to the accumulation of mitochondrial iron92, and the production of ROS and oxidative stress93. Liver-specific knockout of frataxin impairs mitochondrial function and promotes the development of liver tumours in mice94. Conversely, the overexpression of frataxin in human colon cancer cell lines decreases their growth, colony formation in soft agar and tumour growth in xenografts95. As overexpression of frataxin also increased oxidative metabolism (as measured by increased mitochondrial membrane potential, cell respiration and ATP content), it was speculated that frataxin may inhibit tumour formation by reversing the metabolic switch to aerobic glycolysis (the Warburg effect), which occurs in many cancer cells95. Whether frataxin indeed exerts its effects by modifying metabolism and/or by other mechanisms, including the prevention of oxidative DNA damage and fostering DNA repair96, has not yet been definitively addressed.
Iron levels can also regulate hypoxia-inducible factor-α (HIFα) proteins, transcription factor sub units that are central to the regulation of the response to hypoxia (reviewed in REF. 97). There are three HIFα family members: HIF1α, HIF2α and HIF3α. These subunits form heterodimers with HIF1β (also known as ARNT) to form the HIF transcription factors, which transcriptionally induce numerous genes that are important in the response to hypoxia, including vascular endothelial growth factor A (VEGFA), erythropoietin (EPO), glucose transporter 1 (GLUT1; also known as SLC2A1) and survivin (also known as BIRC5). HIFα subunits are post-translationally regulated by prolyl hydroxylases (PHD proteins), which are iron-, 2-oxoglutarate- and oxygen-dependent enzymes that modify HIFα subunits, and this modification targets them for proteasomal degradation. In conditions of normoxia, HIFα is degraded, but under conditions of hypoxia, HIFα is stabilized. Because the PHD proteins require iron for their activity, the HIF transcription factors are also stabilized (and become transcriptionally active) when intracellular iron levels are low.
HIF activity is increased in many tumours98, and upregulation of HIFα is frequently associated with tumour growth and progression97. The stabilization of HIFα in tumours at first seems to be inconsistent with a view of tumour cells as iron-avid cells. However, two sets of observations may reconcile this apparent paradox. First, HIF can be activated by hypoxia even under conditions of high iron levels99. Thus, HIF activation in a tumour environment does not necessarily connote low iron levels. Second, once activated, HIF1 promotes iron uptake. In cell culture, HIF1 induces TFR1 expression, thus increasing iron uptake100,101. HIF1 also induces the expression of haem oxygenase 1 (HO1)102, which degrades haem into biliverdin, carbon monoxide and iron; this intracellular release of iron by HO1 enables iron to be recycled. HIF1 also induces the expression of ceruloplasmin, which oxidizes iron to facilitate the loading of iron onto TF103. In addition to modulating cellular iron levels, HIF2 promotes systemic iron uptake by inducing the expression of ferroportin, DMT1 and duodenal cytochrome b (DCYTB; also known as CYBRD1) in enterocytes104–106. Thus, HIF activation may contribute to enhanced iron accumulation in tumours. Supporting this view, activated HIF2 was recently shown to promote colorectal cancer by inducing DMT1 expression and increasing iron uptake in Apcmin/+ mice107.
Iron regulates DNA metabolism and the cell cycle
The involvement of iron in processes related to DNA replication, maintaining genomic integrity (including DNA repair) and epigenetic regulation contribute both to the tumour-initiating and to the tumour-promoting propensities of iron (FIG. 5). Cells starved of iron accumulate in the G1 phase of the cell cycle108, which is consistent with a crucial role for iron in DNA synthesis. Ribonucleotide reductase catalyses the rate-limiting step in DNA synthesis, the reductive conversion of ribonucleotides to deoxyribonucleotides. This enzyme is essential for cell viability and is a target of the chemotherapeutic agenthydroxyurea. The catalytic activity of ribonucleotide reductase is dependent on a dinuclear iron site in the M2 subunit of the enzyme ribonucleoside-diphosphate reductase subunit M2 (RRM2)109,110. The dependence of ribonucleotide reductase activity on iron has motivated the search for anticancer drugs that inhibit its activity through iron chelation; several of these are currently being tested in preclinical and clinical studies111,112 (dis cussed below) (TABLE 2). More recently, a p53-inducible RRM2 subunit (p53R2; also known as RRM2B) has been discovered113, which is induced in response to DNA damage. p53R2 has a >100-fold increased susceptibility to iron chelation114, possibly owing to a unique pore in the immediate vicinity of the iron-binding site, as demonstrated by X-ray crystallography115. Because p53R2 is less susceptible to inhibition by hydroxyurea than RRM2, but is sensitive to iron chelation, it represents a target for iron chelation therapy in tumours with wild-type p53.
Figure 5. Links between iron, DNA metabolism and genomic integrity.
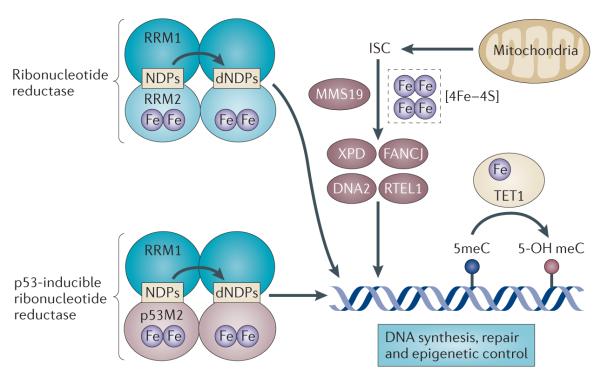
Iron is essential for the activity of the enzymes involved in DNA synthesis, DNA repair and epigenetic regulation. A di-iron site is essential for the catalytic activity of both constitutive and p53-inducible ribonucleotide reductase, the enzyme that catalyses the reductive conversion of ribonucleotides (NDPs) to deoxyribonucleotides (dNDPs) for DNA synthesis. MMS19 serves as a scaffold for the insertion of iron–sulphur clusters (ISCs) into DNA repair enzymes such as Xeroderma pigmentosum group D-complementing protein (XPD), Fanconi anaemia group J protein (FANCJ), DNA replication helicase 2 homologue (DNA2) and regulator of telomere elongation helicase 1 (RTEL1). The ferrous iron- and 2-oxoglutarate-dependent enzyme TET1 catalyses the hydroxylation of methylcytosines (meCs) in the DNA and may have a role in epigenetic control. RRM, ribonucleoside-diphosphate reductase subunit M.
Table 2.
Examples of iron chelators undergoing evaluation as anticancer agents
| Chelator name | Chelator properties | Type of study | Refs* |
|---|---|---|---|
| Ciclopirox | Fungicide | Preclinical and clinical | 137,167,181,183 |
| Tachpyridine | Ferrous iron chelator | Preclinical | 172,182,184,185 |
| Dp44mT | Di-pyridylketone thiosemicarbazone | Preclinical | 151 |
| DpC | Di-pyridylketone thiosemicarbazone analogue | Preclinical | 186 |
| BpT | 2-benzoylpyridine thiosemicarbazone | Preclinical | 187 |
| TSC24 | Thiosemicarbazone | Preclinical | 175 |
| Deferasirox | Orally available iron chelator used in the treatment of iron overload |
Preclinical and single patient case report |
173,188 |
| Triapine | 3-aminopyridine-2-carboxaldehyde thiosemicarbazone |
Preclinical and clinical |
189–192, NCT00941070 |
| Desferoxamine (DFO) | Iron chelator used in the treatment of iron overload |
Preclinical and clinical | 149 |
For clinical trial identification number NCT00941070 see the ClinicalTrials.gov website.
The iron dependence of DNA synthesis may extend beyond RRM2. All four replicative DNA polymerases in yeast were recently shown to contain an iron–sulphur cluster, [4Fe–4S], which has an essential role in their function116. DNA synthesis thus directly depends on components of iron–sulphur cluster biogenesis from mitochondria and the cytosol, which may explain why mitochondrial dysfunction (and thus defective iron–sulphur cluster biogenesis) leads to genome instability117. Defective iron–sulphur cluster biogenesis may also contribute to genome instability by inhibiting the activity of iron–sulphur cluster-dependent enzymes that are involved in DNA repair and recombination, including Xeroderma pigmentosum group D-complementing protein (XPD; also known as ERCC2), Fanconi anaemia group J protein (FANCJ)118, DNA replication helicase 2 homologue (DNA2)119 and regulator of telomere elongation helicase 1 (RTEL1)120,121. Consistent with this concept, defects in the assembly of a subset of iron–sulphur cluster-containing proteins augment the sensitivity of cells to DNA damage121. Additionally, TET1, a ferrous iron- and 2-oxoglutarate-dependent enzyme that is disrupted in some haematological malignancies122, catalyses the conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA. 5-hydroxymethylcytosine is a modified DNA base that is implicated in the early stages of carcinogenesis123. This suggests that epigenetic regulation may represent yet another pathway through which iron can alter DNA function and induce malignant change124.
Proteins that control the cell cycle are also regulated by iron levels (reviewed in REF. 125). Cyclin D1 associates with cyclin-dependent kinase 4 (CDK4) and CDK6 to regulate G1/S progression by phosphorylating RB, which in turn releases the transcription factor E2F from RB. Work with chemically unrelated iron chelators in breast and kidney cancer cell lines demonstrated that iron depletion reduces cyclin D1 expression by stimulating cyclin D1 proteasomal degradation126,127. Thus far, how iron deprivation promotes this degradation pathway remains unknown.
Iron affects WNT signalling
Effects on signalling represent a fairly unexplored mechanism through which iron affects tumour cell proliferation and growth. Although iron has been implicated in several signalling pathways60–62,128–131, the link between iron and WNT signalling may be particularly important in cancer (FIG. 6). Aberrant WNT signalling contributes to many types of cancer (reviewed in REFS 132,133). WNT signalling culminates in the accumulation of β-catenin, which activates the T cell factor (TCF)–lymphoid enhancer factor (LEF) transcription factor complex that induces the expression of target genes such as MYC. WNT signalling is regulated through a destruction complex composed of adenomatous polyposis coli (APC), axin, casein kinase 1 (CK1) and glycogen synthase kinase 3β (GSK3β), which actively targets β-catenin for degradation. Some β-catenin is also sequestered from the destruction complex through an association with E-cadherin. Because inactivating mutations in APC are an early event in colorectal cancer, and because alterations in iron transport proteins have been observed in colorectal cancer tissue, the role of iron in WNT signalling has been investigated134. It was found that iron has two important effects: it augments WNT signalling in cells with aberrant APC or β-catenin, and it also downregulates E-cadherin in an APC-independent manner. These effects of iron on WNT signalling may provide a mechanistic explanation for how iron exacerbates intestinal tumorigenesis, particularly in a background of APC mutation.
Figure 6. A role for iron in canonical WNT signalling.

a | In the absence of a trigger for WNT signalling, β-catenin associates with axin, adenomatous polyposis coli (APC), casein kinase 1 (CK1) and glycogen synthase kinase 3β (GSK3β) (collectively known as the destruction complex) and is targeted for degradation (shown by dashed outline). Simultaneously, β-catenin is sequestered from the destruction complex and associates with E-cadherin. b | In cells with constitutive canonical WNT signalling (such as cells with mutant APC), β-catenin evades destruction, enters the nucleus and promotes T cell factor (TCF)–lymphoid enhancer factor (LEF)-dependent transcription of downstream target genes such as MYC. Iron promotes TCF–LEF-dependent transcription in cells with such constitutive WNT signalling, resulting in the induction of MYC expression. MYC in turn transcriptionally induces transferrin receptor 1 (TFR1) and divalent metal transporter 1 (DMT1) to promote iron uptake. Iron also decreases E-cadherin mRNA and protein levels in an APC-independent manner. c | Iron chelators decrease TCF–LEF signalling in cells with constitutive WNT signalling at a step distal to β-catenin by an unknown mechanism. DVL, disheveled homologue; FZD, frizzled homologue; LRP, low-density lipoprotein receptor-related protein.
These cell culture observations were supported by recent experiments in Apcmin/+ mice50, a mouse model of intestinal cancer in which APC is inactivated. High levels of dietary iron accelerated tumour formation and low iron levels reduced tumour formation in this mouse model. High dietary iron levels induced MYC, TFR1 and DMT1 expression in intestinal polyps from Apcmin/+ mice, as well as human adenomas and carcinomas50. Intriguingly, the stem cell compartment was particularly responsive to iron manipulation. Further, dietary iron, but not systemic iron, was crucial to intestinal tumorigenesis: the reduction in tumour burden in mice fed a low-iron diet was not reversed by raising levels of circulating iron by subcutaneous injection of iron–dextran. In addition, lowering levels of circulating iron through the administration of an iron chelator did not provide the same protection as dietary iron restriction. This suggests that dietary iron may be more effective than systemic iron in modifying WNT signalling, at least in the intestinal epithelium, which is directly exposed to dietary iron in a way that most tissues are not. Supporting the finding that dietary iron increases cancer risk, iron-enriched diets have similarly been shown to increase colorectal tumour incidence in a mouse model of colitis135; a high-iron diet also enhanced proliferation and the formation of large adenomas in an azoxymethane-induced mouse model of colon cancer136. Conversely, low-iron diets reduced the growth of colon cancer (as well as mammary adenocarcinoma and hepatoma) xenografts in mice7.
Reinforcing the dependence of WNT signalling on iron, two independent groups identified iron chelators as top hits in high-throughput screens for WNT inhibitors137,138. In one study137, several acyl hydrazones were identified as inhibitors of WNT signalling that function downstream of the destruction complex. A novel, structurally distinct iron chelator, HQBA, was identified in another study as a potent inhibitor of WNT signalling138. HQBA binds ferrous iron with high affinity (dissociation constant 1.2 × 10−19M) and inhibited the growth of spontaneous mammary tumours in both mouse mammary tumour virus (MMTV)-Wnt1 and MMTV-polyoma middle T (PyMT) mouse models. In line with these findings, leukaemic blasts from seven of nine patients with acute myeloid leukaemia (AML) who were treated in a Phase I trial with another chelator, ciclopirox, exhibited a time-dependent decrease in the expression of AXIN2, a member of the WNT signalling pathway137. Although the iron chelators identified by both groups function downstream of β-catenin to inhibit WNT signalling, the precise target of these chelators remains to be determined.
Aberrant WNT signalling was also observed in an iron-induced mouse model of kidney cancer. In a series of elegant experiments, ferric nitrilotriacetate (FeNTA), a soluble iron salt, was shown to induce nephrotoxicity and kidney cancer in rats and mice139–142. Nitrilotriacetate (NTA) itself was non-carcinogenic, implicating iron as the causative agent in this compound141. Direct analysis of chromatin from the kidneys of rats treated with FeNTA revealed the presence of 8-hydroxy-2′-deoxyguanosine (8-OH-dG) and other modified DNA bases that are hallmarks of oxidatively damaged DNA142. When the mutation range induced by FeNTA in vivo was evaluated, transversions, substitutions and deletions were identified, as well as the loss of CDK inhibitor 2A (Cdkn2a; which encodes the tumour suppressors ARF and INK4A)143,144. High-resolution microarray comparative genomic hybridization of renal tumours that were induced by FeNTA in rats revealed extensive genomic alterations, including amplifications and deletions, with Met amplification and Cdkn2a and Cdkn2b deletion being the most common alterations145. The global pattern of genetic alterations closely paralleled those seen in human renal carcinoma145. FeNTA-induced tumours demonstrated a 16–500-fold increase in the expression of receptor-type tyrosine-protein phosphatase-ζ (RPTPζ; also known as PTPRZ1). RPTPζ participates in multiple signalling pathways146,147. The authors observed that, concomitantly with RPTPζ overexpression, nuclear β-catenin expression was increased, with significant activation of the β-catenin target genes cyclin D1 (Ccnd1), Jun, Myc, fos-like antigen 1 (Fosl1; also known as Fra1) and Cd44. These studies in kidney cancer, together with results indicating that iron potentiates WNT signalling in colorectal cancer134 (discussed above), suggest that the activation of WNT signalling may be a common pathway through which iron contributes to malignant progression.
Iron is a target for cancer therapy and prevention
Iron chelators are natural or synthetic small molecules that bind iron with a high affinity. Several iron chelators, such as desferoxamine (DFO), deferiprone and deferasirox are used clinically for the treatment of patients with iron overload disorders. The avidity of cancer cells for iron has led to the question of whether iron chelators could be used in cancer therapy. Two broad strategies have been explored. The first has been to use iron chelators to deplete cancer cells of iron. A second, more recent strategy has been to use chelators that facilitate the redox cycling of iron to generate cytotoxic ROS within tumours. Both approaches are currently being pursued.
Owing to its safety profile, early studies of the anti-cancer activity of iron chelators used DFO. DFO caused leukaemic cytoreduction in a patient with acute leukaemia148 and a 20% overall response rate in a study of ten patients with treatment-refractory metastatic hepatocellular carcinoma149, prompting an extended search for more effective iron chelators (reviewed in REFS 112,150). Iron chelators that are under preclinical or early clinical investigation as anticancer therapeutics are listed in TABLE 2. An unanticipated recent discovery has been that iron chelators induce the expression of the metastasis suppressor NMYC downstream regulated gene 1 (NDRG1)151. NDRG1 expression is decreased in a number of cancers, and its overexpression reduces the invasion and metastasis of some cancers — for example, prostate and colorectal cancer — but not of all cancers152. The induction of NDRG1 was associated with the inhibition of the epithelial–mesenchymal transition (EMT) in colon and prostate cancer cells153, suggesting the exciting possibility that iron chelators may not only inhibit the growth of primary tumours, but may also potentially inhibit metastatic spread.
New-generation single-chain antibodies targeted towards TFR1 that effectively deplete intracellular iron are also under investigation; these have been shown to effectively antagonize the growth of leukaemia in mice154. The use of TFR1 as a tumour-targeting ligand for the delivery of numerous antitumour cytotoxics is also being actively pursued. Examples include TF–chemotherapeutic drug conjugates, such as TF–doxorubicin, TF–cisplatin and TF–chlorambucil; TF–cytotoxin conjugates such as TF–ricin A chain and TF–diptheria toxin (TF-CRM107); and TF-conjugated micelles, dendrimers and other larger moieties carrying antineoplastic nucleic acids30.
The development of iron chelators or other iron-restrictive strategies as chemopreventives represents additional opportunities. Curcumin, a pigment found in turmeric that is commonly used in curry and with a long history of use in traditional Indian Ayurvedic medicine155, is a cancer chemopreventive and some of its activity may be attributable to its ability to che-late iron156,157. The excellent safety profile of curcumin is congruent with its long-term use as a chemopreventive150,155. Curcumin has also served as a springboard for the design of new and potentially more potent synthetic chemopreventive agents158,159. The iron-binding activity of garcinol, a structurally similar substance that is derived from shrubs native to India and Southeast Asia, may also account for its ability to prevent oral tumours160.
Conclusions
Substantive and transformative evidence, much of it obtained in the past 5 years, implicates changes in the uptake and management of iron as crucial features of cancer, and suggests that altered iron metabolism is a key metabolic ‘hallmark of cancer’ (REF. 161). These advances are both conceptual and factual. The over-arching theme that emerges is that iron has roles in all aspects of cancer development, including the tumour microenvironment and metastasis. Further, the iron biology of a tumour, as evidenced by the expression pattern of ‘iron genes’ in malignant tumours, is not simply associated with cancer, but also has a role of sufficient magnitude to indicate a patient’s chances of survival. In breast cancer, for example, almost 50% of all genes involved in the regulation or maintenance of iron metabolism were significantly associated with clinical outcome71. Thus, iron is more deeply embedded in tumour cell biology than has been previously understood.
Consistent with this scenario, a plethora of iron-regulatory proteins have recently been linked to cancer. The precise contributions of this expanded roster of players are not yet fully understood, but clearly represent new directions for investigation. For example, the recently appreciated role of iron–sulphur cluster biogenesis in DNA repair, replication and modification suggests that this will have an important impact on tumour initiation and progression. Supporting this contention, iron–sulphur cluster scaffold homologue (ISCU), iron–sulphur cluster assembly 1 homologue (ISCA1) and cytosolic iron– sulphur protein assembly 1 (CIAO1) were significantly associated with the survival of cancer patients71.
Clear mechanistic links between iron and signalling in cancer cells are emerging. The identification of the WNT signalling pathway as a key target of iron is perhaps the most important recent example. The identification of this pathway by multiple different laboratories using unrelated approaches suggests that further interrogation of this relationship will yield more detailed mechanistic insights, as well as opportunities for therapeutic intervention.
Just as our biosphere has evolved in an iron-limited environment, living organisms have evolved to tightly partition and limit available iron. Another emerging theme is that tumours create their own iron-rich micro-environment to evade constraints that are imposed by limited systemic iron availability. Compared with non-malignant mammary tissue, breast tumours downregu-late ferroportin and upregulate hepcidin, subverting normal homeostatic controls to acquire more than their fair share of iron71. This recent evidence for autocrine or paracrine regulation of iron in the tumour micro-environment represents a new paradigm in iron biology. Cancer cells can also invert the normal relationship between iron and its regulatory proteins: in the Apcmin/+ mouse model of colorectal cancer, iron induces the expression of TFR1 and DMT1 in the intestine, increasing the iron content of these cells50. This induction of TFR1 expression by iron is opposite to what is observed during the maintenance of normal iron homeostasis (in which iron decreases TFR1 expression).
There are many remaining questions that need to be addressed. For example, are changes to iron-regulatory proteins passengers or drivers in malignant change? Some iron-regulatory proteins may be oncogenic, such as IRP2; others, such as IRP1 and frataxin, may be tumour suppressive. Enhanced iron acquisition and/or retention are characteristics of many tumours, and tumour growth can be directly affected simply by manipulating the expression of TFR1 (REFS 70,154), ferritin162,163 or ferroportin69. Thus, altered levels of at least some iron-regulatory proteins do not merely accompany malignant change, but directly drive tumour growth.
What prognostic and therapeutic opportunities do the connections between iron and cancer provide? Studies in breast cancer suggest that the expression of genes and proteins of iron metabolism could be used to evaluate cancer prognosis and to guide therapy69,71. Ferritin may be a useful prognostic indicator in cancer: although increased ferritin levels in the serum are not tumour-specific164, recent studies have suggested that FTL expression in tumour-associated macrophages may be prognostic in node-negative breast cancer165; ferritin present in breath condensate has even been suggested as a potential prognostic indicator for non-small-cell lung cancer166. Therapeutically, targeting iron-sensitive nodes of growth-regulatory pathways may provide a novel strategy. In addition, iron chelators are a promising approach to targeting iron-regulatory pathways. Although reducing tumour cell iron levels without lowering systemic iron may be a challenge, the conjugation of chelators to tumour-targeting ligands could be used to concentrate chelators at the tumour site to overcome this problem. A complementary strategy would be to focus on chelators that foster oxygen radical formation, as these would be more, rather than less, effective in an environment that is rich in iron112. Further, the discovery that iron chelators target the WNT pathway137,138,167 may offer a specific new way to direct iron chelator therapy: tumours with deregulated WNT signalling may represent particularly vulnerable targets.
Much has been done; and much more needs to be done. A more detailed understanding of specific pathways and proteins that are targeted by iron depletion is needed to optimize therapies targeted towards iron depletion. A more comprehensive picture of exactly how iron metabolism is altered in malignant cells would be of substantial benefit for identifying key points of control that can be therapeutically manipulated; systems biology approaches may be helpful in this regard168–170. The rich vein of iron metabolism will be fully mined when we understand not only normal pathways of iron homeostasis, but also the perturbations of these pathways that initiate and sustain cancer.
At a glance.
Elemental iron is essential for cellular growth and homeostasis but it is potentially toxic to cells and tissues. Excess iron can contribute to tumour initiation and tumour growth.
Epidemiological evidence links increased body iron stores to increased cancer risk. High intake of dietary iron is associated with an increased risk for some cancers, particularly colorectal cancer. Hereditary haemochromatosis, a genetic disease that leads to excess iron accumulation, is associated with increased cancer risk.
Many types of cancer cells reprogramme iron metabolism in ways that result in net iron influx. They upregulate proteins that are involved in iron uptake, such as transferrin receptor 1 (TFR1), STEAP proteins and lipocalin 2 (LCN2), and decrease the expression of iron efflux proteins, such as ferroportin. Other iron-regulatory proteins, such as IRP1 and IRP2, contribute to cancer in ways that are less well understood.
Iron is crucial to many fundamental cellular processes, including DNA synthesis, proliferation, cell cycle regulation and the function of proteins containing iron–sulphur clusters. Iron–sulphur cluster-containing proteins include enzymes that contribute to maintaining genomic stability, as well as respiratory function.
Iron regulates crucial signalling pathways in tumours, including the hypoxia-inducible factor (HIF) and WNT pathways.
Measuring the expression of genes encoding proteins involved in iron metabolism may be useful in cancer prognosis. The expression of ferroportin, hepcidin, TFR1, haemochromatosis (HFE) and other genes involved in iron metabolism is linked to the prognosis of patients with breast cancer.
Iron is a target for cancer therapy. Iron chelators, TFR1 antibodies and cytotoxic ligands conjugated to transferrin (TF) represent some ways in which iron is being exploited therapeutically.
Acknowledgements
Supported in part by grants R01 CA171101 (F.M.T.) and R01DK071892 (S.V.T.) from the US National Institutes of Health.
Glossary
- Fenton reaction
A chemical reaction in which ferrous iron reacts with hydrogen peroxide to produce the hydroxyl radical. Iron oxidized during this reaction can be reduced back to ferrous iron in the presence of superoxide (a by-product of respiration). The sum of these reactions is the iron-catalysed formation of hydroxyl radicals from superoxide (termed the Haber–Weiss reaction).
- Siderophore
A low molecular mass compound that has a high affinity for chelating iron.
- Iron–sulphur clusters
Assemblies of iron and inorganic sulphur that function as protein cofactors.
- Hereditary haemochromatosis
Inherited disorder caused by mutations in several different genes that leads to the accumulation of iron to excess levels in parenchymal tissues.
- Phlebotomy
Drawing or removing blood from the circulation.
- Enterocytes
Intestinal epithelial cells that have major roles in the absorption of nutrients, including iron.
- Iron recycling
Reuse of cellular iron. Typically occurs through the catabolism of senescent red blood cells by macrophages of the liver and spleen.
- Friedreich’s ataxia
Inherited disorder of the neurodegenerative system.
- Warburg effect
The propensity of cancer cells to shift from aerobic respiration to glycolysis for the generation of ATP, even in the presence of adequate oxygen levels. The name derives from the hypothesis proposed by Otto Warburg in 1924 that cancer is driven by the non-oxidative breakdown of glucose.
- Acyl hydrazones
Chemical substances containing oxygen and nitrogen donor ligands that coordinate iron.
- Cytoreduction
Decreasing the number of cancer cells.
Footnotes
Competing interests statement The authors declare no competing financial interests.
DATABASES ClinicalTrials.gov: http://clinicaltrials.gov/NCT00941070
National Cancer Institute Drug Dictionary: http://www.cancer.gov/drugdictionary carmustine | curcumin | deferasirox | doxorubicin |hydroxyurea | TF-CRM107
FURTHER INFORMATION Suzy V. Torti ’s homepage: http://facultydirectory.uchc.edu/profile?profileId=torti-suzy
Frank M. Torti ’s homepage: http://medicine.uchc.edu/about/torti.html
ALL LINKS ARE ACTIVE IN THE ONLINE PDF
References
- 1.Crichton R. Iron Metabolism: from Molecular Mechanisms to Cinical Consequences. John Wiley and Sons; 2009. pp. 17–58. [Google Scholar]
- 2.Inoue S, Kawanishi S. Hydroxyl radical production and human DNA damage induced by ferric nitrilotriacetate and hydrogen peroxide. Cancer Res. 1987;47:6522–6527. [PubMed] [Google Scholar]
- 3.Dizdaroglu M, Rao G, Halliwell B, Gajewski E. Damage to the DNA bases in mammalian chromatin by hydrogen peroxide in the presence of ferric and cupric ions. Arch. Biochem. Biophys. 1991;285:317–324. doi: 10.1016/0003-9861(91)90366-q. [DOI] [PubMed] [Google Scholar]
- 4.Dizdaroglu M, Jaruga P. Mechanisms of free radical-induced damage to DNA. Free Radic. Res. 2012;46:382–419. doi: 10.3109/10715762.2011.653969. [DOI] [PubMed] [Google Scholar]
- 5.Campbell JA. Effects of precipitated silica and of iron oxide on the incidence of primary lung tumours in mice. Br. Med. J. 1940;2:275–280. doi: 10.1136/bmj.2.4156.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Richmond HG. Induction of sarcoma in the rat by iron-dextran complex. Br. Med. J. 1959;1:947–949. doi: 10.1136/bmj.1.5127.947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hann HW, Stahlhut MW, Blumberg BS. Iron nutrition and tumor growth: decreased tumor growth in iron-deficient mice. Cancer Res. 1988;48:4168–4170. [PubMed] [Google Scholar]
- 8.Hann HW, Stahlhut MW, Menduke H. Iron enhances tumor growth. Observation on spontaneous mammary tumors in mice. Cancer. 1991;68:2407–2410. doi: 10.1002/1097-0142(19911201)68:11<2407::aid-cncr2820681113>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 9.Stevens RG, Graubard BI, Micozzi MS, Neriishi K, Blumberg BS. Moderate elevation of body iron level and increased risk of cancer occurrence and death. Int. J. Cancer. 1994;56:364–369. doi: 10.1002/ijc.2910560312. [DOI] [PubMed] [Google Scholar]
- 10.Stevens RG, Jones DY, Micozzi MS, Taylor PR. Body iron stores and the risk of cancer. New Engl. J. Med. 1988;319:1047–1052. doi: 10.1056/NEJM198810203191603. [DOI] [PubMed] [Google Scholar]
- 11.van Asperen IA, Feskens EJ, Bowles CH, Kromhout D. Body iron stores and mortality due to cancer and ischaemic heart disease: a 17-year follow-up study of elderly men and women. Int. J. Epidemiol. 1995;24:665–670. doi: 10.1093/ije/24.4.665. [DOI] [PubMed] [Google Scholar]
- 12.Knekt P, et al. Body iron stores and risk of cancer. Int. J. Cancer. 1994;56:379–382. doi: 10.1002/ijc.2910560315. [DOI] [PubMed] [Google Scholar]
- 13.Wu T, Sempos CT, Freudenheim JL, Muti P, Smit E. Serum iron, copper and zinc concentrations and risk of cancer mortality in US adults. Ann. Epidemiol. 2004;14:195–201. doi: 10.1016/S1047-2797(03)00119-4. [DOI] [PubMed] [Google Scholar]
- 14.Nelson RL. Iron and colorectal cancer risk: human studies. Nutr. Rev. 2001;59:140–148. doi: 10.1111/j.1753-4887.2001.tb07002.x. [DOI] [PubMed] [Google Scholar]
- 15.Kabat GC, Miller AB, Jain M, Rohan TE. Dietary iron and haem iron intake and risk of endometrial cancer: a prospective cohort study. Br. J. Cancer. 2008;98:194–198. doi: 10.1038/sj.bjc.6604110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mursu J, Robien K, Harnack LJ, Park K, Jacobs DR., Jr. Dietary supplements and mortality rate in older women: the Iowa Women’s Health Study. Arch. Intern. Med. 2011;171:1625–1633. doi: 10.1001/archinternmed.2011.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ward MH, et al. Heme iron from meat and risk of adenocarcinoma of the esophagus and stomach. Eur. J. Cancer Prev. 2012;21:134–138. doi: 10.1097/CEJ.0b013e32834c9b6c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cross AJ, Pollock JR, Bingham SA. Haem, not protein or inorganic iron, is responsible for endogenous intestinal N-nitrosation arising from red meat. Cancer Res. 2003;63:2358–2360. [PubMed] [Google Scholar]
- 19.Choi JY, et al. Iron intake, oxidative stress-related genes (MnSOD and MPO) and prostate cancer risk in CARET cohort. Carcinogenesis. 2008;29:964–970. doi: 10.1093/carcin/bgn056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hong CC, et al. Genetic variability in iron-related oxidative stress pathways (Nrf2, NQ01, NOS3, and HO-1), iron intake, and risk of postmenopausal breast cancer. Cancer Epidemiol. Biomarkers Prev. 2007;16:1784–1794. doi: 10.1158/1055-9965.EPI-07-0247. [DOI] [PubMed] [Google Scholar]
- 21.Pietrangelo A. Hereditary hemochromatosis: pathogenesis, diagnosis, and treatment. Gastroenterology. 2010;139:393–408. doi: 10.1053/j.gastro.2010.06.013. [DOI] [PubMed] [Google Scholar]
- 22.Bradbear RA, et al. Cohort study of internal malignancy in genetic hemochromatosis and other chronic nonalcoholic liver diseases. J. Natl Cancer Inst. 1985;75:81–84. [PubMed] [Google Scholar]
- 23.Milman N, et al. Clinically overt hereditary hemochromatosis in Denmark 1948-1985: epidemiology, factors of significance for long-term survival, and causes of death in 179 patients. Ann. Hematol. 2001;80:737–744. doi: 10.1007/s002770100371. [DOI] [PubMed] [Google Scholar]
- 24.Elmberg M, et al. Cancer risk in patients with hereditary hemochromatosis and in their first-degree relatives. Gastroenterology. 2003;125:1733–1741. doi: 10.1053/j.gastro.2003.09.035. [DOI] [PubMed] [Google Scholar]
- 25.Niederau C, et al. Survival and causes of death in cirrhotic and in noncirrhotic patients with primary hemochromatosis. N. Engl. J. Med. 1985;313:1256–1262. doi: 10.1056/NEJM198511143132004. [DOI] [PubMed] [Google Scholar]
- 26.Hsing AW, et al. Cancer risk following primary hemochromatosis: a population-based cohort study in Denmark. Int. J. Cancer. 1995;60:160–162. doi: 10.1002/ijc.2910600204. [DOI] [PubMed] [Google Scholar]
- 27.Osborne NJ, et al. HFE C282Y homozygotes are at increased risk of breast and colorectal cancer. Hepatology. 2010;51:1311–1318. doi: 10.1002/hep.23448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Edgren G, et al. Donation frequency, iron loss, and risk of cancer among blood donors. J. Natl Cancer Inst. 2008;100:572–579. doi: 10.1093/jnci/djn084. [DOI] [PubMed] [Google Scholar]
- 29.Andrews NC. Forging a field: the golden age of iron biology. Blood. 2008;112:219–230. doi: 10.1182/blood-2007-12-077388. Excellent overall review of recent advances in iron biology.
- 30.Daniels TR, et al. The transferrin receptor and the targeted delivery of therapeutic agents against cancer. Biochim. Biophys. Acta. 2012;1820:291–317. doi: 10.1016/j.bbagen.2011.07.016. Summary of past and current strategies used to target TFR1 for anticancer therapy.
- 31.Brooks D, et al. Phase Ia trial of murine immunoglobulin A antitransferrin receptor antibody 42/6. Clin. Cancer Res. 1995;1:1259–1265. [PubMed] [Google Scholar]
- 32.Taetle R, Castagnola J, Mendelsohn J. Mechanisms of growth inhibition by anti-transferrin receptor monoclonal antibodies. Cancer Res. 1986;46:1759–1763. [PubMed] [Google Scholar]
- 33.Ohgami RS, et al. Identification of a ferrireductase required for efficient transferrin-dependent iron uptake in erythroid cells. Nature Genet. 2005;37:1264–1269. doi: 10.1038/ng1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Knutson MD. Steap proteins: implications for iron and copper metabolism. Nutr. Rev. 2007;65:335–340. doi: 10.1111/j.1753-4887.2007.tb00311.x. [DOI] [PubMed] [Google Scholar]
- 35.Leng X, Wu Y, Arlinghaus RB. Relationships of lipocalin 2 with breast tumorigenesis and metastasis. J. Cell. Physiol. 2011;226:309–314. doi: 10.1002/jcp.22403. [DOI] [PubMed] [Google Scholar]
- 36.Zhang Y, Fan Y, Mei Z. NGAL and NGALR overexpression in human hepatocellular carcinoma toward a molecular prognostic classification. Cancer Epidemiol. 2012;36:e294–e299. doi: 10.1016/j.canep.2012.05.012. [DOI] [PubMed] [Google Scholar]
- 37.Leung L, et al. Lipocalin2 promotes invasion, tumorigenicity and gemcitabine resistance in pancreatic ductal adenocarcinoma. PLoS ONE. 2012;7:e46677. doi: 10.1371/journal.pone.0046677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Saha R, Saha N, Donofrio RS, Bestervelt LL. Microbial siderophores: a mini review. J. Basic Microbiol. 2012 Jun 26; doi: 10.1002/jobm.201100552. (doi:10.1002/jobm.201100552) [DOI] [PubMed] [Google Scholar]
- 39.Bao G, et al. Iron traffics in circulation bound to a siderocalin (Ngal)-catechol complex. Nature Chem. Biol. 2010;6:602–609. doi: 10.1038/nchembio.402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Devireddy LR, Hart DO, Goetz DH, Green MR. A mammalian siderophore synthesized by an enzyme with a bacterial homolog involved in enterobactin production. Cell. 2010;141:1006–1017. doi: 10.1016/j.cell.2010.04.040. References 39 and 40 were the first to identify endogenous mammalian siderophores.
- 41.Fernandez CA, et al. The matrix metalloproteinase-9/neutrophil gelatinase-associated lipocalin complex plays a role in breast tumor growth and is present in the urine of breast cancer patients. Clin. Cancer Res. 2005;11:5390–5395. doi: 10.1158/1078-0432.CCR-04-2391. [DOI] [PubMed] [Google Scholar]
- 42.Yang J, McNeish B, Butterfield C, Moses MA. Lipocalin 2 is a novel regulator of angiogenesis in human breast cancer. FASEB J. 2012;27:45–50. doi: 10.1096/fj.12-211730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Berger T, Cheung CC, Elia AJ, Mak TW. Disruption of the Lcn2 gene in mice suppresses primary mammary tumor formation but does not decrease lung metastasis. Proc. Natl Acad. Sci. USA. 2010;107:2995–3000. doi: 10.1073/pnas.1000101107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cramer EP, et al. No effect of NGAL/lipocalin-2 on aggressiveness of cancer in the MMTV-PyMT/FVB/N mouse model for breast cancer. PLoS ONE. 2012;7:e39646. doi: 10.1371/journal.pone.0039646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee HJ, et al. Ectopic expression of neutrophil gelatinase-associated lipocalin suppresses the invasion and liver metastasis of colon cancer cells. Int. J. Cancer. 2006;118:2490–2497. doi: 10.1002/ijc.21657. [DOI] [PubMed] [Google Scholar]
- 46.Sun Y, et al. NGAL expression is elevated in both colorectal adenoma-carcinoma sequence and cancer progression and enhances tumorigenesis in xenograft mouse models. Clin. Cancer Res. 2011;17:4331–4340. doi: 10.1158/1078-0432.CCR-11-0226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bauer M, et al. Neutrophil gelatinase-associated lipocalin (NGAL) is a predictor of poor prognosis in human primary breast cancer. Breast Cancer Res. Treat. 2008;108:389–397. doi: 10.1007/s10549-007-9619-3. [DOI] [PubMed] [Google Scholar]
- 48.Wenners AS, et al. Neutrophil gelatinase-associated lipocalin (NGAL) predicts response to neoadjuvant chemotherapy and clinical outcome in primary human breast cancer. PLoS ONE. 2012;7:e45826. doi: 10.1371/journal.pone.0045826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wu KJ, Polack A, Dalla-Favera R. Coordinated regulation of iron-controlling genes, H-ferritin and IRP2, by c-MYC. Science. 1999;283:676–679. doi: 10.1126/science.283.5402.676. [DOI] [PubMed] [Google Scholar]
- 50.Radulescu S, et al. Luminal iron levels govern intestinal tumorigenesis after apc loss in vivo. Cell Rep. 2012;2:270–282. doi: 10.1016/j.celrep.2012.07.003. This paper provides a mechanistic explanation of how excess iron contributes to intestinal tumorigenesis.
- 51.Tsuji Y, Kwak E, Saika T, Torti SV, Torti FM. Preferential repression of the H subunit of ferritin by adenovirus E1A in NIH-3T3 mouse fibroblasts. J. Biol. Chem. 1993;268:7270–7275. [PubMed] [Google Scholar]
- 52.Kakhlon O, Gruenbaum Y, Cabantchik ZI. Repression of ferritin expression modulates cell responsiveness to H-ras-induced growth. Biochem. Soc. Trans. 2002;30:777–780. doi: 10.1042/bst0300777. [DOI] [PubMed] [Google Scholar]
- 53.Kakhlon O, Gruenbaum Y, Cabantchik ZI. Ferritin expression modulates cell cycle dynamics and cell responsiveness to H-ras-induced growth via expansion of the labile iron pool. Biochem. J. 2002;363:431–436. doi: 10.1042/0264-6021:3630431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang F, Wang W, Tsuji Y, Torti SV, Torti FM. Post-transcriptional modulation of iron homeostasis during p53-dependent growth arrest. J. Biol. Chem. 2008;283:33911–33918. doi: 10.1074/jbc.M806432200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tong WH, et al. The glycolytic shift in fumaratehydratase-deficient kidney cancer lowers AMPK levels, increases anabolic propensities and lowers cellular iron levels. Cancer Cell. 2011;20:315–327. doi: 10.1016/j.ccr.2011.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shpyleva SI, et al. Role of ferritin alterations in human breast cancer cells. Breast Cancer Res. Treat. 2011;126:63–71. doi: 10.1007/s10549-010-0849-4. [DOI] [PubMed] [Google Scholar]
- 57.Liu X, et al. Heavy chain ferritin siRNA delivered by cationic liposomes increases sensitivity of cancer cells to chemotherapeutic agents. Cancer Res. 2011;71:2240–2249. doi: 10.1158/0008-5472.CAN-10-1375. [DOI] [PubMed] [Google Scholar]
- 58.Karin M. Nuclear factor-κB in cancer development and progression. Nature. 2006;441:431–436. doi: 10.1038/nature04870. [DOI] [PubMed] [Google Scholar]
- 59.Torti SV, et al. The molecular cloning and characterization of murine ferritin heavy chain, a tumor necrosis factor-inducible gene. J. Biol. Chem. 1988;263:12638–12644. [PubMed] [Google Scholar]
- 60.Kwak EL, Larochelle DA, Beaumont C, Torti SV, Torti FM. Role for NF-kappa B in the regulation of ferritin H by tumor necrosis factor-alpha. J. Biol. Chem. 1995;270:15285–15293. doi: 10.1074/jbc.270.25.15285. [DOI] [PubMed] [Google Scholar]
- 61.Pham CG, et al. Ferritin heavy chain upregulation by NF-kappaB inhibits TNFalpha-induced apoptosis by suppressing reactive oxygen species. Cell. 2004;119:529–542. doi: 10.1016/j.cell.2004.10.017. [DOI] [PubMed] [Google Scholar]
- 62.Ruddell RG, et al. Ferritin functions as a proinflammatory cytokine via iron-independent protein kinase C zeta/nuclear factor kappaB-regulated signaling in rat hepatic stellate cells. Hepatology. 2009;49:887–900. doi: 10.1002/hep.22716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Alkhateeb AA, Han B, Connor JR. Ferritin stimulates breast cancer cells through an iron-independent mechanism and is localized within tumor-associated macrophages. Breast Cancer Res. Treat. 2013;137:733–744. doi: 10.1007/s10549-012-2405-x. [DOI] [PubMed] [Google Scholar]
- 64.Cortes DF, et al. Differential gene expression in normal and transformed human mammary epithelial cells in response to oxidative stress. Free Radic. Biol. Med. 2011;50:1565–1574. doi: 10.1016/j.freeradbiomed.2011.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nemeth E, et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. 2004;306:2090–2093. doi: 10.1126/science.1104742. Ground-breaking study demonstrating that hepcidin binds to ferroportin and triggers its degradation.
- 66.Ganz T, Nemeth E. Hepcidin and iron homeostasis. Biochim. Biophys. Acta. 2012;1823:1434–1443. doi: 10.1016/j.bbamcr.2012.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ward DM, Kaplan J. Ferroportin-mediated iron transport: expression and regulation. Biochim. Biophys. Acta. 2012;1823:1426–1433. doi: 10.1016/j.bbamcr.2012.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lonnerdal B. Trace element transport in the mammary gland. Annu. Rev. Nutr. 2007;27:165–177. doi: 10.1146/annurev.nutr.27.061406.093809. [DOI] [PubMed] [Google Scholar]
- 69.Pinnix ZK, et al. Ferroportin and iron regulation in breast cancer progression and prognosis. Sci Transl Med. 2010;2:43ra56. doi: 10.1126/scisignal.3001127. This paper demonstrates that levels of ferroportin affect breast cancer cell growth, are altered in patients with breast cancer and affect the prognosis of patients with breast cancer.
- 70.Jiang XP, Elliott RL, Head JF. Manipulation of iron transporter genes results in the suppression of human and mouse mammary adenocarcinomas. Anticancer Res. 2010;30:759–765. [PubMed] [Google Scholar]
- 71.Miller LD, et al. An iron regulatory gene signature predicts outcome in breast cancer. Cancer Res. 2011;71:6728–6737. doi: 10.1158/0008-5472.CAN-11-1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Weiss G, Goodnough LT. Anemia of chronic disease. N. Engl. J. Med. 2005;352:1011–1023. doi: 10.1056/NEJMra041809. [DOI] [PubMed] [Google Scholar]
- 73.Weinberg ED, Miklossy J. Iron withholding: a defense against disease. J. Alzheimers Dis. 2008;13:451–463. doi: 10.3233/jad-2008-13409. [DOI] [PubMed] [Google Scholar]
- 74.Weinberg ED. Iron withholding: a defense against infection and neoplasia. Physiol. Rev. 1984;64:65–102. doi: 10.1152/physrev.1984.64.1.65. [DOI] [PubMed] [Google Scholar]
- 75.Maes K, et al. In anemia of multiple myeloma, hepcidin is induced by increased bone morphogenetic protein 2. Blood. 2010;116:3635–3644. doi: 10.1182/blood-2010-03-274571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hohaus S, et al. Anemia in Hodgkin’s lymphoma: the role of interleukin-6 and hepcidin. J. Clin. Oncol. 2010;28:2538–2543. doi: 10.1200/JCO.2009.27.6873. [DOI] [PubMed] [Google Scholar]
- 77.Hubert N, Hentze MW. Previously uncharacterized isoforms of divalent metal transporter (DMT)-1: implications for regulation and cellular function. Proc. Natl Acad. Sci. USA. 2002;99:12345–12350. doi: 10.1073/pnas.192423399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Galy B, Ferring-Appel D, Kaden S, Grone HJ, Hentze MW. Iron regulatory proteins are essential for intestinal function and control key iron absorption molecules in the duodenum. Cell. Metab. 2008;7:79–85. doi: 10.1016/j.cmet.2007.10.006. [DOI] [PubMed] [Google Scholar]
- 79.Maffettone C, Chen G, Drozdov I, Ouzounis C, Pantopoulos K. Tumorigenic properties of iron regulatory protein 2 (IRP2) mediated by its specific 73-amino acids insert. PLoS ONE. 2010;5:e10163. doi: 10.1371/journal.pone.0010163. This work suggests that IRPs can modify tumour growth in ways that are independent of their effects on iron metabolism.
- 80.Chen G, Fillebeen C, Wang J, Pantopoulos K. Overexpression of iron regulatory protein 1 suppresses growth of tumor xenografts. Carcinogenesis. 2007;28:785–791. doi: 10.1093/carcin/bgl210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454:436–444. doi: 10.1038/nature07205. [DOI] [PubMed] [Google Scholar]
- 82.Recalcati S, et al. Differential regulation of iron homeostasis during human macrophage polarized activation. Eur. J. Immunol. 2010;40:824–835. doi: 10.1002/eji.200939889. [DOI] [PubMed] [Google Scholar]
- 83.Corna G, et al. Polarization dictates iron handling by inflammatory and alternatively activated macrophages. Haematologica. 2010;95:1814–1822. doi: 10.3324/haematol.2010.023879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Cohen LA, et al. Serum ferritin is derived primarily from macrophages through a nonclassical secretory pathway. Blood. 2010;116:1574–1584. doi: 10.1182/blood-2009-11-253815. [DOI] [PubMed] [Google Scholar]
- 85.Han J, et al. Iron uptake mediated by binding of H-ferritin to the TIM-2 receptor in mouse cells. PLoS ONE. 2011;6:e23800. doi: 10.1371/journal.pone.0023800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Li L, et al. Binding and uptake of H-ferritin are mediated by human transferrin receptor-1. Proc. Natl Acad. Sci. USA. 2010;107:3505–3510. doi: 10.1073/pnas.0913192107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Coffman LG, et al. Regulatory effects of ferritin on angiogenesis. Proc. Natl Acad. Sci. USA. 2009;106:570–575. doi: 10.1073/pnas.0812010106. This paper demonstrates that extracellular ferritin can antagonize the activity of endogenous antiangiogenic proteins.
- 88.Tesfay L, Huhn AJ, Hatcher H, Torti FM, Torti SV. Ferritin blocks inhibitory effects of two-chain high molecular weight kininogen (HKa) on adhesion and survival signaling in endothelial cells. PLoS ONE. 2012;7:e40030. doi: 10.1371/journal.pone.0040030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ackroyd R, Shorthouse AJ, Stephenson TJ. Gastric carcinoma in siblings with Friedreich’s ataxia. Eur. J. Surg. Oncol. 1996;22:301–303. doi: 10.1016/s0748-7983(96)80023-0. [DOI] [PubMed] [Google Scholar]
- 90.Kidd A, et al. Breast cancer in two sisters with Friedreich’s ataxia. Eur. J. Surg. Oncol. 2001;27:512–514. doi: 10.1053/ejso.2000.1093. [DOI] [PubMed] [Google Scholar]
- 91.Lill R, et al. The role of mitochondria in cellular iron-sulfur protein biogenesis and iron metabolism. Biochim. Biophys. Acta. 2012;1823:1491–1508. doi: 10.1016/j.bbamcr.2012.05.009. Review of recent advances in mechanisms of iron–sulphur cluster biogenesis.
- 92.Babcock M, et al. Regulation of mitochondrial iron accumulation by Yfh1p, a putative homolog of frataxin. Science. 1997;276:1709–1712. doi: 10.1126/science.276.5319.1709. [DOI] [PubMed] [Google Scholar]
- 93.Shoichet SA, et al. Frataxin promotes antioxidant defense in a thiol-dependent manner resulting in diminished malignant transformation in vitro. Hum. Mol. Genet. 2002;11:815–821. doi: 10.1093/hmg/11.7.815. [DOI] [PubMed] [Google Scholar]
- 94.Thierbach R, et al. Targeted disruption of hepatic frataxin expression causes impaired mitochondrial function, decreased life span and tumor growth in mice. Hum. Mol. Genet. 2005;14:3857–3864. doi: 10.1093/hmg/ddi410. [DOI] [PubMed] [Google Scholar]
- 95.Schulz TJ, et al. Induction of oxidative metabolism by mitochondrial frataxin inhibits cancer growth: Otto Warburg revisited. J. Biol. Chem. 2006;281:977–981. doi: 10.1074/jbc.M511064200. [DOI] [PubMed] [Google Scholar]
- 96.Thierbach R, et al. The Friedreich’s ataxia protein frataxin modulates DNA base excision repair in prokaryotes and mammals. Biochem. J. 2010;432:165–172. doi: 10.1042/BJ20101116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Keith B, Johnson RS, Simon MC. HIF1alpha and HIF2alpha: sibling rivalry in hypoxic tumour growth and progression. Nature Rev. Cancer. 2012;12:9–22. doi: 10.1038/nrc3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Semenza GL. HIF-1: upstream and downstream of cancer metabolism. Curr. Opin. Genet. Dev. 2010;20:51–56. doi: 10.1016/j.gde.2009.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wang GL, Semenza GL. Desferrioxamine induces erythropoietin gene expression and hypoxia-inducible factor 1 DNA-binding activity: implications for models of hypoxia signal transduction. Blood. 1993;82:3610–3615. [PubMed] [Google Scholar]
- 100.Tacchini L, Bianchi L, Bernelli-Zazzera A, Cairo G. Transferrin receptor induction by hypoxia. HIF-1-mediated transcriptional activation and cell-specific post-transcriptional regulation. J. Biol. Chem. 1999;274:24142–24146. doi: 10.1074/jbc.274.34.24142. [DOI] [PubMed] [Google Scholar]
- 101.Lok CN, Ponka P. Identification of a hypoxia response element in the transferrin receptor gene. J. Biol. Chem. 1999;274:24147–24152. doi: 10.1074/jbc.274.34.24147. [DOI] [PubMed] [Google Scholar]
- 102.Lee PJ, et al. Hypoxia-inducible factor-1 mediates transcriptional activation of the heme oxygenase-1 gene in response to hypoxia. J. Biol. Chem. 1997;272:5375–5381. [PubMed] [Google Scholar]
- 103.Mukhopadhyay CK, Mazumder B, Fox PL. Role of hypoxia-inducible factor-1 in transcriptional activation of ceruloplasmin by iron deficiency. J. Biol. Chem. 2000;275:21048–21054. doi: 10.1074/jbc.M000636200. [DOI] [PubMed] [Google Scholar]
- 104.Peyssonnaux C, et al. Regulation of iron homeostasis by the hypoxia-inducible transcription factors (HIFs) J. Clin. Invest. 2007;117:1926–1932. doi: 10.1172/JCI31370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Mastrogiannaki M, et al. HIF-2alpha, but not HIF-1alpha, promotes iron absorption in mice. J. Clin. Invest. 2009;119:1159–1166. doi: 10.1172/JCI38499. This paper demonstrates the role of HIF2α in iron absorption.
- 106.Shah YM, Matsubara T, Ito S, Yim SH, Gonzalez FJ. Intestinal hypoxia-inducible transcription factors are essential for iron absorption following iron deficiency. Cell. Metab. 2009;9:152–164. doi: 10.1016/j.cmet.2008.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Xue X, et al. Hypoxia-inducible factor-2alpha activation promotes colorectal cancer progression by dysregulating iron homeostasis. Cancer Res. 2012;72:2285–2293. doi: 10.1158/0008-5472.CAN-11-3836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Terada N, Or R, Szepesi A, Lucas JJ, Gelfand EW. Definition of the roles for iron and essential fatty acids in cell cycle progression of normal human T lymphocytes. Exp. Cell Res. 1993;204:260–267. doi: 10.1006/excr.1993.1032. [DOI] [PubMed] [Google Scholar]
- 109.Thelander L, Graslund A. Mechanism of inhibition of mammalian ribonucleotide reductase by the iron chelate of 1-formylisoquinoline thiosemicarbazone. Destruction of the tyrosine free radical of the enzyme in an oxygen-requiring reaction. J. Biol. Chem. 1983;258:4063–4066. [PubMed] [Google Scholar]
- 110.Thelander L, Graslund A, Thelander M. Continual presence of oxygen and iron required for mammalian ribonucleotide reduction: possible regulation mechanism. Biochem. Biophys. Res. Commun. 1983;110:859–865. doi: 10.1016/0006-291x(83)91040-9. [DOI] [PubMed] [Google Scholar]
- 111.Martin LK, et al. A dose escalation and pharmacodynamic study of triapine and radiation in patients with locally advanced pancreas cancer. Int. J. Radiat. Oncol. Biol. Phys. 2012;84:e475–e481. doi: 10.1016/j.ijrobp.2012.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Yu Y, et al. Iron chelators for the treatment of cancer. Curr. Med. Chem. 2012;19:2689–2702. doi: 10.2174/092986712800609706. Recent summary of progress and challenges in the development of iron chelators as anticancer therapeutics.
- 113.Tanaka H, et al. A ribonucleotide reductase gene involved in a p53-dependent cell-cycle checkpoint for DNA damage. Nature. 2000;404:42–49. doi: 10.1038/35003506. [DOI] [PubMed] [Google Scholar]
- 114.Shao J, et al. In vitro characterization of enzymatic properties and inhibition of the p53R2 subunit of human ribonucleotide reductase. Cancer Res. 2004;64:1–6. doi: 10.1158/0008-5472.can-03-3048. [DOI] [PubMed] [Google Scholar]
- 115.Smith P, et al. 2.6 A X-ray crystal structure of human p53R2, a p53-inducible ribonucleotide reductase. Biochemistry. 2009;48:11134–11141. doi: 10.1021/bi9001425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Netz DJ, et al. Eukaryotic DNA polymerases require an iron-sulfur cluster for the formation of active complexes. Nature Chem. Biol. 2012;8:125–132. doi: 10.1038/nchembio.721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Veatch JR, McMurray MA, Nelson ZW, Gottschling DE. Mitochondrial dysfunction leads to nuclear genome instability via an iron-sulfur cluster defect. Cell. 2009;137:1247–1258. doi: 10.1016/j.cell.2009.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Rudolf J, Makrantoni V, Ingledew WJ, Stark MJR, White MF. The DNA repair helicases XPD and FancJ have essential iron-sulfur domains. Mol. Cell. 2006;23:801–808. doi: 10.1016/j.molcel.2006.07.019. [DOI] [PubMed] [Google Scholar]
- 119.Karanja KK, Cox SW, Duxin JP, Stewart SA, Campbell JL. DNA2 and EXO1 in replication-coupled, homology-directed repair and in the interplay between HDR and the FA/BRCA network. Cell Cycle. 2012;11:3983–3996. doi: 10.4161/cc.22215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Barber LJ, et al. RTEL1 maintains genomic stability by suppressing homologous recombination. Cell. 2008;135:261–271. doi: 10.1016/j.cell.2008.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Stehling O, et al. MMS19 assembles iron-sulfur proteins required for DNA metabolism and genomic integrity. Science. 2012;337:195–199. doi: 10.1126/science.1219723. Identification of MMS19 as a scaffolding protein involved in the assembly of a subset of iron–sulphur cluster-containing proteins involved in genome integrity, and demonstration of the role of this pathway in the response to DNA damage.
- 122.Lorsbach RB, et al. TET1, a member of a novel protein family, is fused to MLL in acute myeloid leukemia containing the t(10;11)(q22;q23) Leukemia. 2003;17:637–641. doi: 10.1038/sj.leu.2402834. [DOI] [PubMed] [Google Scholar]
- 123.Thomson J, et al. Non-genotoxic carcinogen exposure induces defined changes in the 5-hydroxymethylome. Genome Biol. 2012;13:R93. doi: 10.1186/gb-2012-13-10-r93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Tahiliani M, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930–935. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nature Rev. Cancer. 2009;9:153–166. doi: 10.1038/nrc2602. [DOI] [PubMed] [Google Scholar]
- 126.Kulp KS, Green SL, Vulliet PR. Iron deprivation inhibits cyclin-dependent kinase activity and decreases cyclin D/CDK4 protein levels in asynchronous MDA-MB-453 human breast cancer cells. Exp. Cell Res. 1996;229:60–68. doi: 10.1006/excr.1996.0343. [DOI] [PubMed] [Google Scholar]
- 127.Nurtjahja-Tjendraputra E, Fu D, Phang JM, Richardson DR. Iron chelation regulates cyclin D1 expression via the proteasome: a link to iron deficiency-mediated growth suppression. Blood. 2007;109:4045–4054. doi: 10.1182/blood-2006-10-047753. [DOI] [PubMed] [Google Scholar]
- 128.Ornstein DL, Zacharski LR. Iron stimulates urokinase plasminogen activator expression and activates NF-kappa B in human prostate cancer cells. Nutr. Cancer. 2007;58:115–126. doi: 10.1080/01635580701308265. [DOI] [PubMed] [Google Scholar]
- 129.Tsukamoto H. Iron regulation of hepatic macrophage TNFalpha expression. Free Radic. Biol. Med. 2002;32:309–313. doi: 10.1016/s0891-5849(01)00772-9. [DOI] [PubMed] [Google Scholar]
- 130.Pang H, et al. Crystal structure of human pirin: an iron-binding nuclear protein and transcription cofactor. J. Biol. Chem. 2004;279:1491–1498. doi: 10.1074/jbc.M310022200. [DOI] [PubMed] [Google Scholar]
- 131.Yu Y, Richardson DR. Cellular iron depletion stimulates the JNK and p38 MAPK signaling transduction pathways, dissociation of ASK1-thioredoxin, and activation of ASK1. J. Biol. Chem. 2011;286:15413–15427. doi: 10.1074/jbc.M111.225946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Polakis P. Wnt signaling and cancer. Genes Dev. 2000;14:1837–1851. [PubMed] [Google Scholar]
- 133.Klaus A, Birchmeier W. Wnt signalling and its impact on development and cancer. Nature Rev. Cancer. 2008;8:387–398. doi: 10.1038/nrc2389. [DOI] [PubMed] [Google Scholar]
- 134.Brookes MJ, et al. A role for iron in Wnt signalling. Oncogene. 2008;27:966–975. doi: 10.1038/sj.onc.1210711. One of the first papers demonstrating the connection between iron and WNT signalling.
- 135.Seril DN, et al. Dietary iron supplementation enhances DSS-induced colitis and associated colorectal carcinoma development in mice. Dig. Dis. Sci. 2002;47:1266–1278. doi: 10.1023/a:1015362228659. [DOI] [PubMed] [Google Scholar]
- 136.Ilsley JN, et al. Dietary iron promotes azoxymethane-induced colon tumors in mice. Nutr. Cancer. 2004;49:162–169. doi: 10.1207/s15327914nc4902_7. [DOI] [PubMed] [Google Scholar]
- 137.Song S, et al. Wnt inhibitor screen reveals iron dependence of beta-catenin signaling in cancers. Cancer Res. 2011;71:7628–7639. doi: 10.1158/0008-5472.CAN-11-2745. [DOI] [PubMed] [Google Scholar]
- 138.Coombs GS, et al. Modulation of Wnt/beta-catenin signaling and proliferation by a ferrous iron chelator with therapeutic efficacy in genetically engineered mouse models of cancer. Oncogene. 2012;31:213–225. doi: 10.1038/onc.2011.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Ebina Y, et al. Nephrotoxicity and renal cell carcinoma after use of iron- and aluminum-nitrilotriacetate complexes in rats. J. Natl Cancer Inst. 1986;76:107–113. [PubMed] [Google Scholar]
- 140.Hamazaki S, Okada S, Ebina Y, Fujioka M, Midorikawa O. Nephrotoxicity of ferric nitrilotriacetate. An electron-microscopic and metabolic study. Am. J. Pathol. 1986;123:343–350. [PMC free article] [PubMed] [Google Scholar]
- 141.Li JL, Okada S, Hamazaki S, Ebina Y, Midorikawa O. Subacute nephrotoxicity and induction of renal cell carcinoma in mice treated with ferric nitrilotriacetate. Cancer Res. 1987;47:1867–1869. [PubMed] [Google Scholar]
- 142.Toyokuni S, Mori T, Dizdaroglu M. DNA base modifications in renal chromatin of Wistar rats treated with a renal carcinogen, ferric nitrilotriacetate. Int. J. Cancer. 1994;57:123–128. doi: 10.1002/ijc.2910570122. [DOI] [PubMed] [Google Scholar]
- 143.Jiang L, et al. Deletion and single nucleotide substitution at G.:C in the kidney of gpt delta transgenic mice after ferric nitrilotriacetate treatment. Cancer Sci. 2006;97:1159–1167. doi: 10.1111/j.1349-7006.2006.00301.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Hiroyasu M, et al. Specific allelic loss of p16 (INK4A) tumor suppressor gene after weeks of iron-mediated oxidative damage during rat renal carcinogenesis. Am. J. Pathol. 2002;160:419–424. doi: 10.1016/S0002-9440(10)64860-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Akatsuka S, et al. Fenton reaction induced cancer in wild type rats recapitulates genomic alterations observed in human cancer. PLoS ONE. 2012;7:e43403. doi: 10.1371/journal.pone.0043403. This study establishes a direct connection between iron-induced genomic alterations and cancer.
- 146.Xu Y, et al. Receptor-type protein tyrosine phosphatase beta (RPTP-beta) directly dephosphorylates and regulates hepatocyte growth factor receptor (HGFR/Met) function. J. Biol. Chem. 2011;286:15980–15988. doi: 10.1074/jbc.M110.212597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Yacyshyn OK, et al. Tyrosine phosphatase beta regulates angiopoietin-Tie2 signaling in human endothelial cells. Angiogenesis. 2009;12:25–33. doi: 10.1007/s10456-008-9126-0. [DOI] [PubMed] [Google Scholar]
- 148.Estrov Z, et al. In vitro and in vivo effects of deferoxamine in neonatal acute leukemia. Blood. 1987;69:757–761. [PubMed] [Google Scholar]
- 149.Yamasaki T, Terai S, Sakaida I. Deferoxamine for advanced hepatocellular carcinoma. N. Engl. J. Med. 2011;365:576–578. doi: 10.1056/NEJMc1105726. [DOI] [PubMed] [Google Scholar]
- 150.Hatcher HC, Singh RN, Torti FM, Torti SV. Synthetic and natural iron chelators: therapeutic potential and clinical use. Future Med. Chem. 2009;1:1643–1670. doi: 10.4155/fmc.09.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Whitnall M, Howard J, Ponka P, Richardson DR. A class of iron chelators with a wide spectrum of potent antitumor activity that overcomes resistance to chemotherapeutics. Proc. Natl Acad. Sci. USA. 2006;103:14901–14906. doi: 10.1073/pnas.0604979103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Melotte V, et al. The N-myc downstream regulated gene (NDRG) family: diverse functions, multiple applications. FASEB J. 2010;24:4153–4166. doi: 10.1096/fj.09-151464. [DOI] [PubMed] [Google Scholar]
- 153.Chen Z, et al. The iron chelators Dp44mT and DFO inhibit TGF-beta-induced epithelial-mesenchymal transition via up-regulation of N-Myc downstream-regulated gene 1 (NDRG1) J. Biol. Chem. 2012;287:17016–17028. doi: 10.1074/jbc.M112.350470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Crepin R, et al. Development of human single-chain antibodies to the transferrin receptor that effectively antagonize the growth of leukemias and lymphomas. Cancer Res. 2010;70:5497–5506. doi: 10.1158/0008-5472.CAN-10-0938. [DOI] [PubMed] [Google Scholar]
- 155.Hatcher H, Planalp R, Cho J, Torti FM, Torti SV. Curcumin: from ancient medicine to current clinical trials. Cell. Mol. Life Sci. 2008;65:1631–1652. doi: 10.1007/s00018-008-7452-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Jiao Y, et al. Iron chelation in the biological activity of curcumin. Free Radic. Biol. Med. 2006;40:1152–1160. doi: 10.1016/j.freeradbiomed.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 157.Jiao Y, et al. Curcumin, a cancer chemopreventive and chemotherapeutic agent, is a biologically active iron chelator. Blood. 2009;113:462–469. doi: 10.1182/blood-2008-05-155952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Lin L, et al. Antitumor agents. 250. Design and synthesis of new curcumin analogues as potential anti-prostate cancer agents. J. Med. Chem. 2006;49:3963–3972. doi: 10.1021/jm051043z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Adams BK, et al. Synthesis and biological evaluation of novel curcumin analogs as anti-cancer and anti-angiogenesis agents. Bioorg. Med. Chem. 2004;12:3871–3883. doi: 10.1016/j.bmc.2004.05.006. [DOI] [PubMed] [Google Scholar]
- 160.Chen X, et al. Chemoprevention of 7,12-dimethylbenz[a]anthracene (DMBA)-induced hamster cheek pouch carcinogenesis by a 5-lipoxygenase inhibitor, garcinol. Nutr. Cancer. 2012;64:1211–1218. doi: 10.1080/01635581.2012.718032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 162.Cozzi A, et al. Overexpression of wild type and mutated human ferritin H-chain in HeLa cells: in vivo role of ferritin ferroxidase activity. J. Biol. Chem. 2000;275:25122–25129. doi: 10.1074/jbc.M003797200. [DOI] [PubMed] [Google Scholar]
- 163.Cozzi A, et al. Analysis of the biologic functions of H- and L-ferritins in HeLa cells by transfection with siRNAs and cDNAs: evidence for a proliferative role of L-ferritin. Blood. 2004;103:2377–2383. doi: 10.1182/blood-2003-06-1842. [DOI] [PubMed] [Google Scholar]
- 164.Wang W, Knovich MA, Coffman LG, Torti FM, Torti SV. Serum ferritin: Past, present and future. Biochim. Biophys. Acta. 2010;1800:760–769. doi: 10.1016/j.bbagen.2010.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Jezequel P, et al. Validation of tumor-associated macrophage ferritin light chain as a prognostic biomarker in node-negative breast cancer tumors: A multicentric 2004 national PHRC study. Int. J. Cancer. 2012;131:426–437. doi: 10.1002/ijc.26397. [DOI] [PubMed] [Google Scholar]
- 166.Carpagnano GE, et al. Could exhaled ferritin and SOD be used as markers for lung cancer and prognosis prediction purposes? Eur. J. Clin. Invest. 2012;42:478–486. doi: 10.1111/j.1365-2362.2011.02603.x. [DOI] [PubMed] [Google Scholar]
- 167.Kim Y, et al. Targeting the Wnt/beta-catenin pathway with the antifungal agent ciclopirox olamine in a murine myeloma model. In Vivo. 2011;25:887–893. [PubMed] [Google Scholar]
- 168.Chifman J, et al. The core control system of intracellular iron homeostasis: a mathematical model. J. Theor. Biol. 2012;300:91–99. doi: 10.1016/j.jtbi.2012.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Laubenbacher R, et al. A systems biology view of cancer. Biochim. Biophys. Acta. 2009;1796:129–139. doi: 10.1016/j.bbcan.2009.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Hower V, et al. A general map of iron metabolism and tissue-specific subnetworks. Mol. Biosyst. 2009;5:422–443. doi: 10.1039/b816714c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Sanchez M, Galy B, Muckenthaler MU, Hentze MW. Iron-regulatory proteins limit hypoxiainducible factor-2alpha expression in iron deficiency. Nature Struct. Mol. Biol. 2007;14:420–426. doi: 10.1038/nsmb1222. [DOI] [PubMed] [Google Scholar]
- 172.Abeysinghe RD, et al. p53-independent apoptosis mediated by tachpyridine, an anti-cancer iron chelator. Carcinogenesis. 2001;22:1607–1614. doi: 10.1093/carcin/22.10.1607. [DOI] [PubMed] [Google Scholar]
- 173.Lui GY, et al. The iron chelator, deferasirox, as a novel strategy for cancer treatment: oral activity against human lung tumor xenografts and molecular mechanism of action. Mol. Pharmacol. 2013;83:179–190. doi: 10.1124/mol.112.081893. [DOI] [PubMed] [Google Scholar]
- 174.Liu YT, et al. Chronic oxidative stress causes amplification and overexpression of ptprz1 protein tyrosine phosphatase to activate beta-catenin pathway. Am. J. Pathol. 2007;171:1978–1988. doi: 10.2353/ajpath.2007.070741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Ba Q, et al. Iron deprivation suppresses hepatocellular carcinoma growth in experimental studies. Clin. Cancer Res. 2011;17:7625–7633. doi: 10.1158/1078-0432.CCR-10-3099. [DOI] [PubMed] [Google Scholar]
- 176.Fracanzani AL, et al. Increased cancer risk in a cohort of 230 patients with hereditary hemochromatosis in comparison to matched control patients with non-iron-related chronic liver disease. Hepatology. 2001;33:647–651. doi: 10.1053/jhep.2001.22506. [DOI] [PubMed] [Google Scholar]
- 177.Hann HW, Stahlhut MW, Hann CL. Effect of iron and desferoxamine on cell growth and in vitro ferritin synthesis in human hepatoma cell lines. Hepatology. 1990;11:566–569. doi: 10.1002/hep.1840110407. [DOI] [PubMed] [Google Scholar]
- 178.Boult J, et al. Overexpression of cellular iron import proteins is associated with malignant progression of esophageal adenocarcinoma. Clin. Cancer Res. 2008;14:379–387. doi: 10.1158/1078-0432.CCR-07-1054. [DOI] [PubMed] [Google Scholar]
- 179.Yue J, et al. Transferrin-conjugated micelles: enhanced accumulation and antitumor effect for transferrin-receptor-overexpressing cancer models. Mol. Pharm. 2012;9:1919–1931. doi: 10.1021/mp300213g. [DOI] [PubMed] [Google Scholar]
- 180.Brookes MJ, et al. Modulation of iron transport proteins in human colorectal carcinogenesis. Gut. 2006;55:1449–1460. doi: 10.1136/gut.2006.094060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Eberhard Y, et al. Chelation of intracellular iron with the antifungal agent ciclopirox olamine induces cell death in leukemia and myeloma cells. Blood. 2009;114:3064–3073. doi: 10.1182/blood-2009-03-209965. [DOI] [PubMed] [Google Scholar]
- 182.Torti SV, et al. Tumor cell cytotoxicity of a novel metal chelator. Blood. 1998;92:1384–1389. [PubMed] [Google Scholar]
- 183.Zhou H, et al. The antitumor activity of the fungicide ciclopirox. Int. J. Cancer. 2010;127:2467–2477. doi: 10.1002/ijc.25255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Greene BT, et al. Activation of caspase pathways during iron chelator-mediated apoptosis. J. Biol. Chem. 2002;277:25568–25575. doi: 10.1074/jbc.M110345200. [DOI] [PubMed] [Google Scholar]
- 185.Turner J, et al. Tachpyridine, a metal chelator, induces G2 cell-cycle arrest, activates checkpoint kinases, and sensitizes cells to ionizing radiation. Blood. 2005;106:3191–3199. doi: 10.1182/blood-2005-03-1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Kovacevic Z, Chikhani S, Lovejoy DB, Richardson DR. Novel thiosemicarbazone iron chelators induce up-regulation and phosphorylation of the metastasis suppressor N-myc down-stream regulated gene 1: a new strategy for the treatment of pancreatic cancer. Mol. Pharmacol. 2011;80:598–609. doi: 10.1124/mol.111.073627. [DOI] [PubMed] [Google Scholar]
- 187.Yu Y, Suryo Rahmanto Y, Richardson DR. Bp44mT: an orally active iron chelator of the thiosemicarbazone class with potent anti-tumour efficacy. Br. J. Pharmacol. 2012;165:148–166. doi: 10.1111/j.1476-5381.2011.01526.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Fukushima T, et al. Iron chelation therapy with deferasirox induced complete remission in a patient with chemotherapy-resistant acute monocytic leukemia. Anticancer Res. 2011;31:1741–1744. [PubMed] [Google Scholar]
- 189.Yen Y, et al. A phase I trial of 3-aminopyridine-2-carboxaldehyde thiosemicarbazone in combination with gemcitabine for patients with advanced cancer. Cancer Chemother. Pharmacol. 2004;54:331–342. doi: 10.1007/s00280-004-0821-2. [DOI] [PubMed] [Google Scholar]
- 190.Knox JJ, et al. Phase II study of Triapine in patients with metastatic renal cell carcinoma: a trial of the National Cancer Institute of Canada Clinical Trials Group (NCIC IND. Invest. New Drugs. 2007;25:471–477. doi: 10.1007/s10637-007-9044-9. [DOI] [PubMed] [Google Scholar]
- 191.Ma B, et al. A multicenter phase II trial of 3-aminopyridine-2-carboxaldehyde thiosemicarbazone (3-AP, Triapine) and gemcitabine in advanced non-small-cell lung cancer with pharmacokinetic evaluation using peripheral blood mononuclear cells. Invest. New Drugs. 2008;26:169–173. doi: 10.1007/s10637-007-9085-0. [DOI] [PubMed] [Google Scholar]
- 192.Chao J, et al. A phase I and pharmacokinetic study of oral 3-aminopyridine-2-carboxaldehyde thiosemicarbazone (3-AP, NSC #663249) in the treatment of advanced-stage solid cancers: a California Cancer Consortium Study. Cancer Chemother. Pharmacol. 2012;69:835–843. doi: 10.1007/s00280-011-1779-5. [DOI] [PMC free article] [PubMed] [Google Scholar]