

This work examines the mechanisms by which Chlamydomonas reinhardtii copes with nitrogen (N) limitation, finding transcriptomic and proteomic changes in multiple metabolic pathways and identifying an N-sparing mechanism that prioritizes respiratory metabolism and shifts the proteomic balance toward proteins with lower N contents, a result with implications for engineering of N-use efficiency.
Abstract
Nitrogen (N) is a key nutrient that limits global primary productivity; hence, N-use efficiency is of compelling interest in agriculture and aquaculture. We used Chlamydomonas reinhardtii as a reference organism for a multicomponent analysis of the N starvation response. In the presence of acetate, respiratory metabolism is prioritized over photosynthesis; consequently, the N-sparing response targets proteins, pigments, and RNAs involved in photosynthesis and chloroplast function over those involved in respiration. Transcripts and proteins of the Calvin-Benson cycle are reduced in N-deficient cells, resulting in the accumulation of cycle metabolic intermediates. Both cytosolic and chloroplast ribosomes are reduced, but via different mechanisms, reflected by rapid changes in abundance of RNAs encoding chloroplast ribosomal proteins but not cytosolic ones. RNAs encoding transporters and enzymes for metabolizing alternative N sources increase in abundance, as is appropriate for the soil environmental niche of C. reinhardtii. Comparison of the N-replete versus N-deplete proteome indicated that abundant proteins with a high N content are reduced in N-starved cells, while the proteins that are increased have lower than average N contents. This sparing mechanism contributes to a lower cellular N/C ratio and suggests an approach for engineering increased N-use efficiency.
INTRODUCTION
Inorganic nitrogen (N) is required for the synthesis of the building blocks of protein and nucleic acids. It is an essential nutrient for plants, usually in the form of nitrate or ammonium, and its supply limits plant productivity in soil and in the oceans (reviewed in Zehr and Ward, 2002; Xu et al., 2012). After its uptake and reduction to the level of ammonium, N is further assimilated via Gln synthetase (GS) (Miflin and Lea, 1975). N is transferred from the resulting Gln to 2-oxoglutarate/α-ketoglutarate by reductive transamination catalyzed by Glu oxoglutarate amidotransferase (GOGAT) to yield Glu. Glu is a substrate in many transamination reactions to generate amino acids from the corresponding keto acids. The N atoms of Glu and Gln (amino or amide, respectively) are also used for the synthesis of purines and pyrimidines. The metabolism of N is therefore obligately connected with carbon (C) metabolism (Nunes-Nesi et al., 2010). N is most abundant on Earth in the form of N2, which can be fixed biologically by various bacteria, including in symbiosis with plants, or fixed chemically by the Haber-Bosch process, which is especially energy intensive. The resulting ammonium-containing fertilizers are consequently expensive, increasingly so today as energy costs soar. Therefore, there is considerable interest in understanding N metabolism and N-use efficiency in plants (Xu et al., 2012).
Recently, N metabolism in algae has also captured the attention of researchers. Algae have been recognized for their biotechnological potential in the production of biofuels (H2, ethanol, or biodiesel) and high-value products like carotenoids, fatty acids, or recombinant proteins (Pulz and Gross, 2004; Chisti, 2007; Wijffels and Barbosa, 2010; Georgianna and Mayfield, 2012; Jones and Mayfield, 2012; Merchant et al., 2012), and N fertilizer cost is certainly an important consideration. Nutrient limitation is a key trigger for triacylglycerol (TAG) accumulation in algae, and N limitation is perhaps the best trigger (Rodolfi et al., 2009). This is readily understood intuitively, since TAGs do not contain N atoms and their accumulation diverts C toward storage in a situation where C cannot be used for cell growth and division. We and others have used Chlamydomonas reinhardtii as a reference organism for understanding TAG biosynthesis and N metabolism (Fernandez and Galvan, 2007; Wang et al., 2009; Miller et al., 2010; Boyle et al., 2012; Blaby et al., 2013). Nevertheless, the signaling pathways that effect these metabolic changes are not yet understood and therefore cannot be manipulated in order to control C movement toward TAG for the biodiesel industry.
C. reinhardtii can utilize a number of different N sources, both inorganic (nitrate, nitrite, and ammonium) and organic (purines, urea, urate, and amino acids), although ammonium is preferred (Florencio and Vega, 1983). Genome sequence analysis revealed eight ammonium transporters of the AMT family in the genome, six of which are probably in the plasma membrane to facilitate ammonium uptake and two are predicted to locate to the chloroplast envelope, presumably to facilitate distribution within the cell and assimilation into Gln and Glu via the chloroplast GS/GOGAT cycle (González-Ballester et al., 2004; Fernandez and Galvan, 2007). The GLN1 through GLN4 genes encode GS, but their subcellular location and the contribution of each isozyme to ammonium assimilation has not yet been dissected (Fischer and Klein, 1988). There are two chloroplast-targeted GOGAT enzymes that use either NADH or reduced ferredoxin (Fd) as the reductant (Cullimore and Sims, 1981; Fischer and Klein, 1988). Nitrate and nitrite are taken up via transporters encoded by members of the NRT1, NRT2, and NAR1 families (Quesada et al., 1993; Fernandez and Galvan, 2007). Nitrate assimilation requires energy for reduction to the level of ammonium by enzymes, nitrate reductase in the cytosol, and nitrite reductase in the chloroplast, both encoded by single genes in C. reinhardtii, NIA1/NIT1 and NII1, respectively (Romero et al., 1987; Fernández et al., 1989; Quesada et al., 1998; Fernandez and Galvan, 2008; Hirasawa et al., 2010). Many of the genes involved in nitrate/nitrite uptake and assimilation are under the control of a positive regulator, the NIT2 transcription factor (Fernández and Matagne, 1986; Camargo et al., 2007). Other N-containing compounds that are assimilated include urea (Hodson et al., 1975), acetamide (Gresshoff, 1981), urate, xanthine, hypoxanthine, adenine, guanine (Pineda et al., 1984), and Arg (Kirk and Kirk, 1978). There is no evidence for uptake of amino acids other than Arg, but in the presence of His (and with low ammonium) in the medium, degradative enzymes, histidase and urocanase, are upregulated, implying the existence of a pathway for His uptake and utilization (Hellio et al., 2004). Other amino acids can serve as an N source via the activity of an l-amino acid oxidase in the periplasm (Vallon et al., 1993).
The impact of N deficiency on gene expression and metabolism in C. reinhardtii has been studied for decades. This includes studies of photosynthesis and of the activation of the gametogenesis program in which cells of two mating types (plus and minus) differentiate to express genes that initiate the process of sexual reproduction (Martin and Goodenough, 1975; Martin et al., 1976; Plumley and Schmidt, 1989; Peltier and Schmidt, 1991; Bulté and Wollman, 1992; Giordano et al., 2003; Abe et al., 2004). These studies documented the changes in ribosome abundance, starch accumulation, and organization of the thylakoid membrane in autotrophic conditions. In a classic and elegant study, isotope labeling was used to document the massive degradation of both cytoplasmic and chloroplast ribosomes upon N starvation, with two different temporal profiles with the suggestion that the products were recycled for de novo synthesis of macromolecules required for gametogenesis (Siersma and Chiang, 1971). Ribosomes were not only found to be reduced in abundance, but also compositionally different in N deficiency (Picard-Bennoun and Bennoun, 1985). Interest in these pathways has renewed recently because of the use of C. reinhardtii as a reference organism for understanding TAG accumulation pathways induced by N starvation (Miller et al., 2010; Boyle et al., 2012; Longworth et al., 2012; Merchant et al., 2012; Msanne et al., 2012; Blaby et al., 2013). At the same time, technological advances have made it possible to measure changes in mRNA and protein abundance at a genome-wide scale. As already mentioned, N and C metabolism are intimately interlinked because assimilation of N requires C. Indeed, a large proportion of the total N in a photosynthetic organism is invested in the proteins that are required to perform photosynthesis. Decreases in the N supply typically lead to a decrease in proteins for photosynthesis (see above references), which in turn leads to a decrease in C assimilation, and resulting in a complex metabolic and physiological response in which both N and C availabilities are changing. C. reinhardtii is a facultative autotroph, but also lives on organic C sources like acetate, both in the laboratory and in the field (Harris, 2008). We took advantage of this to study the response to low N in the presence of acetate, thus potentially simplifying the response to minimize secondary responses triggered by changes in photosynthetic rate. Acetate was also successfully used to further enhance TAG accumulation in N-deficient conditions (Goodson et al., 2011) and therefore might become relevant for biodiesel production.
In this work, we use RNA-Seq data to probe the transcriptome and untargeted quantitative liquid chromatography–tandem mass spectrometry (LC-MS/MS) to probe the proteome in acetate-grown C. reinhardtii, after transfer from N-replete to N-limiting conditions. We are able to relate the two studies because of simultaneous sampling for the two types of analyses. In addition, the output is anchored to cellular physiology through parallel measurements of cell growth, chlorophyll content, C and N contents, photosynthetic and respiratory capacity of the cells, and key metabolite pools.
RESULTS
Reduced Chlorophyll and Protein in N-Starved Cells
For a framework and physiological context for the transcriptome and proteome analyses, we analyzed physiological parameters of N-starved cells. Log-phase cells were washed and resuspended in N-free medium and sampled at various time points thereafter to assess growth, chlorophyll content, photosynthetic and respiratory capacity, C and N contents, metabolite abundances, and cellular amino acid composition (Figure 1A).
Figure 1.

Experimental Design and Overview of Output.
(A) Scheme for collecting samples. Each horizontal line indicates an independent experiment, and each vertical line indicates when a sample was taken after transfer to N-free medium. mRNA sequencing experiments for the transcriptome are colored in gray, quantitative proteomics in red, and time points at which samples were taken for analysis of water-soluble metabolites or amino acid composition are indicated in cyan or orange, respectively. The sampling points for protein, chlorophyll, total organic C/N analyses, and cell density are given in dark blue, green, pink, and brown, respectively. Time points at which samples were taken for analyzing the capacity for O2 consumption and evolution are indicated in violet. aa, amino acid.
(B) and (C) Cell density, protein, and chlorophyll content. The number of cells per milliliter of culture was measured for strain CC4532 (dark-gray squares) at the indicated time points after transfer to N-free medium (time point 0). Protein concentrations (red circles) were determined by the Lowry method against a BSA standard, and chlorophyll content (green triangles) was measured spectrometrically. In all experiments, errors bars indicate sd between three replicates.
(D) The N status is evident from Glu and 2-oxoglutarate concentrations in N-replete and -deficient conditions. Glu and 2-oxoglutarate (2-OG) were measured by comparison against a standard at N-replete conditions (+N, white box) and 48 h after transfer to N-free medium (−N, red box). Error bars indicate sd between three biological replicates, and asterisks indicate significant differences between +N and −N (t test, P value < 0.05).
(E) Common responses of the transcriptome to N deprivation in three C. reinhardtii strains. Venn diagram showing overlap of mRNAs found to be changed at any time point within a 48-h N deprivation experiment in CC4532 (blue), CC4349 (red), and CC4348 (sta6) (yellow). The central intersect defines the number of differentially accumulating mRNAs common to all strains (see Supplemental Table 1 for details).
In N-free medium, despite the absence of a N source for growth, the cells do divide once within the first 24 h (from 2 × 106 to 4.4 × 106) before ceasing further division (Figure 1B), suggesting that one round of division occurs at the expense of intracellular N stored in macromolecules. In agreement, there is no net increase in chlorophyll and a small (∼13%) increase in protein content in the culture (because chlorophyll and protein synthesis requires N-containing precursors). The quota of protein and chlorophyll per cell is decreased proportionately, ∼50% less for protein and ∼65% less for chlorophyll (Figure 1C). For cell number, total protein and chlorophyll content acclimation to N deprivation was complete within 24 h, and no further significant changes at a macroscopic level were observed on the second day. The abundance of signature metabolites (Glu and 2-OG or α-ketoglutarate) is consistent with N starvation, since the concentration of the α-keto acid is increased (no N), while that of the corresponding amino acid, Glu, is decreased (Figure 1D), as has been seen in many other studies of the response of algae and plants to low N (Stitt and Krapp, 1999; Foyer et al., 2003; Scheible et al., 2004).
Genome-Wide Changes in mRNA and Protein Abundance in N-Starved Cells
We undertook a multisystem analysis including several independent experiments to characterize the transcriptome of C. reinhardtii cells transferred to N-free medium (Figure 1A). Three time-course experiments included short-term sampling (11 samples, 0 to 60 min [at 0, 2, 4, 8, 12, 18, 24, 30, 45, and 60], in figures and text below abbreviated to S) with the objective of identifying primary responses to nutrient deprivation (such as changes in the levels of transcripts encoding transcription factors involved in N use or assimilatory transporters), medium-term sampling (four samples, 0 to 8 h [at 0, 0.5, 4, and 8], in figures and text below abbreviated to M) with the objective of identifying processes involved in N sparing and recycling during the phase of continued growth and division, and long-term sampling (six samples, 0 to 48 h [at 0, 2, 8, 12, 24, and 48 h], in figures and text below abbreviated to L) to describe the acclimated state, giving a total of 16 time points (Boyle et al., 2012). In addition, we also included long-term sampling of two additional, distinct laboratory strains of C. reinhardtii (Blaby et al., 2013) at equivalent time points (eight samples in biological duplicates, 0 to 48 h, at 0, 0.5, 2, 4, 8, 12, 24, and 48 h) and compared expression (Supplemental Data Set 1). The use of comparative transcriptomics in genetically distinct strains helps to highlight common, “core” responses to removal of N from the medium. In each experiment, multiple samples were taken to provide kinetic information, which is useful for mRNAs whose half-lives are generally shorter than those of polypeptides.
The transcriptome in each sample was determined by RNA-Seq on the Illumina platform, with reads mapped to Au10.2 models, and expression estimates calculated as RPKM (reads per kilobase of gene model per million reads in the data set) (Mortazavi et al., 2008). Transcripts of 12,338 genes (out of 17,301 models) were detected over background in at least one time point in CC4532 using independent filtering. In each of the three experiments, replicate samples were taken at three time points so that the independent experiments could be compared (0 for S, M, and L, 30 min for S and M, and 8 h for M and L) (Figure 1A). When we compared the N-replete samples, we noted that the transcriptomes were indeed highly comparable between the three experiments (Supplemental Figures 1A and 1B) and only a few mRNAs (15 between S and M; 32 between S and L) were significantly (fold change > 2; false discovery rate < 0.05) different in abundance at time point 0. The same is true for comparisons of the 30-min and 8-h points from two experiments (Supplemental Figures 1C and 1D). The variance between the replicate time points was therefore used to estimate and project the variance for the complete data set.
For strain CC4532, we found 4288 genes (34.8% of detected transcripts) whose mRNAs accumulated differentially (fold change > 2; false discovery rate < 0.01) in at least one time point after transfer of cells to N-free medium (3017 for S, 1598 for M, and 1364 for L). In contrast, for strain CC4349, we identified 8482 genes and 8200 genes for CC4348 (Figure 1E). The increased number of significant changes is probably because of increased detection power from the incorporation of independent replicates and a single time course. When all experiments are combined, it is evident that N status has a dramatic impact on the transcriptome. About 85% of all differentially regulated genes were similarly regulated in more than one strain, leaving only 15% uniquely regulated in any one strain (Supplemental Table 1). A total of 2914 genes is significantly regulated in all three strains (Figure 1E), and these were judged to describe the most robust set of genes regulated, both directly and indirectly, by N starvation in C. reinhardtii (Supplemental Data Set 2).
To provide a global view of the most relevant expression patterns that are found in the data, a model-based clustering approach was performed for the RNA-Seq data (Si et al., 2014). Because of the large number of differentially expressed genes, we initially grouped the data in 100 clusters to capture specific responses and to identify coregulated genes (Supplemental Figure 2). Interesting categories and clusters are discussed below (see Supplemental Data Sets for details).
To survey the proteome, we sampled nine time points (+N, 0, 0.5, 2, 4, 8, 12, 24, and 48 h) after transfer to N-free medium in biological triplicates, corresponding to the M and L series in CC4532 and the data sets for CC4349 and CC4348 (sta6) (Figure 1A). Because changes were slower at the protein level, dense sampling in the short time course was not necessary, and the use of triplicates increased the discovery power. Protein samples (separated into soluble versus membrane-associated proteins) were analyzed in a data-dependent mode and quantified against a 15N-labeled universal reference (prepared as a mixture of each time point and spiked into the 14N samples at the level of intact cells; Mühlhaus et al., 2011). This allowed us to determine changes in the abundance of each protein within the proteome during the response to N limitation, independently of the overall 50% decrease in total cellular protein content.
While a total of 11,242 distinct protein groups were identified within the experiment, quantitative information was obtained for 1153 protein groups (10.3%). For the latter, we required that at least two peptides of a protein were reproducibly quantified in each of the experimental triplicates and were found in at least seven of the nine time points (see Supplemental Figure 3 for details). Of these, 635 (55.1%) had changed abundance in at least one sample (one-way ANOVA test for significance of changes). These 635 proteins were clustered into six groups by k-means clustering, while the number of necessary clusters was determined using gap statistics (see Supplemental Data Set 3 for a complete list of the proteins/proteins within each cluster). Visual inspection classified these into increased versus decreased (up versus down) abundance relative to the 0-h time point, with 306 proteins in the former and 329 in the latter, each of the two groups consisting of three different temporal patterns (Supplemental Figure 4). Proteins were mapped into functional categories according to the C. reinhardtii MapMan ontology (Thimm et al., 2004; May et al., 2008) and enrichment within the up- or downregulated groups was assessed (Supplemental Data Set 4).
Bioenergetic Preference for Respiration
A striking visual phenotype of N starvation is chlorosis. Not surprisingly proteins within photosynthesis-related categories (MapMan categories 1.1.1 to 1.1.4, photosystem II [PSII], cytochrome b6f, photosystem I [PSI], and ATP synthase) are less abundant within the proteome of N-starved cells (Figure 2; Supplemental Figure 5 and Supplemental Data Sets 4 and 5). The thylakoid protein complexes respond differently to N deprivation; while the individual proteins of the b6f complex and ATP synthase show similar responses, those of PSII and PSI show more selective responses (Figure 2A). Within PSII, D1 (psbA), D2 (psbD), CP43 (psbC), CP47 (psbB), and both cytochrome b559 subunits (psbE and psbF) are 13 to 45% less abundant after 48 h of N deprivation (Figure 2A; Supplemental Figures 5A and 5B). Subunits of the oxygen-evolving complex are differently affected, with OEE1 (PSBO) being significantly less abundant in the proteome, while OEE2 (PSBP1) retains its abundance. A similar differentiated response was seen for the individual subunits of the light-harvesting complexes, with the proteins encoded by LHCBM1 through LHCBM8 persisting or even increasing and CP26 (LHCB5) and CP29 (LHCB4) decreasing notably (Figure 2A; Supplemental Figure 5A). For PSI, the peripheral proteins and LHCI antenna proteins show a larger decrease, while the reaction center core subunits PsaA and PsaB are more stable under N starvation (Figure 2A; Supplemental Figures 5D and 5E). The cytochrome b6f and plastid ATP synthase complexes are also strongly affected, but in this case, each of the subunits decreases in a quantitatively comparable manner (Figures 2B and 2C; Supplemental Figures 5C and 5F). The decrease of many proteins in the photosynthetic electron transfer complexes and the plastid ATPase points to reduced capacity for photosynthetic electron transport and photophosphorylation. In contrast, the abundance of proteins from the mitochondrial ATP synthase and the analogous mitochondrial cytochrome bc1 complex were found to be increased in N-free conditions (Figures 2B and 2C; Supplemental Figures 6C and 6E), together with other complexes involved in mitochondrial respiration (Figure 2E; Supplemental Figures 6A, 6B, and 6D and Supplemental Data Set 6). This increase occurred even though total protein decreased (see above), which points to maintained or even increased capacity for oxidative phosphorylation. These changes are complete by 24 h after transfer to N-free medium (Figures 2B, 2C, and 2E), by which time the cells have stopped dividing (Figure 1B). When we compared relative mRNA abundances in the same samples, we found that mRNAs encoding the photosynthetic complexes were strongly reduced and the decrease preceded the decrease in the corresponding protein. In contrast, the relative mRNA abundances for the respiratory complexes were maintained (Figures 2D and 2E; Supplemental Figures 5 and 6). There is some variation in the kinetics of mRNA loss in individual C. reinhardtii strains (CC4348 and 4349 complete by 12 h versus CC4532 still decreasing between 12 and 24 h), but the pattern of change is identical (Supplemental Figures 5 and 6).
Figure 2.

Changes in Expression and Abundance of Photosynthetic and Respiratory Complexes.
(A) Overview of changes in abundance of proteins of the photosynthetic electron transfer chain in strain CC4532. Relative changes in protein abundances after 48 h of N deprivation are presented on a log2-transformed scale as a heat map, where green indicates increased and red decreased protein abundance. Proteins are schematically assembled according to their position within the respective complex according to Eberhard et al. (2008) and Allen et al. (2011). Boxes for plastid-encoded proteins are outlined in pink, while nucleus-encoded proteins are outlined in black. The absence of quantitative information is indicated by transparent boxes.
(B) and (C) Relative abundance of chloroplast and mitochondrial ATP synthase subunits (B) and respiratory cytochrome bc1 and photosynthetic cytochrome b6f complex (C) during N deprivation. The log2-transformed relative abundance (14N/15N ratio) at each time point after transfer to N-free medium of individual proteins from mitochondrial (cyan) and chloroplast (pink) ATP synthase complexes (B), respiratory complex III (Cyt bc1, cyan), and cytochrome b6f complex (pink) (C) is shown. Bold lines average the abundance of individual mitochondrial and chloroplast protein complexes; error bars indicate sd between the individual subunits.
(D) Overlay of protein and mRNA abundance of complexes involved in photosynthetic energy generation. Average relative mRNA abundance (percentage of max) for each of the six complexes involved in photosynthetic energy generation during the 48-h N deprivation experiment in C. reinhardtii strain CC4532 are displayed in gray (see Supplemental Figure 5 for individual genes). The maximal RPKM value was set to 1 for each individual gene to compare relative abundances. Average log2-transformed relative abundance (14N/15N ratio) of proteins for the same complexes is given in pink.
(E) Protein and mRNA abundance of complexes of the respiratory electron transfer chain. Relative average abundance of RNAs and log2-transformed relative abundance (14N/15N ratio) of proteins from complexes of mitochondrial respiration as described in (D) (see Supplemental Figure 6 for individual genes).
When we tested O2 consumption (respiration) versus O2 evolution (photosynthesis), the rates recapitulate the pattern of gene expression and protein abundance (Figures 3A to 3D). Respiration was only slightly reduced on a cell basis (Figure 3A), but was actually increased on a protein basis (Figure 3B), like the abundance of mitochondrial respiration complexes and mitochondrial ATP synthase (Figure 2E). Photosynthetic capacity measured as O2 evolution was strongly reduced on a cell (Figure 3C) as well as a protein basis (Figure 3D). O2 evolution starts to decrease ∼8 h after transfer to N-free medium (Figure 3B). In control experiments, when cells were transferred from N-containing medium to fresh N-containing medium (Figures 3A and 3C), neither O2 consumption nor O2 evolution was affected in the long term, but both increased similarly 8 to 12 h after transfer to N-containing medium, probably caused by synchronization of cells upon resuspension in fresh media. We also probed the quantum efficiency of PSII (Fv/Fm), which provides information about the integrity of the photosynthetic apparatus. Fv/Fm decreased in strain CC4532 but the reduction began later, after 12 h (Figure 3E). A decrease of the Fv/Fm ratio was also observed in strain CC4349 and CC4348 (sta6), but like the faster acclimation at the mRNA level (Supplemental Figures 5 and 6), the quantum efficiency of PSII also decreased earlier, by 12 h of N starvation (Figure 3F).
Figure 3.

Selective, Progressive Loss of Photosynthetic versus Respiratory Capacity in N-Starved Cells.
(A) and (B) Oxygen consumption on a per cell (A) or protein (B) basis. Capacity for respiration was measured for 5 min as the rate of O2 consumption in the dark at the indicated time points after the transfer to either normal TAP (+N) or N-free medium (–N) in the presence of 20 mM acetate and 10 mM bicarbonate in strain CC4532 using a Clark-type electrode (Hansatech Oxygraph). Cell density was determined with a hemocytometer, and protein concentrations were determined by the Lowry method against a BSA standard. Error bars indicate sd of three biological replicates.
(C) and (D) Oxygen evolution on a per cell (C) or protein (D) basis. Capacity for photosynthetic O2 evolution was measured in strain CC4532 for 5 min in the light (300 µmol m−2 s−1) as described for oxygen consumption (see above). Net oxygen evolution was calculated as the difference between oxygen consumption in the dark and evolution in the light. Error bars indicate sd of three biological replicates.
(E) Maximal quantum yield of PSII (Fv/Fm): 2 × 106 C. reinhardtii cells of strain CC4532 were spotted on a Whatman GF/A filter and dark-adapted for 10 min. Minimal fluorescence in the dark and maximal fluorescence upon a saturating light pulse were acquired on an imaging fluorometer (FluorCam 700MF; Photon Systems Instruments) and used for calculation of Fv/Fm at the indicated time points after transfer to N-free medium. Error bars indicate sd of three biological replicates.
(F) Maximal quantum yield (Fv/Fm) of PSII in three distinct C. reinhardtii strains. Fv/Fm was determined for CC4532, CC4349, and CC4348 (sta6) at the indicated time points after transfer to N-free medium as described in Figure 2E. Error bars indicate sd of three biological replicates.
Calvin-Benson Cycle Enzymes and mRNAs Are Reduced
Enzymes of the Calvin-Benson cycle, especially Rubisco, are abundant in N-replete cells and were significantly enriched among the proteins whose abundances decreased 48 h after transfer to N-free medium (Figure 4A; Supplemental Data Set 7). On average, the abundances of Calvin-Benson cycle enzymes were reduced by ∼19% 48 h after transfer to N-free medium. There are nevertheless a few exceptions: triose phosphate isomerase (TPIC) and aldolase (FBA3), which are 70% and 12% increased, respectively (Figures 4A and 4B; Supplemental Figures 7A, 7E, and 7F). FBA3 (aldolase) showed a similar decrease as did other Calvin-Benson cycle enzymes during the first hours after transfer to N-free medium but was derepressed between 24 and 48 h. TPIC has a unique response among Calvin-Benson cycle enzymes and steadily increased during N deprivation (Figure 4B; Supplemental Figures 7E and 7F). The abundances of transcripts encoding Calvin-Benson cycle enzymes are more strongly affected than those of the corresponding proteins, being decreased by 2- to 8-fold (Figures 4A and 4C; Supplemental Figure 7). In general, mRNA abundances reach a new steady state 12 to 24 h after transfer to N-free medium. In CC4348 (sta6), the TPIC1 transcript shows a unique pattern in that its abundance remains elevated throughout the course of the experiment, whereas it decreases in the other strains. In previous work, we showed that gluconeogenesis is promoted in N-starved CC4348 (sta6), and this pattern may represent part of that response (Blaby et al., 2013). There is an open debate on the extent to which changes in transcript abundance lead to changes in protein abundance (de Sousa Abreu et al., 2009; Vogel et al., 2010). As shown in Supplemental Figure 7, there is a remarkably close agreement between the changes in transcript and protein abundance for several Calvin-Benson cycle enzymes, including Rubisco, glyceraldehyde 3-phosphate dehydrogenase (GAP3), transketolase (TRK), sedoheptulose 1,7-bisphosphatase (SEBP1), phosphopentose isomerase (RPI1), and phosphoribulokinase (PRK1) (Supplemental Figures 7B, 7D, 7H to 7J, and 7L), weaker agreement for fructose 1,6-bisphosphatase (FBP1) and FBA3 (Supplemental Figures 7F and 7G), and poor or even opposing responses for phosphoglycerate kinase (PGK1), phosphopentose epimerase (RPE1), and especially TPIC (Supplemental Figures 7C, 7E, and 7K). This points to there being very different contributions of transcriptional and posttranscriptional mechanisms to the regulation of protein abundance, even within a single metabolic pathway.
Figure 4.

Changes in mRNA Abundance Precede Changes in Abundance of Calvin-Benson Cycle Enzymes and Intermediate Metabolites.
(A) Overview of expression of Calvin-Benson cycle genes after 48 h of N depletion. Summary of changes in abundances of Calvin-Benson cycle transcripts and proteins. Arrows indicate reactants and products of the catalyzed reaction for each of the enzymes catalyzing the respective reaction. Given are the maximum expression estimates within all the experiments in strain CC4532 [mRNA (RPKM)], the log2 fold change of mRNA abundance [mRNA (FC)] between N-replete and 48-h N deficiency in C. reinhardtii strain CC4532 (48 h experiment), CC4349, and the CC4348 (sta6) mutant, as well as the log2 fold change of protein abundance (Protein) between N-replete and 48-h N deficiency in C. reinhardtii strain CC4532. Box color intensity (green increase; red decrease) indicates magnitudes of changes within the transcriptome and proteome data set. NA indicates absence of quantitative information from the data sets. For each enzyme, only the highest expressed potentially encoding gene and corresponding protein is shown; for a complete list of candidates, see Supplemental Data Set 8.
(B) Relative changes of enzymes. Shown are log2-transformed relative changes in abundance of individual enzymes (14N/15N ratio) of the Calvin-Benson cycle at the indicated time points after transfer to N-free medium.
(C) Relative changes of mRNAs. mRNA abundance (percentage of max) for each of the genes encoding Calvin-Benson cycle enzymes during the 48-h experiment of N depletion C. reinhardtii strain CC4532.
(D) Concentration of metabolites before and 48 h after the transfer to N-free medium. Water-soluble metabolites were analyzed via LC-MS/MS at N-replete conditions (+N, white) and after 48 h of N deprivation (−N, red). Concentrations are given for RuBP, GAP, DHAP, fructose-6-phosphate (F6P), Xu5P, sedohepulose-1,7-bisphosphate (SBP), sedoheptulose-7-phosphate (S7P), ribose-5-phosphate (R5P), and ribulose-5-phosphate (Ru5P). Error bars indicate sd between three biological replicates, each measured in two technical replicates. Asterisks indicate significance (t test, P value < 0.05).
Measurement of Calvin-Benson cycle metabolites (Figure 4D) indicated higher abundance of all intermediates, but in particular ribulose-1,5-bisphosphate (RuBP, substrate of Rubisco), xylulose-5-phosphate (Xu5P), and ribulose-5-phosphate (Ru5P) (the substrate of phosphoribulokinase whose level is decreased) and triose phosphates dihydroxyacetone-phosphate (DHAP) and glyceraldehyde-3-phosphate (GAP), interconverted by TPIC and substrates for FBA3 and TRK. The higher metabolite abundance may be a consequence of the decrease in protein abundance of Calvin-Benson cycle enzymes. In particular the increase in RuBP is consistent with Rubisco exerting an increased constraint on flux in the Calvin-Benson cycle, with accumulation of other intermediates occurring due to feedback regulatory loops in the Calvin-Benson cycle (Scheibe, 1991; Stitt et al., 2010), for example, feedback inhibition of PRK by RuBP (Gardemann et al., 1983). It is also possible that the increase in metabolite levels (which do not contain any N) partially compensates for the decrease in enzyme abundance, by allowing the remaining complement of enzymes to operate closer to substrate saturation. Curiously, the large increase in triose-phosphate abundance (DHAP and GAP) was accompanied by an increase in TPIC, which (see above) was due to posttranscriptional regulation. It might be noted that the 4-fold increase in DHAP abundance may increase the rate of chemical decay of this metabolite to form the toxic metabolite methylglyoxal (Kalapos, 1999).
The Tetrapyrrole Pathway Is an Early Casualty of the N Starvation Response
The N requirement of chlorophyll biosynthesis (beginning from Glu) and the fact that photosynthetic capacity is reduced in N starvation prompted us to curate the mRNAs encoding enzymes of chlorophyll biosynthesis and degradation (Figure 5A; Supplemental Figure 8 and Supplemental Data Set 8). Not surprisingly, in all three strains, the mRNAs for each enzyme involved in tetrapyrrole biosynthesis are reduced within 2 to 4 h and reach a lower steady state 4 to 8 h after transfer to N free medium (Figure 5B; Supplemental Figure 8A). The weakly expressed CPX2 and PBGD2, encoding alternative isoforms of the corresponding enzymes, are less affected, which is consistent with the utilization of more abundant proteins for N scavenging (see below). The rapid response of these transcripts is notable because it precedes the loss of the transcripts encoding subunits of the photosynthetic apparatus (compare Figures 5B and 2 and Supplemental Figure 5). While mRNA abundances remained low until the end of day 2 in strain CC4532, the mRNA abundance recovered to a variable extent in strains CC4349 and the CC4348 (sta6), sometimes to levels comparable to those in N-replete conditions (Supplemental Figure 8A). Among the tetrapyrrole metabolism-related enzymes, the proteomics experiment yielded excellent data on CTH1, encoding the major form of the aerobic cyclase (Moseley et al., 2002), which showed a parallel loss of protein and mRNA (Figure 5C).
Figure 5.

Reciprocal Expression of Tetrapyrrole Biosynthesis versus Degradation Enzymes.
(A) Overview of mRNAs encoding enzymes involved in tetrapyrrole biosynthesis during N deprivation. Summary of relative mRNA abundances of genes involved in tetrapyrrole biosynthesis. Arrows indicate reactants and products of the catalyzed reactions. Maximal expression estimates [mRNA (RPKM)] derive from all the experiments in strain CC4532. Box color intensity (green increase; red decrease) indicates log2-transformed fold change of transcript abundances [mRNA (FC)] between N-replete and 48-h N deficiency in C. reinhardtii strain CC4532 (48 h experiment), CC4349, and the CC4348 (sta6) mutant.
(B) Relative abundance of RNAs. Relative mRNA abundance (percentage of max) in C. reinhardtii strain CC4532 for all genes encoding enzymes involved in tetrapyrrole biosynthesis at the indicated time after transfer to N-free medium.
(C) Overlay of protein and mRNA abundance for CTH1. The relative abundance of CTH1 mRNA (percentage of max) within the 48-h N deprivation experiment in C. reinhardtii strain CC4532 is displayed in gray. The relative abundance (14N/15N, log2 transformed) of the corresponding protein is overlaid in blue.
(D) Relative abundance of mRNAs encoding candidate enzymes involved in chlorophyll degradation. Relative mRNA abundance (percentage of max) within the 1 h of N deprivation experiment in C. reinhardtii strain CC4532 for a select number of genes potentially involved in chlorophyll degradation (names of corresponding homologs in Arabidopsis are denoted in the label with a “_” in front of the name).
Although chlorophyll degradation has not yet been investigated in C. reinhardtii, we identified orthologs of Arabidopsis thaliana proteins (Hörtensteiner and Kräutler, 2011) (based on mutual best BLAST hit of protein sequences; Supplemental Figure 9) and found that the corresponding mRNAs start to increase within an hour after transfer to N-free medium (Figure 5D; Supplemental Figures 8B to 8F). This is surprising because the chlorophyll content of the culture does not change rapidly (Figures 1B and 1C). Consistent with maintenance of respiration in N-starved cells, mRNAs of enzymes specifically involved in heme biosynthesis accumulated and enzymes involved in degradation were not upregulated (Supplemental Figures 10A and 10B).
Regulation of Cytosolic and Plastid Ribosome Abundance Occurs via Different Mechanisms
Decreased abundance of ribosomes under N limitation was noted decades earlier in both biochemical studies and ultrastructural microscopy studies (Siersma and Chiang, 1971; Martin and Goodenough, 1975). Our data set offered the opportunity to distinguish the responses of individual subunits and isoforms. Because of their abundance, ribosomes are well covered in proteomics data sets; in this study, 47 cytoplasmic and eight plastid ribosomal proteins were obtained consistently (Supplemental Data Set 9). The abundances of all of these were substantially reduced (see Supplemental Data Set 4, bin 29.2), but with a different timing and extent, and differing relationships between the changes in protein and mRNA abundance (Figures 6A and 6B; Supplemental Figures 11A to 11D and 12A). The cytoplasmic ribosomal proteins were rapidly reduced in abundance, reaching a minimum by 12 h after N removal (∼40% less than in N-replete conditions). Interestingly, in all three strains, mRNAs encoding the cytoplasmic 80S ribosomes increased gradually rather than decreasing (Figure 6B; Supplemental Figures 11C and 11D). The chloroplast ribosomal protein abundances decreased to an even lower level than the cytosolic ones (∼75% reduction in average), but gradually over the 48-h time course (Figures 6A and 6B; Supplemental Figures 11A, 11B, and 12A). The mRNAs corresponding to the chloroplast ribosomal subunits decreased very quickly within 2 h after transfer to N-free medium in all strains (Figure 6A; Supplemental Figures 11A and 11B). Later, the mRNA abundances recovered to some extent (depending on the strain, to ∼40% in CC4532 and CC4348 [sta6] and ∼75% of expression levels in N-replete conditions in CC4349), perhaps reflecting different phases of the N starvation response. These results suggest distinct mechanisms for decreasing ribosome abundance in the two compartments, with degradation possibly playing a larger role in the cytosol versus decreased synthesis in the plastid. We therefore inspected expression of genes involved in degradative pathways and found that genes involved in autophagy were upregulated shortly after transfer to N-free medium (Supplemental Figures 13A and 13B and Supplemental Data Set 10). Mitochondrial ribosome subunits were not identified in these experiments, possibly because of their much lower abundance, but their mRNAs increased in the early time points after transfer of cells to N-free medium and the abundances remained high for 24 h (Supplemental Figures 11E and 11F).
Figure 6.

Impact of N Starvation on Capacity of Protein Synthesis and N Content of the Cell and Proteome.
(A) Protein and mRNA abundance of plastidic ribosomal proteins. The relative average abundance of RNAs (percentage of max) encoding subunits of plastidic ribosomes within the 48-h N deprivation experiment in C. reinhardtii strain CC4532 is displayed in gray. The average of the 30S SSU (light gray) and the 50S LSU (dark gray) are denoted in dashed lines. Average log2-transformed relative protein abundances (14N/15N) for the same complexes are given in green. Error bars indicate sd from averages of all subunits.
(B) Protein and mRNA abundance of cytosolic ribosomal proteins. Average relative mRNA and log2-transformed protein abundances (14N/15N ratio) as described in (A) for cytosolic ribosomal proteins.
(C) Changes in cellular C/N upon N deprivation. Molar C/N in C. reinhardtii CC4532 at the indicated time points after transfer to N-containing (+N) or N-free (−N) media. Error bars indicate sd of three biological replicates.
(D) Comparison of N content between up- and downregulated proteins. Quantile distributions of N atoms in amino acid side chains (Karlin and Brendel, 1992) of upregulated (green) or downregulated (red) proteins. The significance of the differences of the distributions was assessed by two-tailed Student’s t test (Baudouin-Cornu et al., 2001). The whole proteome based on Augustus 10.2 gene models was used as a reference (gray).
(E) Amino acid composition in N-replete and deficient conditions. Whole-cell lysates were purified from polysaccharides, pigments, and lipids, and the amino acid composition (w/w) of hydrolyzed samples was analyzed on an amino acid analyzer. Red labels indicate amino acids containing N in their side chains. Asx and Glx indicate the combined abundance of Asn and Asp or Gln and Glu, respectively. Error bars indicate sd of three biological replicates. Asterisks indicate significance (t test, P value < 0.01).
N Economy at the Proteome Level
When we measured the total cellular N content (as total organic N per cell), we noted that the amount of cellular N decreased in the first 24 h in N-free medium (Supplemental Figure 12B). The ratio of total organic C to N is ∼5 in N-replete cultures but almost triples to ∼14 in cells subjected to N starvation for 48 h (Figure 6C). The change is detectable already within 2 h after the shift (t test, P value < 0.01). Since the cells stop dividing after 24 h of N starvation, the C/N ratio does not change in the second day. Two factors contribute to the change in ratio: first, the reduction of N on a per cell basis within the first 12 h (Supplemental Figure 12B); second, the increase in C, which doubles in cells in N-free medium versus N-replete medium later (Supplemental Figure 12C), obviously attributable to the accumulation of storage carbohydrate and neutral lipid (Wang et al., 2009; Siaut et al., 2011; Msanne et al., 2012). Analysis of the proteome of N-starved cells revealed that abundant proteins like Rubisco and ribosomal subunits whose levels decreased under N deficiency are rich in amino acids with N-containing side chains (Supplemental Figure 12D). When we plotted the distribution of N atoms in side chains of significantly reduced or induced proteins separately and compared them to the distribution in all C. reinhardtii proteins, we found that the proteins that are decreased in abundance in N-free medium are those with a higher N content than average (Figure 6D, red triangles). In contrast, proteins whose abundances increase in N-starved medium have less N in the side chains than average, speaking to a specific adaptation of the proteome to the N deficiency state (Figure 6D, green triangles; two tailed t test, P value < 0.01). The adaptation acts at the level of N use efficiency rather than stress because proteins whose abundances change in response to S or P deficiency do not show this effect, nor does the number of S or C atoms in the side chains differ between up- versus downregulated proteins (Supplemental Figures 12E and 12F).
We used the empirical protein abundance index of the proteins identified in N-replete conditions and at 48 h after transfer to N-free medium to calculate the contribution to absolute N-sparing resulting from the reduction of amino acids that contain N in their side chains (see Methods). We found that the proteome induced upon N starvation contains an absolute amount of 6% (±1% sd) less N in side chains compared with the proteins whose abundance decreased (Figure 6D). When inspecting the increasing/decreasing proteins for differences in individual amino acid levels, we found that Arg was significantly reduced in the group of proteins whose abundance increases (Supplemental Figure 12G).
The above observation is based on the relative changes in protein abundance and reflects the N-sparing due to changes in the proteome-wide amino acid composition (Figure 6D). To take this observation further on an absolute level, we compared the amino acid composition of total protein extracted from N-replete and N-deficient conditions (Figure 6E). Arg especially, containing three N atoms in its side chain, and to lesser extent also Lys were significantly decreased upon N deprivation, with 13 and 7% reduction compared with N-replete conditions, respectively (Figure 6E). His, which also contains N in its side chains, is not affected. This is consistent with His function in catalysis, which can be indispensable or at least not easily replaced, while a fraction of Arg/Lys, delivering electrostatic interactions required for protein structure, may be easier to replace.
N Assimilation Pathways
Ammonium Assimilation
N assimilation and metabolism in C. reinhardtii is well described and has been studied for decades (Fernandez and Galvan, 2007; see Figure 7 and Supplemental Data Sets 11 and 12 for an overview). Of the eight ammonium transporter (AMT) proteins, six are likely plasma membrane localized, while two are localized to the plastid (Figure 7A; González-Ballester et al., 2004). In the N-replete cultures, the most abundant AMT-encoding mRNAs were AMT7 (∼20 to 50 RPKM in all three strains), AMT3 (∼8 to 35 RPKM), and AMT6 (∼2 to 12 RPKM) (Figure 7B; Supplemental Figure 14A). In N-free medium, the combined abundances of the AMT mRNAs are strongly increased within the first 2 h after transfer, from ∼50 to ∼1570 RPKM. While expression of the constitutively expressed genes (AMT7, AMT3, and AMT6) is reduced, expression of the other AMT transporter genes, AMT1, AMT4, AMT5, and AMT8, is strongly increased, and this is true in all three strains (Figure 7A; Supplemental Figure 14B). Therefore, this subset becomes more relevant with respect to the template that is used to synthesize new transporter molecules (Figure 7B). While transporters tend to contain less N in side chains relative to the whole proteome (reflecting most likely the hydrophobic transmembrane segments; Acquisti et al., 2009), the N assimilation–related transporters contained even less N, with the induced ones, AMT1, AMT4, AMT5, and AMT8, tending to be on the lower end of the distribution relative to the constitutive ones (Figure 7C). Furthermore, AMT1 (105 amino acids) is the smallest transporter, while AMT6 (778 amino acids) is the largest one, which would further reduce the N quota.
Figure 7.

Adjustment of N Assimilation.
(A) Overview of changes in the abundance of mRNAs involved in N acquisition upon N depletion. The fold change (log2) of mRNA abundance between N-replete and 2-h N-starved cultures from C. reinhardtii strain CC4532 (black outline), CC4349 (blue contours), and CC4348 (sta6) (red contours) are summarized; green color indicates increased and red color decreased relative mRNA abundances. Genes are schematically sorted to the compartment to which the corresponding protein is predicted to be targeted (Fernandez and Galvan, 2007; Harris, 2008). Black arrows indicate transport processes and gray arrows enzymatic reactions catalyzed by the gene product. Transparent boxes with gray contours indicate proteins/genes necessary but unidentified or absent from Au10.2 gene models.
(B) Composition of the mRNA pool encoding potential NH4+ transporters of the AMT family. Expression estimates (RPKM) for C. reinhardtii strain CC4532 for each of the potential NH4+ transporters of the AMT family were used to determine pie sizes in N-replete conditions and after 1 h of N deprivation. The N-replete pie is also shown enlarged to allow an assessment of its constituents.
(C) N content of NH4+ transporters. Range (from the 10 to 90% quantile) of average number of N atoms in amino acid side chains in all proteins of the proteome, in transporters facilitating the acquisition/distribution of nitrogen, phosphorous, and sulfur and in candidate NH4+ transporters (AMT and RHP families). The color indicates (average) log2 fold changes of mRNA abundance (green, increase; red, decrease) between N-replete conditions and 60 min after the transfer to N-free medium in C. reinhardtii strain CC4532. The N content in side chains is based on Augustus 10.2 gene models.
Intracellular ammonium is assimilated via GS and GOGAT (Miflin and Lea, 1975; Cullimore and Sims, 1981; Fernandez and Galvan, 2007). Of four GS-encoding genes in C. reinhardtii, GLN1 and GLN4 encode cytosolic isoforms, while GLN2 and GLN3 the plastid-targeted ones (Fischer and Klein, 1988). GLN2 is dominant in terms of abundance in all three strains (e.g., 1574 versus 0.4 RPKM in CC4532), but for both, abundance increases in N-starved cells (Figure 7A; Supplemental Figures 15A and 15B). Of the plastid isoforms, only the GLN2-product (GS2) was identified and quantitated in the proteomics study, and its abundance decreased rather than increased in N-starved cells (Supplemental Figures 15C and 15D). The mRNAs encoding the two cytosolic isoforms are more evenly expressed in all three strains (e.g., 139 to 234 RPKM in CC4532), but only GLN1 transcript increases notably in all strains in N starvation (Figure 7A; Supplemental Figures 15E and 15F). Its gene product, GS1, was consistently identified in the proteome study, and its abundance increased in parallel with the corresponding mRNA (Supplemental Figure 15C), again reflecting distinct behavior of proteins in the plastid versus the cytosol. The two GOGATs, NADH-dependent encoded by GSN1 and Fd-dependent encoded by GSF1, were quantified in the proteomics study as also the corresponding mRNAs, and based on the increase in abundance, both forms must be relevant for N metabolism in N-starved cells (Supplemental Figure 16).
Alternative N Sources
Nitrate is another key source of N in nature, but it requires greater input of energy for assimilation because it needs to be reduced to the level of ammonium (Bloom et al., 1992; Fernandez and Galvan, 2008). Both NIA1/NIT1 and NII1, encoding respectively cytosolic nitrate reductase and plastid nitrite reductase, are highly induced within 1 h after transfer of cells to N-free medium, with the mRNAs peaking by 2 h and then remaining high throughout the experiment in all three strains (Figure 7A; Supplemental Figure 17A). Nitrate and nitrite are brought into the cell by transporters of the NRT1, NRT2, and NAR1 families (Fernandez and Galvan, 2008). The NRT1.1 gene is expressed already in N-replete medium in all three strains but further upregulated early upon transfer to N-free medium with transcripts reaching maximal abundance between 12 and 24 h (Supplemental Figure 17B). Of the NRT2 family, there are strain-specific differences, but, similar to the AMT NH4+ transporters, genes expressed in N-replete conditions (NRT2.4 and NRT2.5 in CC4349 and CC4348 [sta6]) are repressed, while others are induced (Supplemental Figure 17C). Upon transfer to N-free medium, in all three strains, NRT2.1 was strongly induced within an hour after transfer to N-free medium and constituted the main portion of the NRT2 mRNA pool (Supplemental Figure 17C). The NAR2 gene product is required for NRT2.1 function (Galván et al., 1996), and in all three strains, NAR2 is coordinately expressed with NRT2.1, supporting a common, conserved regulatory mechanism (Supplemental Figure 17C). Among the NAR1 transporters, NAR1.2 is strongly increased in abundance in N-starved cells; at some time points, it constitutes more than 90% of the different NAR1 transporter mRNAs (Supplemental Figure 17D), and interestingly, among the various proteins encoded by NAR1 genes, NAR1.2 contains the least amount of N in the side chains.
C. reinhardtii can also use other N sources, and genes for those pathways were also rapidly induced within the first hours after transfer to N-free medium. For instance, the genes responsible for uptake and metabolism of urea, DUR3A-C, DUR1, and DUR2, were strongly induced in all three strains (Figure 7A) as also was AMI1, encoding an acetamidase. Transporters and enzymes for uptake (UAPA1-UAPA6) and metabolism of purines (XDH1 and UOX1) are also dramatically upregulated in all strains (Figure 7A). The most highly induced mRNA (from near 0 RPKM in replete to 1 to 3 × 103 RPKM in N-starved CC4532) is LAO1, encoding an l-amino acid oxidase that facilitates the extracellular deamination of amino acids (Vallon et al., 1993).
Candidate Regulators
There is a cluster of genes involved in nitrate/nitrite assimilation on chromosome 9; none of those is expressed to high levels in replete medium, but all are strongly induced upon transfer to N-free medium, consistently in all strains, and consistent with their regulator NIT2 (Schnell and Lefebvre, 1993; Camargo et al., 2007), whose mRNA was not detected in N-replete medium and only detected under N starvation conditions (Supplemental Figure 18A). Similar expression profiles were found for other regulators as well, for example, NRR1 (Supplemental Figure 18A), a previously identified transcription factor involved in TAG accumulation upon N deprivation (Boyle et al., 2012), and GLB1 (Supplemental Figure 18B), encoding the PII protein, a well-known regulator of N metabolism in bacteria (Uhrig et al., 2009; Ermilova et al., 2013). We therefore further inspected two transcription factor databases (PlnTFDB [Pérez-Rodríguez et al., 2010] and PlantTFDB 2.0 [Zhang et al., 2011]) for the presence of DNA binding proteins with similar expression patterns to known genes and regulators involved in N assimilation (Supplemental Data Set 13). Cre01.g011150, present in both databases and containing a basic helix-loop-helix motif, is highly induced upon N deficiency, consistently in all three strains, and clusters together with NRR1 and 12 other genes involved in N assimilation in cluster 25 (Supplemental Figure 18C). Its N content in amino acid side chains is very low (0.21 N atoms/side chain compared with 0.34 in the whole proteome), which is especially remarkable for a nucleic acid binding protein (NRR1 is also below average with 0.26 N atoms/side chain), suggesting a role in transcriptional regulation during N deficiency (Supplemental Figure 18D). Two other DNA binding proteins were identified in cluster 25 (Supplemental Figures 18C and 18D), both also containing less N than average, each present in one of the two databases, Cre04.g216200, another basic helix-loop-helix–type transcription factor, and Cre16.g673250, a protein with Squamosa promoter binding domain. Although nearly the entire genome is impacted by starvation from N, we focused attention on these regulators because of the timing of their response, the lower than average N content (which probably facilitates their de novo synthesis when N-rich amino acids like Arg, Lys, and His are scarce), and the similar pattern of their response in the three different strains (Supplemental Figure 18C).
Fatty Acid Metabolism
To investigate the acclimation of lipid metabolism to N deprivation, we curated a set of genes (Supplemental Data Set 14) that was suggested to be involved in the pathways according to the previous literature and by similarity of protein sequence to curated genes in other organisms (Harris, 2008; Moellering et al., 2010; Boyle et al., 2012; Merchant et al., 2012; Blaby et al., 2013). Proteins involved in lipid metabolism (MapMan category 11: lipid metabolism, as well as the subcategory 11.1: fatty acid synthesis and fatty acid elongation) were found to be significantly enriched among the proteins that decreased in N-starved cells in the proteomics experiment in CC4532 (Supplemental Data Set 4), particularly proteins involved in early steps of fatty acid biosynthesis. Two protein constituents of the tetrameric, chloroplast-localized acetyl-CoA carboxylase (ACCase) complex were identified and found to be strongly reduced upon N deprivation at the protein level: the β-carboxyltransferase (BCX1) and the biotin carboxyl carrier protein (BCC1). In addition, all mRNAs encoding the different ACCase subunits were repressed similarly and early upon N deprivation in the three strains (Supplemental Data Set 14). ACP2, by far the most highly expressed acyl-carrier protein in N-replete conditions, was found to be strongly reduced in the proteomics experiment, and its mRNA was also depleted in all three strains upon N deprivation (Supplemental Data Set 14). ENR1, the enoyl-ACP reductase, which is part of the fatty acid synthase (FAS) complex, was reduced at the protein level, while mRNA abundances of all components of the FAS were also reduced early in the three strains. This emphasizes the role of TAG synthesis via recycling of preexisting thylakoid membrane lipids (Miller et al., 2010; Li et al., 2012). Interestingly, the observed reduction of mRNAs of all the components involved early in fatty acid biosynthesis (ACCase, ACP, and FAS) was only transient, especially in CC4349 and CC4348 (sta6), and mRNA levels recovered to N-replete level between about 12 and 24 h after transfer to N-free medium, resulting in an overall almost constant expression in the 48-h time course or even a slight increase for some components (Supplemental Data Set 14). mRNAs encoding two of the three glycerol-3-phosphate dehydrogenase isozymes that were found to be increased within CC4349 and CC4348 (sta6) (Blaby et al., 2013) were also induced in CC4532 (Supplemental Data Set 14), including the already highlighted GPD2 mRNA. Acyltransferases specific for TAG biosynthesis were consistently found to be increasingly expressed in all three strains, especially DGAT1 and DGTT1, which accumulate specifically in N-deprived conditions, supporting earlier findings (Boyle et al., 2012). Besides these exceptions, genes from lipid metabolism are expressed rather stably, if changes are observed, and gene expression is more likely reduced than increased, consistent with previous findings (Miller et al., 2010; Boyle et al., 2012). In the absence of phosphatidylserine and phosphatidylcholine, phosphatidylethanolamine and diacylglyceryl-N,N,N-trimethylhomoserine are the only lipids containing N within their head groups. The genes involved in their biosynthesis were not found to be regulated differently from genes involved in the biosynthesis of other lipids (Supplemental Data Set 14).
Comparison of Acclimation Strategies between Different Macronutrient Limitations
Responses to macronutrient limitation include general global responses that are common for several nutrients (stress response, growth inhibition, protease upregulation, and lipid body accumulation) and also ones specific for an individual nutrient (selective transport and selective sparing). We took advantage of comparable RNA-Seq experiments involving the S and P limitation response of C. reinhardtii (6 and 24 h after transfer to nutrient-free medium, in strain D66 [CC4425] and 21 gr [CC1690], respectively) to distinguish these common versus distinct responses (González-Ballester et al., 2010; this work). The response to P starvation involves fewer changes in the transcriptome (407 transcripts whose abundance changes) compared with S (1578); consequently, there are more genes with a common response in the N deficiency and the S deficiency transcriptome (673; Supplemental Data Set 15) than between N and P deprivation (156; Supplemental Data Set 16), representing ∼23 and ∼5% of the N-responsive genes, respectively (Figure 8A). We compared the list of differentially accumulating transcripts at each individual time point in each strain within the N starvation data set to those in the S or P starvation data sets in order to capture the largest overlap (Figures 8B and 8C). In general, the changes tended to be in the same direction (increase or decrease) for all strains at all time points (Figures 8B and 8C; Supplemental Table 2). The number of transcripts in common in these responses reached a maximum by ∼30 to 60 min after transfer of cells to N-free medium.
Figure 8.

Conserved Aspects of S, P, and N Deprivation.
(A) Common responses of the transcriptome to N, S, and P deprivation. Venn diagram showing overlap of mRNAs found to be changed at any time point within a 48-h N deprivation experiment in all three strains (red) and changed in C. reinhardtii strain CC4425 upon 6 h of S deprivation (yellow) or 6 h P deprivation (blue). The central intersect defines the number of differentially accumulating mRNAs common to all strains.
(B) and (C) mRNAs with common responses in S (B) or P (C) and N deprivation. Comparison of the number of mRNAs that differentially accumulate in S- or P-deprived cells to each time point and each strain in N deficiency and the number of mRNAs whose abundances changes in the same direction (upward triangles) or opposite direction (downward triangles) between the conditions are plotted for each strain on a time axis (see Supplemental Table 2 for details).
(D) Comparison of overlap and individual macronutrient deprivations. Overview of all increased (green) or decreased (pink) mRNAs (No.) found in 8-h N-deprived (–N) cells of C. reinhardtii strain CC4532 and those additionally increasing or decreasing (log2 fold increase or decrease > 2) in S or P deprivation.
(E) Comparison of functional categories affected similarly by either N and S or N and P deprivation. The number of transcripts that increase (green) or decrease (pink/red) in both N and S deprivation (left scale, dark green, pink) or N and P deprivation (right scale, light green, red) determines the length of the bar. Genes were grouped according to the functional annotation of their deduced protein sequence according to the MapMan annotation (Thimm et al., 2004; May et al., 2008); the respective MapMan category is denoted in the figure and given in parentheses in the legend. Categories from left to right are indicated in the legend. Only categories with more than two changing genes in either condition are summarized.
The largest overlap was between 8 h of N deprivation in strain CC4532 (495 genes) and among these, more of them represent transcripts whose abundance is decreased (65%; Figure 8D; Supplemental Data Set 15). This indicates that genes that are upregulated tend to be nutrient specific. Photosynthesis and tetrapyrrole synthesis represent an important area of overlap, but there are differences between N and S starvation. For instance, the magnitude of the change is less dramatic in the –S conditions compared with the –N condition (Supplemental Data Set 5; see fold change in CC4532, 8 h –N versus –S), and the abundances of transcripts for the chloroplast ATP synthase are not reduced in –S, whereas they are in –N. Some changes were specific for an individual nutrient, like the upregulation of LHCBM9, which is part of a S-sparing response. It has reduced Cys and Met, but its N content is not different from other proteins in this family and, hence, it offers no advantage in the N deficiency situation. Other specific responses include the genes for assimilatory transporters and nutrient acquisition: The expression of N assimilation components is not increased in S deficiency and vice versa, and these genes are not in the overlap set (Figure 8E). The ribosomes represent a substantial source of both N and S, but their reduction is less pronounced in S deficiency at the transcript level.
For the P deficiency response, the overlap was greatest with the list of genes whose expression is changed in the sta6 mutant after 48 h in –N medium, with 130 transcripts changing abundance in a similar fashion (Figure 8C; Supplemental Data Set 16). In this case, most (∼70%) of these represent transcripts whose abundances increase. Consistent with the absence of chlorosis and no impact on photosynthesis 24 h after P depletion, we do not find genes encoding the proteins of photosynthetic apparatus and enzymes of tetrapyrrole biosynthesis in the overlap set (Figure 8E). A few genes of plastid ribosomes were found to be consistently reduced between the conditions, as ribosomes are also considered a large reservoir for P in cells (Veneklaas et al., 2012).
Ninety-five genes were found to be similarly regulated between N, P, and S deprivation (Supplemental Data Set 17). Among the genes induced in N, P, and S deprivation, we found both LHCSR3 genes, LHCSR3.1 (Cre08.g367500) and LHCSR3.2 (Cre08.g367400), indicating that there is a common mechanism for handling excess excitation energy resulting from macronutrient deficiencies. Additionally, reduced expression of a limited set of genes encoding chloroplast ribosomal proteins could be observed, indicative for a common mechanism for ribosome reduction. Additional experiments, especially time courses of P and S deprivation, might allow a more comprehensive comparison of the common regulated mechanisms in macronutrient deficiency.
DISCUSSION
Based on its quantitative contribution to biomass, N is the most important nutrient for algal and plant growth (besides C), representing typically ∼3 to 5% of total dry weight and contributing substantially to the cost of fertilizer (Boyle and Morgan, 2009; Robertson and Vitousek, 2009; Masclaux-Daubresse et al., 2010). Hence, there is considerable interest in nitrogen use efficiency (NUE) in plants, and now in algae, because of growing interest in biotechnological applications (Scott et al., 2010; Merchant et al., 2012; Xu et al., 2012; Wase et al., 2014). Molecular analyses of the responses to N nutrition have been investigated and detailed for decades, including (especially in C. reinhardtii) the complex circuitry of ammonium versus nitrate utilization (Fernandez and Galvan, 2007). In this work, we exploit deep sequencing of mRNA with parallel quantitative proteomics to understand the high-level mechanisms operating in N-limited C. reinhardtii cells. For the transcriptome, our use of three genetically distinct strains helps to reduce noise and focus on conserved common responses that are present despite quite strong genetic differences, including the occurrence or absence of a cell wall or starch biosynthesis. Comparative transcriptomics across several ecotypes has proven very useful recently, for example, to identify a handful of key iron deficiency responses in Arabidopsis (Stein and Waters, 2012; Urzica et al., 2012; Waters et al., 2012).
The immediate response of the transcriptome is the upregulation of transporters for acquiring N: those for the preferred N source, ammonium, and the more complex N-containing organic compounds, which require further metabolism (Figure 7A; Supplemental Figures 14 to 18). In the absence of an external source of N, the reuse and recycle mode kicks into operation, also almost immediately within the first hour (Merchant and Helmann, 2012) in which N from abundant molecules (ribosomes, Rubisco, and chlorophyll) is released by upregulation of degradation pathways (chlorophyll degradation: Figure 5D, Supplemental Figures 8B to 8F, and Supplemental Data Set 8; autophagy: Supplemental Figure 13 and Supplemental Data Set 10 [Wang et al., 2009]; ribosomes: Figure 6, Supplemental Figures 11 and 12, and Supplemental Data Set 9 [Siersma and Chiang, 1971; Martin and Goodenough, 1975]). This results in an overall reduction of the total protein content per cell by ∼50% (Figure 1C) and of total RNA per cell by ∼60% (rRNA constitutes the major fraction of cellular RNA) (Plumley and Schmidt, 1989), which allows continued growth and division at lower ratios of total organic N/C than in N-replete cells (Figures 1B and 6C). In addition, reduction of ribosomes reduces the capacity for protein synthesis, especially in the chloroplast, where we observed an ∼75% reduction of ribosomal proteins. This decrease in ribosome abundance presumably reflects the decrease in the rate of protein synthesis and might also contribute to it, thus saving N by decreasing investment of N in ribosomes, while the decrease in ribosome abundance may also contribute to a decrease in the use of N for the synthesis of other proteins. A similar strategy was recently discussed for adaptation of southwestern Australia Proteaceae species that grow on extremely low P soils and contain very low levels of ribosomes in their young leaves (Sulpice et al., 2013). The resulting switch from well-balanced (mixotrophic) energy production between photosynthesis and respiration in N-replete conditions to mainly mitochondrial energy generation in N-starved C. reinhardtii (Figures 2 and 3; Plumley and Schmidt, 1989; Peltier and Schmidt, 1991; Bulté and Wollman, 1992; Wang et al., 2009; Longworth et al., 2012; Wase et al., 2014; Wei et al., 2014) allows extraction of N from abundant and N-rich chloroplast proteins, like the photosystems or Calvin-Benson cycle enzymes (especially Rubisco). It is accompanied by a particular large decrease of plastid ribosomal proteins compared with cytosolic ribosomal proteins; this presumably reflects the fact that the former are almost exclusively involved in synthesis of proteins in the thylakoid complexes and the large subunit of Rubisco, whereas the synthesis of photosynthesis proteins is only one of many tasks performed by the latter. A program of sexual reproduction is also induced upon N deprivation with the production of mating competent gametes (Martin and Goodenough, 1975). Several genes found to be involved in this process in previous studies were also identified and found to be induced upon N deprivation here (Supplemental Data Set 12) (Merchán et al., 2001; Abe et al., 2004).
Besides the acclimation mechanisms, we show evidence of long-term adaptation of the genome, specifically the operation of N sparing at the level of protein sequence in which the amino acid composition of the N deficiency proteome has been optimized by reduction of Arg and Lys content to reduce the demand for N (reviewed in Merchant and Helmann, 2012). Although the benefit in terms of the N quota is small (additional 6% ± 1%), its existence (E-values: 9.7 e-08 [up versus down], 2.0 e-06 [up versus reference], and 2.5 e-03 [down versus reference]) suggests evolutionary pressure and, hence, a benefit from recoding the protein composition. Consequently, the overall protein content can be slightly increased (∼13%), despite the absence of external N sources. Each of these N-sparing strategies is discussed further below.
The Role of Respiration Is Increasingly Important in N-Deficient Conditions
The reciprocal loss of components of the photosynthetic apparatus versus increased abundance of respiratory chain components within the proteome is striking (Figure 2) and indicates that the degradation of proteins of photosystems (presumably for N salvage) is a selective process. The photosynthetic apparatus is abundant in C. reinhardtii and, hence, a substantial reservoir of N. In contrast, the respiratory complexes are found at much lower abundances. The chloroplast constitutes ∼40% of the C. reinhardtii cell, while mitochondria take up only ∼3% of the cells volume (Schötz et al., 1972) and, hence, commitment to energy generation via respiration represents an economical use of N, especially when degradative processes generate substrates for respiration. This is true not only in mixotrophically grown cells as in this work, but was also noted in autotrophically grown cells in previous work (Plumley and Schmidt, 1989). This contrasts with the response of C. reinhardtii to iron limitation where photoautotrophically grown cells maintain their photosynthetic apparatus, while mixotrophically grown ones sacrifice photosynthesis in favor of respiration (Naumann et al., 2007; Terauchi et al., 2010). The bioenergetics remodeling response is therefore specific for −N (or any individual nutrient stress), rather than a default choice. Although there are certain similar aspects in comparison to different (macro)nutrient deficiencies, reduced photosynthetic activity, for example, is found also early in –S and later in –P medium (Wykoff et al., 1998), several aspects show the distinct features of N deprivation, resulting in differential accumulation of storage compounds (Cakmak et al., 2012) or unique metabolite profiles in N, S, and P deprivation (Bölling and Fiehn, 2005). When taken together, the changes suggest that in S deficiency the earlier proposed (Wykoff et al., 1998; Melis et al., 2000) remodeling from linear to cyclic electron flow might be the more likely adjustment while in –N the focus is shifted toward a heterotrophic lifestyle.
In this work, we observed a coordinate loss of each of the photosynthetic complexes, cytochrome b6f, ATP synthase, and the two photosystems, although not all subunits have the same half-life (Supplemental Figure 5 and Supplemental Data Set 5), whereas in other work, preferential loss of the cytochrome b6f complex occurred (Bulté and Wollman, 1992; Wei et al., 2014). Three factors may contribute to this: variation in strain genotype in individual laboratories (CC4532 here versus wild-type S24 in their works), variation in photon flux density (90 µmol m−2 s−1 here and 5 to 10 µmol m−2 s−1 in the other works), and different experimental setup. The global LC-MS/MS approach allowed sensitive and accurate analysis of a larger number of proteins simultaneously and enabled us to determine also less pronounced changes. We observed a lower, but still significant reduction of LHCII (Supplemental Figure 5A) as well as D1 and D2 reaction center subunits (Supplemental Figure 5B). In addition, specific previously not assessed subunits from PSI and PSII are included in our analysis, especially peripheral subunits, like LHCA8 and LHCA9, the PSAE, F, G, H, J, and L proteins (PSI), or the oxygen-evolving complex (OEE1,2,3), both cytochrome b559 subunits, and CP43, CP26, and CP29 (Supplemental Figure 5) from PSII, all of them being substantially reduced upon transfer to N-free medium (Bulté and Wollman, 1992; Wei et al., 2014). This points to a much more general reorganization of the photosynthetic complexes upon N deprivation than was previously recognized, supported also by recent, similar findings of reduced proteins of PSI (PSAG, PSAH, LHCA5, and LHCA9) and PSII (LHCB4,5 and OEE1,2,3) in a proteomics study of N deprivation in C. reinhardtii strain CC125 (Wase et al., 2014). However, much lower light intensity was used in experiments describing the selective loss of the cytochrome b6f complex; therefore, we cannot rule out the possibility that photodamage also contributes to the decreased abundance of the photosystems in this work.
Since chlorophyll breakdown has not been studied in C. reinhardtii, we do not know whether the N is recycled. The main breakdown products (nonfluorescent chlorophyll catabolites) that are produced in plants still contain all the N atoms of the original chlorophyll molecule (Kräutler and Hörtensteiner, 2006; Hörtensteiner and Kräutler, 2011) and are transported into the vacuole; hence, chlorophyll degradation may simply be necessary during breakdown of the chlorophyll-protein complexes to avoid production of toxic reactive oxygen species. Nevertheless, cessation of chlorophyll synthesis, reflected by decreased mRNA for nearly all the enzymes of the tetrapyrrole biosynthesis pathway (Figures 5A and 5B; Supplemental Figure 8A and Supplemental Data Set 8), is rapid and is an early event, concomitant with decreased mRNA for components of the photosynthetic apparatus.
N Sparing at the Level of the Proteome
A limiting nutrient growth environment creates selective pressure so that cellular demand for the limiting nutrient is minimized (Baudouin-Cornu et al., 2001). The elements used by cells for structural constituents and catalysis reflect their availability on earth and the impact of geochemical cycles over time (Mazel and Marlière, 1989; Merchant and Helmann, 2012). For instance, the proteins and enzymes involved in sulfur uptake and metabolism in Escherichia coli and Saccharomyces cerevisiae are significantly depleted of S-containing amino acids, while those involved in C metabolism have fewer C atoms in their side chains (Baudouin-Cornu et al., 2001). In S. cerevisiae in a situation of high demand for S for glutathione biosynthesis, abundant enzymes are replaced by isozymes with lower content of Cys and Met (Fauchon et al., 2002). We noted this previously in C. reinhardtii as well: Proteins that are upregulated in S deficiency have very few Cys and Met (González-Ballester et al., 2010). Elemental sparing has not been studied as extensively in the case of N. The proteins of N assimilation in S. cerevisiae (including those involved in conversion of ammonium, urea, allantoate, and Pro) do have fewer N atoms in the amino acid side chains, although the impact of N sparing is not as dramatic as for S (Baudouin-Cornu et al., 2001).
When we examined the N content of the proteins whose abundances are increased (Supplemental Data Set 3, Cluster 1-3) upon transfer of cells to N-free medium (or the proteins encoded by mRNAs whose abundances are increased; data not shown), the decreased fraction of N is notable and significant (Figure 6D). In contrast, proteins whose abundances decrease have a very high N content (Figure 6D; Supplemental Data Set 3, Cluster 4-6). This includes nucleic acid binding proteins, which contain more positively charged amino acids and therefore contain more N than average (Acquisti et al., 2009); among these, the ribosomal proteins constitute a substantial fraction. The decrease in ribosome abundance in N-starved C. reinhardtii cells was noted already by microscopy and metabolic labeling experiments decades ago (Siersma and Chiang, 1971; Martin and Goodenough, 1975) and, in fact, is a common response in eukaryotic cells to other elemental deficiencies as well because of their cellular abundance (Kraft et al., 2008; Sulpice et al., 2013). For instance, ribosomes are a reservoir of P (in rRNA) and Zn (up to 10% of cellular Zn is ribosome associated) (Hensley et al., 2011; Veneklaas et al., 2012). In the case of N deficiency, the decrease in ribosome abundance may also serve to reduce protein synthesis, which is naturally N dependent and N consuming.
Interestingly, the ribosome content is adjusted by different mechanisms: The chloroplast ribosomes are more actively targeted with the mRNAs encoding ribosomal proteins strongly reduced 30 to 45 min after cells are transferred to N-free medium (Figure 6A; Supplemental Figures 11A and 11B and Supplemental Data Set 9), but the mRNAs for the cytosolic ones are maintained (Figure 6B; Supplemental Figures 11C and 11D and Supplemental Data Set 9) (Miller et al., 2010; Longworth et al., 2012). In S. cerevisiae, the 80S ribosomes are targeted by an autophagy-related process called ribophagy in N-starved cells (Kraft et al., 2008), which could be a mechanism operating in C. reinhardtii as well. Indeed, marker genes for autophagy are upregulated in N-deficient C. reinhardtii cells (Supplemental Figure 13 and Supplemental Data Set 10) (Díaz-Troya et al., 2008; Wang et al., 2009). Besides ribosome content, ribosome composition was also noted to be distinct between N-replete versus N-starved C. reinhardtii cells (Picard-Bennoun and Bennoun, 1985), a phenomenon that also occurs in land plants between different environmental conditions (Hummel et al., 2012; Wang et al., 2013). However, while ribosomal proteins are encoded by multigene families in land plants, this is not the case in C. reinhardtii (Barakat et al., 2001; Manuell et al., 2005), and all of these genes showed a highly similar expression pattern upon N deficiency (Supplemental Figure 11 and Supplemental Data Set 9). Therefore, the observed changes in electrophoretic mobility of C. reinhardtii ribosomal proteins upon N deficiency likely reflect posttranslational modifications.
Compared with the proteome median (0.34 N atoms in amino acid side chains), Rubisco large (0.37) and small (0.35) subunits have slightly increased N content. Given their abundance and necessity for photosynthetic carbon fixation, a substantial amount of cellular N is localized within this enzyme complex. Therefore, Rubisco might be a valuable target to improve NUE. The N content of RBCL from different organisms is highly comparable, suggesting that RBCL may already have been targeted by natural selection for NUE. However, nucleus-encoded small subunits have a much higher variability and would allow one to substantially increase NUE.
Adjustment of N Metabolism
Among the proteins that are upregulated in N deficiency are those involved in the uptake and metabolism of N-containing compounds (Figure 7A; Supplemental Figures 14 to 18 and Supplemental Data Set 11). Some of these likely become quite abundant, given their abundance of the upregulated mRNAs (see above); hence, their synthesis would draw on the internal N reservoir. Therefore, the lower N content of these proteins (Figure 7C) would have a physiological advantage. Although transporters in general have lower than average N content (presumably because of the prevalence of hydrophobic side chains in the transmembrane segments), those involved in N transport are even more depleted of N-containing side chains (Figure 7C). In the case of the AMT family, AMT3, AMT6, and AMT7 are expressed in N-replete cells (González-Ballester et al., 2005; Kim et al., 2005). We found that these genes are not upregulated upon transfer to N-free medium and in fact may even be repressed (Supplemental Figure 14). They contain the highest proportion of N-containing amino acids within this group.
Changes in RNA and protein abundance reveal coordinate expression of previously described pathways for utilization of alternate N sources, such as amino acids, urea, urate, or purines (Figure 7A). The mRNAs encoding the relevant transporters, for example, DUR3A-C for urea or UAPA1-6 for urate and purines, and enzymes to release N from these compounds, especially LAO1, encoding an extracellular l-amino acid oxidase releasing NH4+ from a range of amino acids (Vallon et al., 1993), DUR1 and DUR2, encoding ATP:urea amidolyase required for urea breakdown to CO2 and NH3, and XDH1 and UOX1, encoding xanthine dehydrogenase and urate oxidase required for purine breakdown (Harris, 2008), are coordinately upregulated very soon (within 30 to 60 min) after transfer of cells to N-free medium. The relative abundances of the mRNAs can reach levels corresponding to those of the most abundant mRNAs in the cell; for example, expression of LAO1 after transfer to N-free medium (3793 RPKM, 48 h N-deprived) exceeds PSAD expression (3643 RPKM) in N-replete conditions in C. reinhardtii strain CC4532 (see Supplemental Data Sets 5 and 11 for details). Thirteen of the genes involved in N metabolism are found in cluster 25, six of them in cluster 2, and four of them in cluster 1, all resembling clusters strongly induced between 30 and 60 min after transfer to N-free medium (Supplemental Figure 2). Interestingly, transcript levels are not maintained in all cases in the absence of these alternate N sources, consistent with the operation of a robust N-sparing response (see below). “Redundant” genes from certain pathways are found in different clusters, an indication that the response can be further dissected. For example, a complete set of genes capable of urea uptake and assimilation (DUR1, DUR2, and DUR3A) reached maximal expression ∼2 to 4 h after transfer to N-free medium and was subsequently maintained at a lower level. DUR3C, which encodes an alternative urea transporter, reaches maximal expression at 12 to 24 h, and DUR3B at 48 h in the three strains. We made similar observation for nitrate assimilation, where we found a subset of genes that affect N uptake and assimilation (NRT2.1/NAR2, NIA1/NIT1, NII1, NIT2, and MoCo biosynthesis enzymes), peaking 1 to 4 h after transfer to N-free medium, while a number of redundantly present transporters displayed different temporal profiles, with NRT1.1 peaking 12 to 24 h (Supplemental Figure 17B) and NAR1.6 peaking 24 to 48 h after transfer to N-free medium (Supplemental Figure 17D).
METHODS
Strains and Culture Conditions
Chlamydomonas reinhardtii strains 2137 (CC4532, wild-type nit2 mt+), cw15 (CC4349 nit1 NIT2 mt+), and sta6 (CC4348 cw15 nit1 NIT2 arg7-7 sta6-1::ARG7 mt+) were grown in either ammonium-containing (+N, N replete) or ammonium-free (−N, N deficient) Tris-acetate-phosphate (TAP) medium (Harris, 1989) in an Innova incubator (180 rpm; New Brunswick Scientific) at 24°C in continuous light (90 µmol m−2 s−1), provided by cool white fluorescent bulbs (4100K) and warm white fluorescent bulbs (3000K) in the ratio of 2:1, unless stated otherwise. Cell density (number of cells per milliliter of culture) was determined with a hemocytometer.
Protein and Chlorophyll Concentration Measurement
To determine protein concentrations, the Lowry method was used and the absorption at 750 nm at three different concentrations was measured and compared with a BSA standard (Lowry et al., 1951). Chlorophyll was extracted from whole cells using an 80/20 (v/v) acetone/methanol mixture, and chlorophyll content was measured spectrometrically at 646.6 and 663.6 nm according to Porra (1989) on a Perkin-Elmer LAMBDA 25 UV/Vis spectrometer. The absorption at 750 nm was used as a reference.
mRNA Preparation and RNA-Seq Analysis in N Deficiency
With respect to the identification of a conserved N-sparing mechanism, the choice of more distantly related strains is crucial, as it excludes more specific modifications made to the metabolism restricted to a single genotype or different experimental conditions, and all sparing strategies present in the overlap of these experiments and strains might be more universal. The presence (CC4532) or absence (CC4349, CC4348 [sta6]) of a proteinaceous cell wall might affect N acclimation, as it is either a large sink or a large reservoir for N. The presence (CC4532 and CC4349) or absence (CC4348 [sta6]) of a functional starch biosynthesis pathway might also affect N acclimation rather broadly (for example, see Blaby et al. [2013] on differences in gluconeogenesis and glyoxylate pathway), as one of the main energy storage compounds used in N deficiency is not accessible anymore. Strains CC4532, CC4349, and CC4348 (sta6) were grown in TAP medium to a density of 4 × 106 cells mL−1, collected by centrifugation (5 min at 2170g, 22°C), washed, and resuspended in N-free TAP medium to 2 × 106 cells mL−1 as described previously (Boyle et al., 2012; Blaby et al., 2013). Fifty milliliters of the culture was collected (5 min at 3440g, 22°C) at the indicated time points after transfer to N-free medium and resuspended in 2 mL of RNase free water. Samples for RNA-Seq analysis of CC4349 and CC4348 (sta6) were taken in duplicates. The resuspended cells were added to 2 mL of 50°C prewarmed lysis buffer (0.1 M Tris-HCl, pH 7.5, 0.3 M NaCl, 30 mM EDTA, 4% SDS, and 80 µg/mL Proteinase K) in 14 mL of RNA-free Sarstedt tubes (catalog numbers 55-538 [tube] and 65-816 [cap]), and the suspension was rocked for 20 min at room temperature, flash-frozen in liquid N2, and stored at −80°C. Frozen samples were subsequently heated at 65°C for 3 min before mRNA was isolated after four extractions with an equal volume of phenol/chloroform/isoamyl alcohol (25:24:1), followed by two extractions of the resultant aqueous phase with an equal volume of chloroform/isoamyl alcohol (24:1). RNA was precipitated overnight at −20°C after the addition of 2.5 volumes of −20°C precooled 100% ethanol. The precipitate was collected by centrifugation (30 min at 7840g, 4°C) and washed with 70% ethanol.
After removal of the final traces of ethanol, the pellet was resuspended in 100 to 200 μL water. mRNA concentration was determined on a NanoDrop 2000 (Thermo Scientific), and the quality was assessed on an Agilent 2100 bioanalyzer and by blot hybridization for CBLP (RACK1) mRNA (Quinn and Merchant, 1998). RNAs of all strains were sequenced at Illumina and Los Alamos National Laboratory on a GAIIx system for assessing transcript abundance in N deficiency time-course experiments and analyzed as described previously (Boyle et al., 2012; Blaby et al., 2013). Sequencing reads were aligned to the C. reinhardtii genome (Merchant et al., 2007) using GSNAP (Wu and Nacu, 2010). For each read, we imposed a minimum alignment similarity of 80% to the reference, and this threshold was used to differentiate between unique and ambiguous alignments. Only unique alignments were used to compute expression estimates. Per-sample normalized coverage vectors were built from the final set of high-quality disambiguated alignments, which are displayed in the form of per base graphs on our local installation of the UCSC genome browser (http://genomes.mcdb.ucla.edu/). Sequence alignments were summarized for Au10.2 gene models to generate matrices of counts per gene. For each sample and gene, expression estimates were obtained after normalizing the counts by the gene mappable length and the sample’s sequencing depth and are given in units of RPKM (Mortazavi et al., 2008). Independent filtering (Anders and Huber, 2010) was subsequently applied to remove genes with low expression across the entire CC4532 data set. The remaining 12,338 genes were used for differential expression analysis using DESeq (Anders and Huber, 2010). In the absence of biological replicates in the CC4532 data, the biological variance within the experiment was estimated from the variance at time point 0 in the 1-, 8-, and 48-h experiment. Genes were classified as differentially expressed using a minimum of 2-fold regulation and a false discovery rate of <1% for the CC4349 and CC4348 (sta6) data sets and <5% for the CC4532 data set. Afterwards, genes were classified according to their temporal expression profiles. For clustering, the data from the three independent experiments in C. reinhardtii strain CC4532 were combined into a single, global data set, including all time points from the 1-h experiment (+N, 0, 2, 4, 8, 12, 18, 24, 30, 45, and 60 min after transfer to N-free medium), the 2-h time point from the 48-h experiment, the 4-h time point from the 8-h experiment, and the 8-, 12-, 24-, and 48-h time point from the 48-h experiment. Hierarchical clustering was performed using the R package MBCluster.Seq (Si et al., 2014) on the 4288 differentially expressed genes in strain CC4532 in order to provide a global view of the most relevant expression patterns.
Nano-LC-MS/MS Protein Identification, Quantification, and Analysis
15N quantitative shotgun proteomics was mainly performed as previously described by Mühlhaus et al. (2011). Differences are mentioned in the following brief summary of the experimental workflow. C. reinhardtii cells were grown at constant illumination in TAP medium containing 7.5 mM 15NH4Cl (>99%; Cambridge Isotope Laboratories; NLM-467-25) as a N source and constantly were subcultured into 15NH4Cl -containing TAP medium for at least 10 generations to fully 15N-label the cells. The labeled cells were depleted from N as described above, were collected at similar time points (+15N and 0, 0.5, 2, 4, 8, 12, 24, and 48 h after transfer to N-free medium), and were combined based on a similar number of cells to generate a 15N-labeled reference standard. This reference standard of 15N-labeled cells was then added to unlabeled samples from three biological replicates of the N deficiency time course at a 15N/14N ratio of 0.8 based on total protein content determined by the Lowry method. Mixed 15N/14N cells were ruptured by multiple freeze/thaw cycles and separated by centrifugation into soluble and membrane-enriched fractions. The supernatant was recovered, considered as soluble protein fraction, while the pellet, containing membrane-associated proteins, was washed once in ammonium bicarbonate buffer, centrifuged again, and resuspended in the same buffer. In each case, 200 µg of soluble or membrane-associated proteins as determined by the Lowry method were precipitated in 80% acetone overnight at −80°C, solubilized/denatured in urea/thiourea, and digested to peptides by endopeptidase trypsin and LysC.
The extracted peptides were subjected to the mass spectrometer by an ACQUITY nanoUPLC (Waters). Peptides were trapped on a symmetry C18 trap column (5-µm particle size; 180-µm × 20-mm column dimension) and separated on a BEH 130 C18 column (1.7-µm particle size; 75-µm × 150-mm column dimension). For separation, the dual gradient ramped from 100% buffer A (0.4% acetic acid, 1% 2-propanole, and 2% acetonitrile) to 40% buffer B (0.4% acetic acid, 1% 2-propanole, and 90% acetonitrile) within 2 h and then to 90% B within 5 min, stayed at 90% buffer B for 15 min, and the column was equilibrated for 15 min with 100% buffer A again. The employed hybrid LTQ XL-Orbitrap mass spectrometer (Thermo Scientific) operated in the data-dependent mode in a cycle of one full scan mass spectrum (300 to 1500 m/z or 300 to 2000 m/z; Orbitrap) at a set resolution of 60,000 at 400 m/z followed by six consecutive data-dependent MS2 scans (LTQ). Parent ions used for MS2 analysis were set on an exclusion list for 20 s. Each sample was analyzed three times, excluding single charged parent ions from MS2 analysis and one time including single charged parent ions for MS2 analysis.
Proteins were identified and quantified using the IOMIQS framework (integration of mass spectrometry identification and quantification software) (Mühlhaus et al., 2011). Four different search engines for peptide identifications were used, namely, Mascot (version 2.2.04) (Perkins et al., 1999), Sequest (version 28) (Eng et al., 1994), OMSSA (version 2.1.4) (Geer et al., 2004), and X!Tandem (version 2009.04.01.1) (Craig and Beavis, 2004). Precursor mass tolerance was set to 10 ppm (OMSSA: 0.015 D), and the fragment ion tolerance was set to 0.8 D. Up to three missed cleavages were allowed for tryptic peptides. As variable modifications, carbamidomethylation of Cys and oxidation of Met were selected for Mascot, OMSSA, and X!Tandem. The protein sequence database included translated sequences of Augustus gene models (version 10.2) of the C. reinhardtii genome sequence as well as mitochondrial and chloroplast proteins (downloaded from http://chlamycyc.mpimp-golm.mpg.de/files/sequences/protein/). False discovery rate was determined using shuffled protein sequences added to the search database to perform the target decoy approach (Elias and Gygi, 2007). The significance threshold was set to a P value ≤ 0.05. 15N-labeled peptides were identified with exactly the same settings but in 15N mode. For peptide quantification, the algorithm ASAPRatio (Li et al., 2003) was used with peptide mass tolerance set to 0.8 D. The median of peptides light-to-heavy ratios was used to determine the relative abundance for the corresponding proteins. Ratios were corrected for unequal mixing of labeled and unlabeled proteins in each sample by the geometric median of light-to-heavy ratios of all peptides in that sample. Peptides were classified according to their information content according to Qeli and Ahrens (2010). Class 1a peptides unambiguously identify a single unique protein sequence, class 1b is only ambiguous regarding protein isoforms, class 2a and 2b identify a distinct gene model, while class 2a peptides additionally identifies a proper subset. Class 3a peptides unambiguously identify a protein sequence that can be encoded by several gene models from distinct loci, and class 3b peptides are totally ambiguous. The Occam’s Razor approach (Nesvizhskii et al., 2003) was applied to each peptide class to report a minimal set of proteins explaining the identified peptides by proteins and protein groups. Protein groups were classified according to their peptide(s) with the maximal information content.
Statistical analyses for mass spectrometry data were performed using Microsoft F# functional programming language with the mathematical library Math.NET Numerics (http://numerics.mathdotnet.com/) and the graphical library FSharpChart (http://code.msdn.microsoft.com/windowsdesktop/FSharpChart-b59073f5). Significance was tested using one-way ANOVA over the time points of log-transformed protein kinetics. P values were adjusted according to the Benjamini and Hochberg procedure (Benjamini and Hochberg, 1995), and the significance threshold was set to a P value ≤ 0.05. Identified proteins were functionally annotated using MapMap for C. reinhardtii (Thimm et al., 2004), including changes made by the community within the IOMIQS functional annotation tool (http://iomiqsweb1.bio.uni-kl.de/). The centered protein changes were clustered using k-means algorithm with euclidean distance metric, and the number of clusters was determined using gap statistics taking into account the original data set's shape (Tibshirani et al., 2001). Clusters were categorized as “up” and “down,” and their significant functional enrichment is tested using hypergeometric testing. The null hypothesis is that the change in protein abundance and its presence within a certain protein category is statistically independent. Hypergeometric formulation is directly derived from the problem statement and was shown to be appropriate even for a small number of proteins (Rivals et al., 2007). P values were adjusted according to the Benjamini and Hochberg procedure (Benjamini and Hochberg, 1995), and the significance threshold was set to a P value ≤ 0.05. Hierarchical Treemap diagrams of enriched functional categories were plotted using IOMIQS functional annotation tool.
LC-MS/MS Metabolite Analysis
Cells were directly sprayed into precooled (< −60°C) quenching solution by a syringe (1:2 culture:quenching solution), or precooled methanol was added to the cells as described previously (Bölling and Fiehn, 2005; Kempa et al., 2009). The temperature was checked regularly to assure it remained below −20°C during the whole procedure. The material was frozen in liquid nitrogen and stored at −80°C or in liquid nitrogen before metabolite extraction. Water-soluble metabolites were extracted using a method based on previously published protocols (Lunn et al., 2006; Arrivault et al., 2009). In brief, 525 μL of quenched algal material was added to 105 μL of precooled chloroform and was vigorously mixed by vortexing in the presence of glass beads (final ratio C:W:M = 1:5.3:2.3). Authentic analyte standards were added to the quenched material before adding chloroform. Cell lysis was achieved using three cycles of thaw-freeze. The aqueous fraction was collected after centrifugation (5 min at 13,500 rpm, 4°C), and the chloroform fraction was washed twice with 560 μL of ice-cold water. The three aqueous fractions were unified and freeze-dried overnight (Alpha 2-4; Christ GmbH). Samples were resuspended and filtered with a multiscreen filter plates (Ultracel-10; Millipore) before LC-MS/MS analysis. A total of 70 to 80 μL of culture equivalent (cell density 3 to 5 × 106 cells mL−1) was analyzed per LC-MS/MS run. LC-MS/MS was performed on a Dionex HPLC system coupled to a Finnnigan TSQ Quantum Discovery MS-Q3 (Thermo Scientific) equipped with an electrospray ionization interface. Runs were performed as described (Arrivault et al., 2009) with slight modifications to the LC gradient. Chromatographic separation was performed by passing aliquots through a Gemini (C18) 4 × 2-mm precolumn (Phenomenex), before separation on a Gemini C18 column (150 × 2-mm inner diameter, 5-μm 110 Å particle; Phenomenex) at 35°C using a multistep gradient with online-degassed eluent A (10 mM tributylamine aqueous solution, adjusted to pH 5.0 with 15 mM acetic acid and 5% methanol) and eluent B (100% methanol): 0 to 5 min, 100% A; 5 to 15 min, 100 to 95% A; 15 to 22 min, 95 to 90% A; 22–37 min, 90 to 85% A; 37 to 40 min, 85 to 70% A, and maintained for 3 min; 43 to 47 min, 70 to 45% A, and maintained for 3 min; 50 min, 10% A, and maintained for 8 min; 58 min, 100% A, and maintained for 8 min. The flow rate was 0.2 mL min−1 and was increased to 0.3 mL min−1 between 22 and 58 min. After separation, compounds were ionized by electrospray ionization and detected by a triple quadrupole that was operated in negative ion mode with selected reaction monitoring, using an ion spray voltage of 4000 V and a capillary temperature of 230°C. Finnigan XCALIBUR 2.5 software (Thermo Scientific) was used for both instrument control and data acquisition. Prior to injection (100 µL), a mixture of 15 stable isotope reference compounds of known concentrations was added to the sample to correct for matrix effects on these analytes in the analysis. Metabolites were quantified by comparison of the integrated MS-Q3 signal peak area with a calibration curve obtained using authentic standards by the LCQuan software (Thermo Scientific). Further analysis was done by Windows Excel and R statistics software (R version 2.9.2 and 2.14.1 provided by the CRAN project; http://www.R-project.org).
mRNA Preparation and RNA-Seq Analysis in P Deficiency
C. reinhardtii strain 21 gr (CC1690, wild-type) was grown in TAP medium (Harris, 1989) in a rotary incubator (200 rpm) at 25°C in continuous light (70 µmol m−2 s−1). P deprivation was achieved by washing cells twice in mid-logarithmic growth phase with liquid TAP medium without P (TAP-P), and cells were resuspended at a density of 2.5 mg/mL chlorophyll in TAP or TAP-P. Cell aliquots were collected for mRNA isolation 24 h after being transferred either to TAP or TAP-P medium. Isolation of total RNA was realized using a phenol-chloroform procedure as previously described (Schloss et al., 1984). cDNA libraries were prepared and sequenced as 35-mers at Illumina on a GAIIx system for assessing transcript abundance. Sequencing reads were aligned and analyzed as described above for the N deprivation experiments.
Oxygen Consumption and Evolution
Oxygen evolution rates were measured on a standard Clark-type electrode (Hansatech Oxygraph with a DW-1 chamber). All rates were recorded and analyzed with the Hansatech OxyLab software version 1.15. Experiments were performed with 2 mL of culture (strain CC4532) at a density of ∼2 × 106 cells mL−1 in the presence of 20 mM acetate and 10 mM KHCO3. Respiration rate was measured as oxygen consumption over a period of 5 min in the dark. The rate of photosynthetic O2 evolution was measured for 5 min in the light (300 µmol photons m−2 s−1) after a 5-min acclimation period and was calculated as the difference between oxygen evolution in the light and oxygen consumption in the dark.
N, C, and S Contents of the Proteome
To compare N, C, and S atom composition in different protein sets, the quantile distributions of these atoms in amino acid residues were calculated for each set (Karlin and Brendel, 1992). The significance of the differences of the distributions was assessed by two-tailed Student’s t test (Baudouin-Cornu et al., 2001). The protein sets were defined by the contrast between time points 0 and 48 h also assessed by two-tailed Student’s t test and divided in up- and downregulated proteins among the differential changed proteins. The whole proteome based on Augustus 10.2 gene models was used as the reference set. To determine the absolute amount of N that is spared by the reduction of proteins containing a high number of amino acids with N-containing side chain, for each protein the average number of N in site chain was weighted by the particular empirical protein abundance index of each detectable protein as an estimate for the absolute abundance, according to:
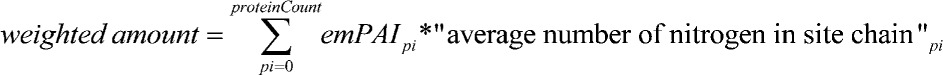 |
The weighted amount at time point 0 was set 100% and compared with the 48-h time point to calculate the percentage of decrease. The values are estimated under the assumption that the amount of protein within the cell stays constant and so only take into account the sparing resulting from the sequence differences.
Total Organic C and N Measurement
A total of 3 × 107 cells (CC4532) were collected by centrifugation (2 min at 3100g, 4°C) in an Eppendorf 5810R centrifuge at different time points after transfer to N-free medium. The cells were washed once in 50 mL deionized water and then resuspended in 900 μL of 3 M HCl (3.33 × 107 cells mL−1). Cells were incubated for 16 h at 65°C with constant agitation before being subjected to nonpurgeable organic C and total N analysis on a Shimadzu TOC-L CSH instrument. Two hundred microliters of a 3 × 105 cells mL−1 diluted sample was cleared of inorganic C by sparging, and the remaining nonpurgeable organic C was oxidized to CO2 at 720°C in the presence of a catalyst. The nonpurgeable organic C was measured in technical triplicates with a nondispersive infrared gas analyzer, while total N was analyzed in triplicates on a chemiluminescence gas analyzer. The peak area was calculated and compared with a standard curve from 0.5 to 25 ppm C (from potassium hydrogen phthalate) or N (from potassium nitrate) using the TOC-Control L software version 1.0 (Shimadzu). Measured concentrations did not exceed the range of 4 to 15 ppm for C and 0.7 to 2.5 for N.
Amino Acid Analysis
A total of 2 × 107 cells (CC4532) was collected by centrifugation (2 min at 3100g, 4°C) in an Eppendorf 5810R centrifuge 48 h after transfer to either N-free (−N) or N-containing (+N) medium. The cells were washed twice in 50 mL deionized water and then resuspended in 2% SDS. After three freeze/thaw cycles, the broken cells were centrifuged for 1 h at 104,300g, 4°C, TLA110 to remove polysaccharides and unsolubilized matter. Four volumes of −20°C precooled 100% acetone was added to the supernatants, and after 1 h incubation at −20°C, the proteins were collected (30 min at 16,100g, 4°C). The protein pellet was washed twice with 80% acetone and dried before being resuspended in 2% acetonitrile. Acetone precipitation was repeated twice to ensure complete pigment and lipid extraction. The resuspended precipitates were hydrolyzed in vapor phase in 6 n HCl for 22 h at 110°C before amino acid composition was determined on an amino acid analyzer at the protein/peptide chemistry core facility at UCLA (Biopolymer Laboratory, under the supervision of David B. Teplow). The amino acid samples were quantified using AccQTag Chemistry (Waters), a precolumn derivatization method, followed by reverse-phase HPLC using a binary pump system (Waters 1500 Series), a scanning fluorescence detector (Waters Model 474), and a Waters 717 Plus Automated Injector. Sample amino acid quantification was done by comparison to amino acid standards analyzed in an identical manner.
Accession Numbers
Sequence data from this article concerning N deficiency are deposited in the sequence read archive (http://sra.dnanexus.com) under accession number SRX038871 (CC4532) and in the National Center for Biotechnology Information Gene Expression Omnibus (NCBI GEO) database (http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE51602 (CC4348 and CC4349). Sequence data from this article concerning P deprivation are deposited in the NCBI GEO database under accession number GSE56505.
Supplemental Data
The following materials are available in the online version of this article.
Supplemental Figure 1. Comparison of mRNA Expression Estimates between Different Biological Replicates of C. reinhardtii Strain CC4532.
Supplemental Figure 2. Clustering of 4288 Differentially Expressed Genes Identified in the CC4532 RNA-Seq Experiments.
Supplemental Figure 3. Summary of the Experimental Data Gathered from the Proteomics Time-Course Experiment.
Supplemental Figure 4. Clustering of 635 Differentially Accumulating Proteins upon Transfer to N-Free Media.
Supplemental Figure 5. mRNA and Protein Abundances for Components of the Photosynthetic Complexes.
Supplemental Figure 6. mRNA and Protein Abundance of Components of the Respiratory Complexes.
Supplemental Figure 7. mRNA and Protein Abundance of Enzymes Involved in Calvin-Benson Cycle.
Supplemental Figure 8. mRNA Abundances of Enzymes Involved in Tetrapyrrole Biosynthesis and Degradation.
Supplemental Figure 9. Alignment of Putative Chlorophyll b Reductases from Oryza sativa, Arabidopsis thaliana, and Chlamydomonas reinhardtii.
Supplemental Figure 10. mRNA Abundance of Enzymes Involved in Heme Biosynthesis and Degradation.
Supplemental Figure 11. Abundance of Ribosomal Proteins and Corresponding mRNAs.
Supplemental Figure 12. Nitrogen-Sparing Mechanisms in C. reinhardtii.
Supplemental Figure 13. Abundance of Autophagy-Related Genes.
Supplemental Figure 14. mRNA Abundance of Transporters Involved in Ammonium Import.
Supplemental Figure 15. Abundance of Glutamine Synthases and GLN RNAs.
Supplemental Figure 16. Abundance of Glutamate Oxoglutarate Amidotransferases and Corresponding RNAs.
Supplemental Figure 17. Abundance of RNAs Encoding Nitrate or Nitrite Metabolism Functions.
Supplemental Figure 18. Abundance of RNAs Encoding (Potential) Regulators of N Assimilation.
Supplemental Table 1. Overlap of Differentially Accumulating mRNAs between All and Individual Strains.
Supplemental Table 2. Common Patterns of Changing mRNA Abundances in Nitrogen, Sulfur, and Phosphorus Deprivation.
Supplemental Data Set 1. Overview of RNA-Seq and Quantitative Proteomics Experiments (All Genes).
Supplemental Data Set 2. Overview of RNA-Seq and Quantitative Proteomics Experiments (Conserved N-Regulated Genes).
Supplemental Data Set 3. Significantly Altered Proteins upon Transfer to N-Free Media.
Supplemental Data Set 4. Significantly Regulated MapMan Categories.
Supplemental Data Set 5. Overview of RNA-Seq and Quantitative Proteomics Experiments (Photosynthesis).
Supplemental Data Set 6. Overview of RNA-Seq and Quantitative Proteomics Experiments (Respiration).
Supplemental Data Set 7. Overview of RNA-Seq and Quantitative Proteomics Experiments (Calvin-Benson Cycle).
Supplemental Data Set 8. Overview of RNA-Seq and Quantitative Proteomics Experiments (Tetrapyrrole Metabolism).
Supplemental Data Set 9. Overview of RNA-Seq and Quantitative Proteomics Experiments (Ribosomes).
Supplemental Data Set 10. Overview of RNA-Seq and Quantitative Proteomics Experiments (Stress and Autophagy-Related Genes).
Supplemental Data Set 11. Overview of RNA-Seq and Quantitative Proteomics Experiments (N Metabolism).
Supplemental Data Set 12. Overview of RNA-Seq and Quantitative Proteomics Experiments (Gametogenesis).
Supplemental Data Set 13. Overview of RNA-Seq and Quantitative Proteomics Experiments (Transcription Factors).
Supplemental Data Set 14. Overview of RNA-Seq and Quantitative Proteomics Experiments (Fatty Acid Metabolism).
Supplemental Data Set 15. Overview of RNA-Seq and Quantitative Proteomics Experiments (Similar Changing Genes in N and S Deficiency).
Supplemental Data Set 16. Overview of RNA-Seq and Quantitative Proteomics Experiments (Similar Changing Genes in N and P Deficiency).
Supplemental Data Set 17. Overview of RNA-Seq and Quantitative Proteomics Experiments (Similar Changing Genes in N, S, and P Deficiency).
Supplementary Material
Acknowledgments
This work was supported by Department of Energy Contract DE-EE0003046 (to S.S.M. and M.P. via the National Alliance for Advance Biofuels and Bioproducts Consortium), NSF 0951094 (to A.R.G.), and in part by the National Institutes of Health R24 GM092473 to S.S.M. This work was also supported by the Max Planck Society and the Bundesministerium für Bildung und Forschung (Systems Biology Initiative FORSYS, Project GoFORSYS). I.K.B is supported by a training grant from the National Institutes of Health (T32 ES015457), and D.S. is supported by an EMBO fellowship (ALTF 653-2013). We thank Ursula Goodenough for forwarding strains cw15 (CC4349), sta6 (CC4348), STA6-C2 (CC4565), STA6-C4 (CC4566), and STA6-C6 (CC4567) and David Dauvillée for an independent cw15 (CC-4568).
AUTHOR CONTRIBUTIONS
S.S.M., I.K.B., N.R.B., T. Mettler, T. Mühlaus, S.S., and D.C. designed the experiments. N.R.B., D.H., and F.S. performed the proteomics experiments. N.R.B. and I.K.B. performed the RNA-Seq experiments. J.L.M. and A.R.G. designed and performed the –P RNA-Seq study. J.K., S.S., D.S., and T. Mettler performed other experiments. D.C. performed the bioinformatic analysis of RNA-Seq data. T. Mühlaus performed the bioinformatic analysis of the proteomics data. M.P. supervised all bioinformatic analysis. M. Schroda supervised the proteomics analysis. M. Stitt supervised the metabolite analysis. S.S.M., T. Mühlaus, S.S., and I.K.B. analyzed the data. S.S.M. and S.S. prepared and edited the article. All authors commented on and revised the article.
Glossary
- GS
Gln synthetase
- GOGAT
Glu oxoglutarate amidotransferase
- TAG
triacylglycerol
- LC-MS/MS
liquid chromatography–tandem mass spectrometry
- RPKM
reads per kilobase of gene model per million reads
- PSII
photosystem II
- PSI
photosystem I
- Fv/Fm
quantum efficiency of PSII
- TPIC
triose phosphate isomerase
- ACCase
acetyl-CoA carboxylase
- FAS
fatty acid synthase
- NUE
nitrogen use efficiency
- TAP
Tris-acetate-phosphate
Footnotes
Online version contains Web-only data.
References
- Abe J., Kubo T., Takagi Y., Saito T., Miura K., Fukuzawa H., Matsuda Y. (2004). The transcriptional program of synchronous gametogenesis in Chlamydomonas reinhardtii. Curr. Genet. 46: 304–315 [DOI] [PubMed] [Google Scholar]
- Acquisti C., Kumar S., Elser J.J. (2009). Signatures of nitrogen limitation in the elemental composition of the proteins involved in the metabolic apparatus. Proc. Biol. Sci. 276: 2605–2610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen J.F., de Paula W.B.M., Puthiyaveetil S., Nield J. (2011). A structural phylogenetic map for chloroplast photosynthesis. Trends Plant Sci. 16: 645–655 [DOI] [PubMed] [Google Scholar]
- Anders S., Huber W. (2010). Differential expression analysis for sequence count data. Genome Biol. 11: R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrivault S., Guenther M., Ivakov A., Feil R., Vosloh D., van Dongen J.T., Sulpice R., Stitt M. (2009). Use of reverse-phase liquid chromatography, linked to tandem mass spectrometry, to profile the Calvin cycle and other metabolic intermediates in Arabidopsis rosettes at different carbon dioxide concentrations. Plant J. 59: 826–839 [DOI] [PubMed] [Google Scholar]
- Barakat A., Szick-Miranda K., Chang I.F., Guyot R., Blanc G., Cooke R., Delseny M., Bailey-Serres J. (2001). The organization of cytoplasmic ribosomal protein genes in the Arabidopsis genome. Plant Physiol. 127: 398–415 [PMC free article] [PubMed] [Google Scholar]
- Baudouin-Cornu P., Surdin-Kerjan Y., Marlière P., Thomas D. (2001). Molecular evolution of protein atomic composition. Science 293: 297–300 [DOI] [PubMed] [Google Scholar]
- Benjamini Y., Hochberg Y. (1995). Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. B 57: 289–300 [Google Scholar]
- Blaby I.K., et al. (2013). Systems-level analysis of nitrogen starvation-induced modifications of carbon metabolism in a Chlamydomonas reinhardtii starchless mutant. Plant Cell 25: 4305–4323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloom A.J., Sukrapanna S.S., Warner R.L. (1992). Root respiration associated with ammonium and nitrate absorption and assimilation by barley. Plant Physiol. 99: 1294–1301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bölling C., Fiehn O. (2005). Metabolite profiling of Chlamydomonas reinhardtii under nutrient deprivation. Plant Physiol. 139: 1995–2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle N.R., Morgan J.A. (2009). Flux balance analysis of primary metabolism in Chlamydomonas reinhardtii. BMC Syst. Biol. 3: 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle N.R., et al. (2012). Three acyltransferases and nitrogen-responsive regulator are implicated in nitrogen starvation-induced triacylglycerol accumulation in Chlamydomonas. J. Biol. Chem. 287: 15811–15825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulté L., Wollman F.A. (1992). Evidence for a selective destabilization of an integral membrane protein, the cytochrome b6/f complex, during gametogenesis in Chlamydomonas reinhardtii. Eur. J. Biochem. 204: 327–336 [DOI] [PubMed] [Google Scholar]
- Cakmak T., Angun P., Demiray Y.E., Ozkan A.D., Elibol Z., Tekinay T. (2012). Differential effects of nitrogen and sulfur deprivation on growth and biodiesel feedstock production of Chlamydomonas reinhardtii. Biotechnol. Bioeng. 109: 1947–1957 [DOI] [PubMed] [Google Scholar]
- Camargo A., Llamas Á., Schnell R.A., Higuera J.J., González-Ballester D., Lefebvre P.A., Fernández E., Galván A. (2007). Nitrate signaling by the regulatory gene NIT2 in Chlamydomonas. Plant Cell 19: 3491–3503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chisti Y. (2007). Biodiesel from microalgae. Biotechnol. Adv. 25: 294–306 [DOI] [PubMed] [Google Scholar]
- Craig R., Beavis R.C. (2004). TANDEM: matching proteins with tandem mass spectra. Bioinformatics 20: 1466–1467 [DOI] [PubMed] [Google Scholar]
- Cullimore J.V., Sims A.P. (1981). Pathway of ammonia assimilation in illuminated and darkened Chlamydomonas reinhardii. Phytochemistry 20: 933–940 [Google Scholar]
- de Sousa Abreu R., Penalva L.O., Marcotte E.M., Vogel C. (2009). Global signatures of protein and mRNA expression levels. Mol. Biosyst. 5: 1512–1526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Díaz-Troya S., Pérez-Pérez M.E., Florencio F.J., Crespo J.L. (2008). The role of TOR in autophagy regulation from yeast to plants and mammals. Autophagy 4: 851–865 [DOI] [PubMed] [Google Scholar]
- Eberhard S., Finazzi G., Wollman F.A. (2008). The dynamics of photosynthesis. Annu. Rev. Genet. 42: 463–515 [DOI] [PubMed] [Google Scholar]
- Elias J.E., Gygi S.P. (2007). Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods 4: 207–214 [DOI] [PubMed] [Google Scholar]
- Eng J.K., McCormack A.L., Yates J.R., III (1994). An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 5: 976–989 [DOI] [PubMed] [Google Scholar]
- Ermilova E., Lapina T., Zalutskaya Z., Minaeva E., Fokina O., Forchhammer K. (2013). PII signal transduction protein in Chlamydomonas reinhardtii: localization and expression pattern. Protist 164: 49–59 [DOI] [PubMed] [Google Scholar]
- Fauchon M., Lagniel G., Aude J.C., Lombardia L., Soularue P., Petat C., Marguerie G., Sentenac A., Werner M., Labarre J. (2002). Sulfur sparing in the yeast proteome in response to sulfur demand. Mol. Cell 9: 713–723 [DOI] [PubMed] [Google Scholar]
- Fernandez E., Galvan A. (2007). Inorganic nitrogen assimilation in Chlamydomonas. J. Exp. Bot. 58: 2279–2287 [DOI] [PubMed] [Google Scholar]
- Fernandez E., Galvan A. (2008). Nitrate assimilation in Chlamydomonas. Eukaryot. Cell 7: 555–559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández E., Matagne R.F. (1986). In vivo complementation analysis of nitrate reductase-deficient mutants in Chlamydomonas reinhardtii. Curr. Genet. 10: 397–403 [DOI] [PubMed] [Google Scholar]
- Fernández E., Schnell R., Ranum L.P.W., Hussey S.C., Silflow C.D., Lefebvre P.A. (1989). Isolation and characterization of the nitrate reductase structural gene of Chlamydomonas reinhardtii. Proc. Natl. Acad. Sci. USA 86: 6449–6453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer P., Klein U. (1988). Localization of nitrogen-assimilating enzymes in the chloroplast of Chlamydomonas reinhardtii. Plant Physiol. 88: 947–952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florencio F.J., Vega J.M. (1983). Utilization of nitrate, nitrite and ammonium by Chlamydomonas reinhardii: Photoproduction of ammonium. Planta 158: 288–293 [DOI] [PubMed] [Google Scholar]
- Foyer C.H., Parry M., Noctor G. (2003). Markers and signals associated with nitrogen assimilation in higher plants. J. Exp. Bot. 54: 585–593 [DOI] [PubMed] [Google Scholar]
- Galván A., Quesada A., Fernández E. (1996). Nitrate and nitrate are transported by different specific transport systems and by a bispecific transporter in Chlamydomonas reinhardtii. J. Biol. Chem. 271: 2088–2092 [DOI] [PubMed] [Google Scholar]
- Gardemann A., Stitt M., Heldt H.W. (1983). Control of CO2 fixation. Regulation of spinach ribulose-5-phosphate kinase by stromal metabolite levels. Biochim. Biophys. Acta 722: 51–60 [Google Scholar]
- Geer L.Y., Markey S.P., Kowalak J.A., Wagner L., Xu M., Maynard D.M., Yang X., Shi W., Bryant S.H. (2004). Open mass spectrometry search algorithm. J. Proteome Res. 3: 958–964 [DOI] [PubMed] [Google Scholar]
- Georgianna D.R., Mayfield S.P. (2012). Exploiting diversity and synthetic biology for the production of algal biofuels. Nature 488: 329–335 [DOI] [PubMed] [Google Scholar]
- Giordano M., Norici A., Forssen M., Eriksson M., Raven J.A. (2003). An anaplerotic role for mitochondrial carbonic anhydrase in Chlamydomonas reinhardtii. Plant Physiol. 132: 2126–2134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- González-Ballester D., Camargo A., Fernández E. (2004). Ammonium transporter genes in Chlamydomonas: the nitrate-specific regulatory gene Nit2 is involved in Amt1;1 expression. Plant Mol. Biol. 56: 863–878 [DOI] [PubMed] [Google Scholar]
- González-Ballester D., Casero D., Cokus S., Pellegrini M., Merchant S.S., Grossman A.R. (2010). RNA-seq analysis of sulfur-deprived Chlamydomonas cells reveals aspects of acclimation critical for cell survival. Plant Cell 22: 2058–2084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- González-Ballester D., de Montaigu A., Higuera J.J., Galván A., Fernández E. (2005). Functional genomics of the regulation of the nitrate assimilation pathway in Chlamydomonas. Plant Physiol. 137: 522–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodson C., Roth R., Wang Z.T., Goodenough U. (2011). Structural correlates of cytoplasmic and chloroplast lipid body synthesis in Chlamydomonas reinhardtii and stimulation of lipid body production with acetate boost. Eukaryot. Cell 10: 1592–1606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gresshoff P.M. (1981). Amide metabolism of Chlamydomonas reinhardi. Arch. Microbiol. 128: 303–306 [Google Scholar]
- Harris, E.H. (1989). The Chlamydomonas Sourcebook: A Comprehensive Guide to Biology and Laboratory Use. (San Diego, CA: Academic Press). [DOI] [PubMed] [Google Scholar]
- Harris, E.H. (2008). The Chlamydomonas Sourcebook: Introduction to Chlamydomonas and Its Laboratory Use, 2nd ed. (San Diego, CA: Elsevier/Academic Press). [Google Scholar]
- Hellio C., Veron B., Le Gal Y. (2004). Amino acid utilization by Chlamydomonas reinhardtii: specific study of histidine. Plant Physiol. Biochem. 42: 257–264 [DOI] [PubMed] [Google Scholar]
- Hensley M.P., Tierney D.L., Crowder M.W. (2011). Zn(II) binding to Escherichia coli 70S ribosomes. Biochemistry 50: 9937–9939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirasawa M., Tripathy J.N., Sommer F., Somasundaram R., Chung J.S., Nestander M., Kruthiventi M., Zabet-Moghaddam M., Johnson M.K., Merchant S.S., Allen J.P., Knaff D.B. (2010). Enzymatic properties of the ferredoxin-dependent nitrite reductase from Chlamydomonas reinhardtii. Evidence for hydroxylamine as a late intermediate in ammonia production. Photosynth. Res. 103: 67–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodson R.C., Williams S.K., II, Davidson W.R., Jr (1975). Metabolic control of urea catabolism in Chlamydomonas reinhardi and Chlorella pyrenoidosa. J. Bacteriol. 121: 1022–1035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hörtensteiner S., Kräutler B. (2011). Chlorophyll breakdown in higher plants. Biochim. Biophys. Acta 1807: 977–988 [DOI] [PubMed] [Google Scholar]
- Hummel M., Cordewener J.H., de Groot J.C., Smeekens S., America A.H., Hanson J. (2012). Dynamic protein composition of Arabidopsis thaliana cytosolic ribosomes in response to sucrose feeding as revealed by label free MSE proteomics. Proteomics 12: 1024–1038 [DOI] [PubMed] [Google Scholar]
- Jones C.S., Mayfield S.P. (2012). Algae biofuels: versatility for the future of bioenergy. Curr. Opin. Biotechnol. 23: 346–351 [DOI] [PubMed] [Google Scholar]
- Kalapos M.P. (1999). Methylglyoxal in living organisms: chemistry, biochemistry, toxicology and biological implications. Toxicol. Lett. 110: 145–175 [DOI] [PubMed] [Google Scholar]
- Karlin S., Brendel V. (1992). Chance and statistical significance in protein and DNA sequence analysis. Science 257: 39–49 [DOI] [PubMed] [Google Scholar]
- Kempa S., Hummel J., Schwemmer T., Pietzke M., Strehmel N., Wienkoop S., Kopka J., Weckwerth W. (2009). An automated GCxGC-TOF-MS protocol for batch-wise extraction and alignment of mass isotopomer matrixes from differential 13C-labelling experiments: a case study for photoautotrophic-mixotrophic grown Chlamydomonas reinhardtii cells. J. Basic Microbiol. 49: 82–91 [DOI] [PubMed] [Google Scholar]
- Kim K.S., Feild E., King N., Yaoi T., Kustu S., Inwood W. (2005). Spontaneous mutations in the ammonium transport gene AMT4 of Chlamydomonas reinhardtii. Genetics 170: 631–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirk D.L., Kirk M.M. (1978). Carrier-mediated uptake of arginine and urea by Chlamydomonas reinhardtii. Plant Physiol. 61: 556–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraft C., Deplazes A., Sohrmann M., Peter M. (2008). Mature ribosomes are selectively degraded upon starvation by an autophagy pathway requiring the Ubp3p/Bre5p ubiquitin protease. Nat. Cell Biol. 10: 602–610 [DOI] [PubMed] [Google Scholar]
- Kräutler, B., and Hörtensteiner, S. (2006). Chlorophyll catabolites and the biochemistry of chlorophyll breakdown. In Chlorophylls and Bacteriochlorophylls, B. Grimm, R.J. Porra, W. Rüdiger, and H. Scheer, eds (Dordrecht, The Netherlands: Springer), pp. 237–260. [Google Scholar]
- Li X., Moellering E.R., Liu B., Johnny C., Fedewa M., Sears B.B., Kuo M.H., Benning C. (2012). A galactoglycerolipid lipase is required for triacylglycerol accumulation and survival following nitrogen deprivation in Chlamydomonas reinhardtii. Plant Cell 24: 4670–4686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X.J., Zhang H., Ranish J.A., Aebersold R. (2003). Automated statistical analysis of protein abundance ratios from data generated by stable-isotope dilution and tandem mass spectrometry. Anal. Chem. 75: 6648–6657 [DOI] [PubMed] [Google Scholar]
- Longworth J., Noirel J., Pandhal J., Wright P.C., Vaidyanathan S. (2012). HILIC- and SCX-based quantitative proteomics of Chlamydomonas reinhardtii during nitrogen starvation induced lipid and carbohydrate accumulation. J. Proteome Res. 11: 5959–5971 [DOI] [PubMed] [Google Scholar]
- Lowry O.H., Rosebrough N.J., Farr A.L., Randall R.J. (1951). Protein measurement with the Folin phenol reagent. J. Biol. Chem. 193: 265–275 [PubMed] [Google Scholar]
- Lunn J.E., Feil R., Hendriks J.H.M., Gibon Y., Morcuende R., Osuna D., Scheible W.R., Carillo P., Hajirezaei M.R., Stitt M. (2006). Sugar-induced increases in trehalose 6-phosphate are correlated with redox activation of ADPglucose pyrophosphorylase and higher rates of starch synthesis in Arabidopsis thaliana. Biochem. J. 397: 139–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manuell A.L., Yamaguchi K., Haynes P.A., Milligan R.A., Mayfield S.P. (2005). Composition and structure of the 80S ribosome from the green alga Chlamydomonas reinhardtii: 80S ribosomes are conserved in plants and animals. J. Mol. Biol. 351: 266–279 [DOI] [PubMed] [Google Scholar]
- Martin N.C., Goodenough U.W. (1975). Gametic differentiation in Chlamydomonas reinhardtii. I. Production of gametes and their fine structure. J. Cell Biol. 67: 587–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin N.C., Chiang K.S., Goodenough U.W. (1976). Turnover of chloroplast and cytoplasmic ribosomes during gametogenesis in Chlamydomonas reinhardi. Dev. Biol. 51: 190–201 [DOI] [PubMed] [Google Scholar]
- Masclaux-Daubresse C., Daniel-Vedele F., Dechorgnat J., Chardon F., Gaufichon L., Suzuki A. (2010). Nitrogen uptake, assimilation and remobilization in plants: challenges for sustainable and productive agriculture. Ann. Bot. (Lond.) 105: 1141–1157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- May P., Wienkoop S., Kempa S., Usadel B., Christian N., Rupprecht J., Weiss J., Recuenco-Munoz L., Ebenhöh O., Weckwerth W., Walther D. (2008). Metabolomics- and proteomics-assisted genome annotation and analysis of the draft metabolic network of Chlamydomonas reinhardtii. Genetics 179: 157–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazel D., Marlière P. (1989). Adaptive eradication of methionine and cysteine from cyanobacterial light-harvesting proteins. Nature 341: 245–248 [DOI] [PubMed] [Google Scholar]
- Melis A., Zhang L., Forestier M., Ghirardi M.L., Seibert M. (2000). Sustained photobiological hydrogen gas production upon reversible inactivation of oxygen evolution in the green alga Chlamydomonas reinhardtii. Plant Physiol. 122: 127–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merchán F., van den Ende H., Fernández E., Beck C.F. (2001). Low-expression genes induced by nitrogen starvation and subsequent sexual differentiation in Chlamydomonas reinhardtii, isolated by the differential display technique. Planta 213: 309–317 [DOI] [PubMed] [Google Scholar]
- Merchant S.S., Helmann J.D. (2012). Elemental economy: microbial strategies for optimizing growth in the face of nutrient limitation. Adv. Microb. Physiol. 60: 91–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merchant S.S., Kropat J., Liu B., Shaw J., Warakanont J. (2012). TAG, you’re it! Chlamydomonas as a reference organism for understanding algal triacylglycerol accumulation. Curr. Opin. Biotechnol. 23: 352–363 [DOI] [PubMed] [Google Scholar]
- Merchant S.S., et al. (2007). The Chlamydomonas genome reveals the evolution of key animal and plant functions. Science 318: 245–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miflin B.J., Lea P.J. (1975). Glutamine and asparagine as nitrogen donors for reductant-dependent glutamate synthesis in pea roots. Biochem. J. 149: 403–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller R., et al. (2010). Changes in transcript abundance in Chlamydomonas reinhardtii following nitrogen deprivation predict diversion of metabolism. Plant Physiol. 154: 1737–1752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moellering, E.R., Miller, R., and Benning, C. (2010). Molecular genetics of lipid metabolism in the model green alga Chlamydomonas reinhardtii In Lipids in Photosynthesis, H. Wada and N. Murata, eds (Dordrecht, The Netherlands: Springer), pp. 139–155. [Google Scholar]
- Mortazavi A., Williams B.A., McCue K., Schaeffer L., Wold B. (2008). Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 5: 621–628 [DOI] [PubMed] [Google Scholar]
- Moseley J.L., Page M.D., Alder N.P., Eriksson M., Quinn J., Soto F., Theg S.M., Hippler M., Merchant S. (2002). Reciprocal expression of two candidate di-iron enzymes affecting photosystem I and light-harvesting complex accumulation. Plant Cell 14: 673–688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Msanne J., Xu D., Konda A.R., Casas-Mollano J.A., Awada T., Cahoon E.B., Cerutti H. (2012). Metabolic and gene expression changes triggered by nitrogen deprivation in the photoautotrophically grown microalgae Chlamydomonas reinhardtii and Coccomyxa sp. C-169. Phytochemistry 75: 50–59 [DOI] [PubMed] [Google Scholar]
- Mühlhaus, T., Weiss, J., Hemme, D., Sommer, F., and Schroda, M. (2011). Quantitative shotgun proteomics using a uniform 15N-labeled standard to monitor proteome dynamics in time course experiments reveals new insights into the heat stress response of Chlamydomonas reinhardtii Mol. Cell. Proteomics 10: M110.004739. [DOI] [PMC free article] [PubMed]
- Naumann B., Busch A., Allmer J., Ostendorf E., Zeller M., Kirchhoff H., Hippler M. (2007). Comparative quantitative proteomics to investigate the remodeling of bioenergetic pathways under iron deficiency in Chlamydomonas reinhardtii. Proteomics 7: 3964–3979 [DOI] [PubMed] [Google Scholar]
- Nesvizhskii A.I., Keller A., Kolker E., Aebersold R. (2003). A statistical model for identifying proteins by tandem mass spectrometry. Anal. Chem. 75: 4646–4658 [DOI] [PubMed] [Google Scholar]
- Nunes-Nesi A., Fernie A.R., Stitt M. (2010). Metabolic and signaling aspects underpinning the regulation of plant carbon nitrogen interactions. Mol. Plant 3: 973–996 [DOI] [PubMed] [Google Scholar]
- Peltier G., Schmidt G.W. (1991). Chlororespiration: an adaptation to nitrogen deficiency in Chlamydomonas reinhardtii. Proc. Natl. Acad. Sci. USA 88: 4791–4795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez-Rodríguez P., Riaño-Pachón D.M., Corrêa L.G., Rensing S.A., Kersten B., Mueller-Roeber B. (2010). PlnTFDB: updated content and new features of the plant transcription factor database. Nucleic Acids Res. 38: D822–D827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins D.N., Pappin D.J.C., Creasy D.M., Cottrell J.S. (1999). Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 20: 3551–3567 [DOI] [PubMed] [Google Scholar]
- Picard-Bennoun M., Bennoun P. (1985). Change in cytoplasmic ribosome properties during gametogenesis in the alga Chlamydomonas reinhardtii. Curr. Genet. 9: 239–243 [DOI] [PubMed] [Google Scholar]
- Pineda M., Fernández E., Cárdenas J. (1984). Urate oxidase of Chlamydomonas reinhardii. Physiol. Plant. 62: 453–457 [Google Scholar]
- Plumley F.G., Schmidt G.W. (1989). Nitrogen-dependent regulation of photosynthetic gene expression. Proc. Natl. Acad. Sci. USA 86: 2678–2682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porra R.J., Thompson W.A., Kriedemann P.E. (1989). Determination of accurate extinction coefficients and simultaneous equations for assaying chlorophylls a and b extracted with four different solvents: verification of the concentration of chlorophyll standards by atomic absorption spectroscopy. Biochim. Biophys. Acta 975: 384–394 [Google Scholar]
- Pulz O., Gross W. (2004). Valuable products from biotechnology of microalgae. Appl. Microbiol. Biotechnol. 65: 635–648 [DOI] [PubMed] [Google Scholar]
- Qeli E., Ahrens C.H. (2010). PeptideClassifier for protein inference and targeted quantitative proteomics. Nat. Biotechnol. 28: 647–650 [DOI] [PubMed] [Google Scholar]
- Quesada A., Gómez I., Fernández E. (1998). Clustering of the nitrite reductase gene and a light-regulated gene with nitrate assimilation loci in Chlamydomonas reinhardtii. Planta 206: 259–265 [DOI] [PubMed] [Google Scholar]
- Quesada A., Galván A., Schnell R.A., Lefebvre P.A., Fernández E. (1993). Five nitrate assimilation-related loci are clustered in Chlamydomonas reinhardtii. Mol. Gen. Genet. 240: 387–394 [DOI] [PubMed] [Google Scholar]
- Quinn J.M., Merchant S. (1998). Copper-responsive gene expression during adaptation to copper deficiency. Methods Enzymol. 297: 263–279 [DOI] [PubMed] [Google Scholar]
- Rivals I., Personnaz L., Taing L., Potier M.C. (2007). Enrichment or depletion of a GO category within a class of genes: which test? Bioinformatics 23: 401–407 [DOI] [PubMed] [Google Scholar]
- Robertson G.P., Vitousek P.M. (2009). Nitrogen in agriculture: Balancing the cost of an essential resource. Annu. Rev. Environ. Resour. 34: 97–125 [Google Scholar]
- Rodolfi L., Chini Zittelli G., Bassi N., Padovani G., Biondi N., Bonini G., Tredici M.R. (2009). Microalgae for oil: strain selection, induction of lipid synthesis and outdoor mass cultivation in a low-cost photobioreactor. Biotechnol. Bioeng. 102: 100–112 [DOI] [PubMed] [Google Scholar]
- Romero L.C., Galván F., Vega J.M. (1987). Purification and properties of the siroheme-containing ferredoxin-nitrite reductase from Chlamydomonas reinhardtii. Biochim. Biophys. Acta 914: 55–63 [Google Scholar]
- Scheibe R. (1991). Redox-modulation of chloroplast enzymes: a common principle for individual control. Plant Physiol. 96: 1–3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheible W.R., Morcuende R., Czechowski T., Fritz C., Osuna D., Palacios-Rojas N., Schindelasch D., Thimm O., Udvardi M.K., Stitt M. (2004). Genome-wide reprogramming of primary and secondary metabolism, protein synthesis, cellular growth processes, and the regulatory infrastructure of Arabidopsis in response to nitrogen. Plant Physiol. 136: 2483–2499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schloss J.A., Silflow C.D., Rosenbaum J.L. (1984). mRNA abundance changes during flagellar regeneration in Chlamydomonas reinhardtii. Mol. Cell. Biol. 4: 424–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnell R.A., Lefebvre P.A. (1993). Isolation of the Chlamydomonas regulatory gene NIT2 by transposon tagging. Genetics 134: 737–747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schötz F., Bathelt H., Arnold C.G., Schimmer O. (1972). The architecture and organization of the Chlamydomonas cell. Results of serial-section electron microscopy and a three-dimensional reconstruction. (in English). Protoplasma 75: 229–254 [DOI] [PubMed] [Google Scholar]
- Scott S.A., Davey M.P., Dennis J.S., Horst I., Howe C.J., Lea-Smith D.J., Smith A.G. (2010). Biodiesel from algae: challenges and prospects. Curr. Opin. Biotechnol. 21: 277–286 [DOI] [PubMed] [Google Scholar]
- Si Y., Liu P., Li P., Brutnell T.P. (2014). Model-based clustering for RNA-seq data. Bioinformatics 30: 197–205. [DOI] [PubMed] [Google Scholar]
- Siaut M., Cuiné S., Cagnon C., Fessler B., Nguyen M., Carrier P., Beyly A., Beisson F., Triantaphylidès C., Li-Beisson Y., Peltier G. (2011). Oil accumulation in the model green alga Chlamydomonas reinhardtii: characterization, variability between common laboratory strains and relationship with starch reserves. BMC Biotechnol. 11: 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siersma P.W., Chiang K.S. (1971). Conservation and degradation of cytoplasmic and chloroplast ribosomes in Chlamydomonas reinhardtii. J. Mol. Biol. 58: 167–185 [DOI] [PubMed] [Google Scholar]
- Stein R.J., Waters B.M. (2012). Use of natural variation reveals core genes in the transcriptome of iron-deficient Arabidopsis thaliana roots. J. Exp. Bot. 63: 1039–1055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stitt M., Krapp A. (1999). The interaction between elevated carbon dioxide and nitrogen nutrition: the physiological and molecular background. Plant Cell Environ. 22: 583–621 [Google Scholar]
- Stitt M., Lunn J., Usadel B. (2010). Arabidopsis and primary photosynthetic metabolism - more than the icing on the cake. Plant J. 61: 1067–1091 [DOI] [PubMed] [Google Scholar]
- Sulpice R., et al. (2013). Low levels of ribosomal RNA partly account for the very high photosynthetic phosphorus-use efficiency of Proteaceae species. Plant Cell Environ. http://dx.doi.org/10.1111/pce.12240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terauchi A.M., Peers G., Kobayashi M.C., Niyogi K.K., Merchant S.S. (2010). Trophic status of Chlamydomonas reinhardtii influences the impact of iron deficiency on photosynthesis. Photosynth. Res. 105: 39–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thimm O., Bläsing O., Gibon Y., Nagel A., Meyer S., Krüger P., Selbig J., Müller L.A., Rhee S.Y., Stitt M. (2004). MAPMAN: a user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant J. 37: 914–939 [DOI] [PubMed] [Google Scholar]
- Tibshirani R., Walther G., Hastie T. (2001). Estimating the number of clusters in a data set via the gap statistic. J. R. Stat. Soc. B 63: 411–423 [Google Scholar]
- Uhrig R.G., Ng K.K., Moorhead G.B. (2009). PII in higher plants: a modern role for an ancient protein. Trends Plant Sci. 14: 505–511 [DOI] [PubMed] [Google Scholar]
- Urzica E.I., Casero D., Yamasaki H., Hsieh S.I., Adler L.N., Karpowicz S.J., Blaby-Haas C.E., Clarke S.G., Loo J.A., Pellegrini M., Merchant S.S. (2012). Systems and trans-system level analysis identifies conserved iron deficiency responses in the plant lineage. Plant Cell 24: 3921–3948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallon O., Bulté L., Kuras R., Olive J., Wollman F.A. (1993). Extensive accumulation of an extracellular L-amino-acid oxidase during gametogenesis of Chlamydomonas reinhardtii. Eur. J. Biochem. 215: 351–360 [DOI] [PubMed] [Google Scholar]
- Veneklaas E.J., Lambers H., Bragg J., Finnegan P.M., Lovelock C.E., Plaxton W.C., Price C.A., Scheible W.R., Shane M.W., White P.J., Raven J.A. (2012). Opportunities for improving phosphorus-use efficiency in crop plants. New Phytol. 195: 306–320 [DOI] [PubMed] [Google Scholar]
- Vogel C., Abreu Rde.S., Ko D., Le S.Y., Shapiro B.A., Burns S.C., Sandhu D., Boutz D.R., Marcotte E.M., Penalva L.O. (2010). Sequence signatures and mRNA concentration can explain two-thirds of protein abundance variation in a human cell line. Mol. Syst. Biol. 6: 400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Lan P., Gao H., Zheng L., Li W., Schmidt W. (2013). Expression changes of ribosomal proteins in phosphate- and iron-deficient Arabidopsis roots predict stress-specific alterations in ribosome composition. BMC Genomics 14: 783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z.T., Ullrich N., Joo S., Waffenschmidt S., Goodenough U. (2009). Algal lipid bodies: stress induction, purification, and biochemical characterization in wild-type and starchless Chlamydomonas reinhardtii. Eukaryot. Cell 8: 1856–1868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wase N., Black P.N., Stanley B.A., Dirusso C.C. (February 26, 2014). Integrated quantitative analysis of nitrogen stress response in Chlamydomonas reinhardtii using metabolite and protein profiling. J. Proteome Res. http://dx.doi.org/10.1021/pr400952z [DOI] [PubMed] [Google Scholar]
- Waters B.M., McInturf S.A., Stein R.J. (2012). Rosette iron deficiency transcript and microRNA profiling reveals links between copper and iron homeostasis in Arabidopsis thaliana. J. Exp. Bot. 63: 5903–5918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei, L., Derrien, B., Gautier, A., Houille-Vernes, L., Boulouis, A., Saint-Marcoux, D., Malnoë, A., Rappaport, F., de Vitry, C., Vallon, O., Choquet, Y., and Wollman, F.A. (2014). Nitric oxide–triggered remodeling of chloroplast bioenergetics and thylakoid proteins upon nitrogen starvation in Chlamydomonas reinhardtii Plant Cell 26: 353–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wijffels R.H., Barbosa M.J. (2010). An outlook on microalgal biofuels. Science 329: 796–799 [DOI] [PubMed] [Google Scholar]
- Wu T.D., Nacu S. (2010). Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics 26: 873–881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wykoff D.D., Davies J.P., Melis A., Grossman A.R. (1998). The regulation of photosynthetic electron transport during nutrient deprivation in Chlamydomonas reinhardtii. Plant Physiol. 117: 129–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu G., Fan X., Miller A.J. (2012). Plant nitrogen assimilation and use efficiency. Annu. Rev. Plant Biol. 63: 153–182 [DOI] [PubMed] [Google Scholar]
- Zehr J.P., Ward B.B. (2002). Nitrogen cycling in the ocean: new perspectives on processes and paradigms. Appl. Environ. Microbiol. 68: 1015–1024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H., Jin J., Tang L., Zhao Y., Gu X., Gao G., Luo J. (2011). PlantTFDB 2.0: update and improvement of the comprehensive plant transcription factor database. Nucleic Acids Res. 39: D1114–D1117 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.