

Abstract
The syntheses of 3-(1-methyl-1H-imidazol-2-ylthio)acrylic acid and 3-(1-methyl-1H-imidazol-2-ylthio)propanoic acid, mitochondria-targeted prodrugs of the antioxidant methimazole, are described. The method of Fan et al. (Fan et al., Synthesis 2006, 2286) for the reaction of phenols with propiolic acid and propiolate esters was modified to synthesize (E)-3-(1-methyl-1H-imidazol-2-ylthio)acrylic acid. The intermediate tert-butyl (E)-3-(1-methyl-1H-imidazol-2-ylthio)acrylate was prepared by the reaction of tert-butyl propiolate with methimazole; the use of tert-butyl propiolate rather than methyl propiolate gave tert-butyl (E)-3-(1-methyl-1H-imidazol-2-ylthio)acrylate as the predominant isomer. Acid hydrolysis of the intermediate ester afforded the target compound. 3-(1-Methyl-1H-imidazol-2-ylthio)propanoic acid was synthesized from 3-bromopropanoic acid and methimazole under conditions that gave preferential substitution on sulfur and minimized substitution on nitrogen.
Keywords: methimazole, thiols, alkyne activation, antioxidants, β-oxidation
Introduction
Mitochondria are the major cellular source of reactive oxygen species and also play a central role in the life and death of cells; hence, there is considerable contemporary interest in targeting antioxidant compounds to mitochondria.[1, 2] A strategy to exploit the high negative internal potential of mitochondria allows targeting of lipophilic cationic antioxidants to their ultimate site of action where a unique mitochondrial localization of specific enzymes exists to catalyze the release of drugs from prodrugs.[1, 3–6]
We recently reported the successful mitochondrial targeting of alkanoic- and alkenoic-acid-based prodrugs of phenolic and thiol antioxidants; these compounds are biotransformed by the fatty acid β-oxidation pathway to release antioxidant methyl-substituted phenols or methimazole. [6] 3-(2,6-Dimethylphenoxy)propanoic acid and 3-(2,6-dimethylphenoxy)acrylic acid as well as 3- and 5-(1-methyl-1H-imidazol-2-ylthio)alkanoic acids conferred significant cytoprotection against hypoxia-reoxygenation injury in isolated rat cardiomyocytes. These studies also showed that the medium-chain acyl-CoA dehydrogenase apparently imposes significant structural limitations on substrates.[6] To bypass this limitation, (E)-3-(2,6-dimethylphenoxy)acrylic acid, which was predicted to be a substrate for enoyl-CoA hydratase after conversion to its CoA thioester, was prepared, tested and found to undergo a much higher rate of biotransformation than the alkanoic acid-based analog.[6] Moreover, 3-(aryloxy)- and 3-(arylthio)acrylic acids and their esters are of interest as insecticides, larvicides, and synthons and more recently as prodrugs (Fig. 1).[6–9]
Figure 1.
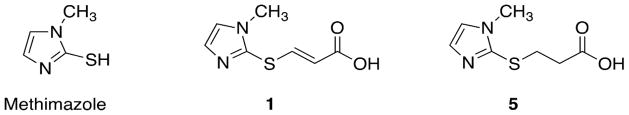
Structures of methimazole and of methimazole-based mitochondria-targeted prodrugs.
Two general synthetic pathways for the preparation of 3-(aryloxy)- and 3-(arylthio)acrylic acids have been reported: phenols or aryl thiols are reacted with propiolic acids or propiolate esters or with 3-haloacrylic acids or 3-haloacrylate esters.[7–17] The addition of phenols and arylthiols to propiolic acids and propiolate esters gives a mixture of E and Z isomers. Recently published reaction conditions make use of alkyl amine catalysis to control E:Z ratios.
Fan et al. reported reaction conditions that yield high E:Z product ratios from the reaction of phenols with methyl propiolates.[18, 19] We report herein the successful adaptation of the procedure of Fan et al. to the preparation of the new compound (E)-3-(1-methyl-1H-imidazol-2-ylthio)acrylic acid 1 with high E:Z product ratios. Given that enoyl-CoA hydratase catalyzes the stereospecific hydration of (E)-α,β-unsaturated acyl-CoA thioesters, [20] we focused on optimizing the E:Z ratios in the preparation of the intermediate esters. We also report an improved synthesis of 3-(1-methyl-1H-imidazol-2-ylthio)propanoic acid 5.
Results and Discussion
E-isomer of tert-Butyl 3-(1-methyl-1H-imidazol-2-ylthio)acrylate 2
Based on the work of Fan et al., it was hypothesized that the reaction of methyl propiolate with methimazole at RT in the presence of triethylamine would yield predominately the E-isomer of methyl ester 3 (Scheme 1); however, the reaction yielded a mixture of the E:Z isomers of methyl esters 3 and 4 that slightly favored the E-isomer, as shown by analytical [high-performance liquid chromatography (HPLC) Table 1].[18, 19] Various conditions were investigated in an attempt to find conditions favouring only the E-isomer including use of 1,4-diazabicyclo[2.2.2]octane (DABCO) rather than triethylamine as the catalyst. An increased E:Z ratio of methyl esters 3 and 4 was obtained (Table 1). Addition of the methyl propiolate dropwise to the methimazole and DABCO solution yielded a 7.4:1 E:Z ratio of methyl esters 3 and 4; lowering the reaction temperature to −20 °C gave an improved E:Z ratio of 8:1 (Table 1). Due to the difficulty in separating the E:Z isomers 3 and 4, as well as the acyl-CoA hydratase for the E-isomer, reaction conditions were sought to optimize the E:Z ratio in favor of the E isomer 3.
Scheme 1.
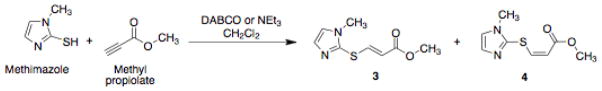
Synthesis of methyl (E)-3-(1-methyl-1H-imidazol-2-ylthio)acrylate 3 and methyl (Z)-3-(1-methyl-1H-imidazol-2-ylthio)acrylate 4.
Table 1.
Relationship between reaction conditions and E/Z ratios for the addition of methimazole to propiolate esters.
| Reactant | Reaction Conditions | E/Z ratio |
|---|---|---|
| Methyl propiolate | RT, Et3N | 1.1:1 |
| Methyl propiolate | RT, DABCO | 8:1 |
| Methyl propiolate | RT, DABCO, dropwise addition of ester | 7.4:1 |
| Methyl propiolate | −20°C, DABCO | 8:1 |
| tert-Butyl propiolate | RT, DABCO | 60:1 |
| tert-Butyl propiolate | −15°C, DABCO | 70:1 |
Yavari et al. showed that the reaction of phenols with tert-butyl propiolate in presence of triphenylphosphine afforded only tert-butyl (E)-3-(aryloxy)acrylates.[21] The reaction of tert-butyl propiolate with methimazole at RT in the presence of DABCO yielded one major peak on HPLC, which was confirmed to be the E-isomer, tert-Butyl (E)-3-(1-methyl-1H-imidazol-2-ylthio)acrylate 2 by 1H NMR (Table 1). When the reaction of methimazole and tert-butyl propiolate with DABCO was carried out at −15 °C (ammonium chloride/ice water), the minor peak diminished even further (Scheme 2, Table 1). tert-Butyl (Z)-3-(1-methyl-1H-imidazol-2-ylthio)acrylate was identified by 1H NMR spectroscopy as the minor peak (data not shown). The results of the several reaction conditions used are compiled summarized in Table 1 and show that the greatest E:Z ratio was achieved with tert-butyl propiolate with DABCO at −15 °C.
Scheme 2.
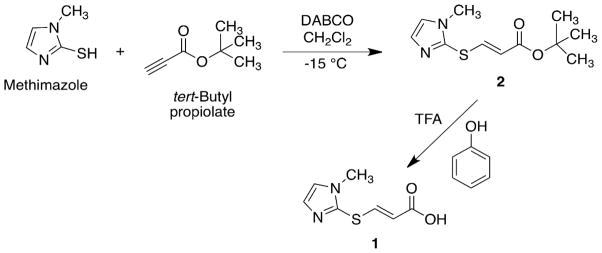
Synthesis of tert-butyl (E)-3-(1-methyl-1H-imidazol-2-ylthio)acrylate (2) and subsequent conversion to the acid (1).
Hydrolysis of tert-Butyl (E)-3-(1-methyl-1H-imidazol-2-ylthio)acrylate 2
tert-Butyl (E)-3-(1-methyl-1H-imidazol-2-ylthio)acrylate 2 was readily hydrolyzed to (E)-3-(1-methyl-1H-imidazol-2-ylthio)acrylic acid 1 (Scheme 2) in a mixture of trifluoroacetic acid and phenol.[22]
Synthesis of 3-(1-Methyl-1H-imidazol-2-ylthio)propanoic acid 5
3-(1-Methyl-1H-imidazol-2-ylthio)propanoic acid 5 was previously prepared by a modification of the route described for 5-(1-methyl-1H-imidazol-2-ylthio)pentanoic acid.[23] In our hands, however, this route gave poor yields and significant substitution occurred on N-3 rather than on S-2 of methimazole. A more efficient synthesis of acid 1 that yielded substitution on S-2 was achieved by the reaction of 3-bromopropanoic acid and methimazole under the conditions shown in Scheme 3. This procedure is analogous to the protocol reported for the synthesis of 5-(2-carboxyethylthio)-2,3-diphenyltetrazolium bromide and 3-(4,5-di(p-chlorophenyl)imidazoline-2-ylthio)propanoic acids.[24, 25]
Scheme 3.
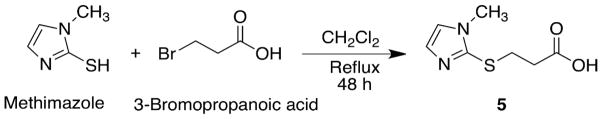
Synthesis of 3-(1-methyl-1H-imidazol-2-ylthio)propanoic acid 5.
Herein we present descriptions of the syntheses of 3-(1-methyl-1H-imidazol-2-ylthio)acrylic acid 1 and 3-(1-methyl-1H-imidazol-2-ylthio)propanoic acid 5.
Experimental
Methimazole, 3-bromopropanoic acid, methyl propiolate, tert-butyl propiolate, and 1,4-diazabicyclo[2.2.2]octane (DABCO) were purchased from Sigma-Aldrich (St. Louis, MO).
tert-Butyl (E)-3-(1-methyl-1H-imidazol-2-ylthio)acrylate 2
A modification of the method of Fan et al. for the DABCO-catalyzed reaction of phenols with alkynes was adapted to the synthesis of tert-butyl ester 2 (Scheme 2) and methyl esters 3 and 4 (Scheme 1).[18, 19] tert-Butyl propiolate (11.8 mmol, 1.49 g) was added to a stirred solution of methimazole (11.8 mmol, 1.35 g) in 93 mL of dichloromethane at −15 °C (ammonium chloride/ice water). Addition of tert-butyl propiolate was followed by addition of DABCO (1.18 mmol, 132 mg). The progress of the reaction was followed by analytical HPLC (Beckman-Coulter Ultrasphere® 4.6mm × 25cm ODS, 5 μm) using a binary gradient of acentonitile/water 0/1% TFA, the gradient was as follows: 9–17% acetonitrile (0.1%TFA) over 13 min at 1 mL/min followed by 17–95% acetonitrile over 5 min or by thin-layer chromatography (TLC, ethyl acetate:hexanes 60:40). Eluting peaks were detected by absorbance at 280 nm. After a 1 h reaction, the dichloromethane was removed under reduced pressure to give an oily white semi-solid product. The crude product was dissolved in methanol, combined with dry silica gel, and loaded onto a solid-load precolumn of a flash chromatography system (CombiFlash Rf; ISCO, Lincoln, NE). Ester 2 was purified on a normal-phase flash silica-gel column (24 gram column ISCO RediSep Rf®, 69-2203-324). The column was eluted with a gradient of 40 to 60% ethyl acetate in hexanes over 15 min at 35 mL/min, and the absorbance of the eluate was measured at 280 nm. With tert-butyl propiolate rather than methyl propiolate, only the E isomer of ester 2 was obtained. The fractions containing the product were collected, and the eluent was removed under reduced pressure. The purified tert-butyl (E)-3-(1-methyl-1H-imidazol-2-thioyl)acrylate 2, 1.4 grams (50%), was obtained as a white solid. 1H NMR (400 MHz, CDCl3): δ 1.44 (s, 9H), 3.70 (s, 3H), 5.48 (d, J = 15.0 Hz, 1H), 7.10 (s, 1H), 7.20 (s, 1H), 7.49 (d, J = 15.0 Hz, 1H); 13C NMR (400 MHz, CDCl3): δ 28.25, 34.93, 80.93, 119.77, 124.21, 130.64, 135.28, 141.58, 164.03; HRESI-MS: m/z 241.1005 [M + H]+.
(E)-3-(1-methyl-1H-imidazol-2-ylthio)acrylic acid 1
Ester 2 (0.13 mmol, 31 mg) was dissolved in a mixture of trifluoroacetic acid (0.13 mol) and phenol (5.6 mmol) and heated to 60 °C with constant stirring to cleave the tert-butyl ester.[22] The reaction mixture was stirred at 60 °C for 1 h. Trifluoracetic acid was removed from the crude product under reduced pressure. The crude product was then combined with 250 μL water and 50 μL acetonitrile and purified by HPLC (Ultrasphere® Si column, no. 235340, 10 mm × 25 cm, 5 μm particle size; Beckman-Coulter, Brea, CA) with a gradient of 5–95% acetonitrile (0.1% TFA) over 30 minutes at 3 mL/min with water (0.1% TFA) as a the second mobile phase. Detection was by absorbance at 280 nm). (E)-3-(1-Methyl-1H-imidazol-2-ylthio)acrylic acid 1 (2.1 mg, 9%) was obtained as a slightly yellow oil. 1H NMR (300 MHz, D2O): δ 3.91 (s, 3H), 5.86 (d, J = 12.0 Hz, 1H), 7.50 (s, 1H), 7.58 (s, 1H), 7.66 (s, J = 12.0 Hz, 1H); 13C NMR (400 MHz, CDCl3): δ 33.82, 118.48, 125.64, 127.60, 134.21, 142.26, 165.07; HRESI-MS: m/z 185.0380 [M + H]+.
Methyl (E)-3-(1-methyl-1H-imidazol-2-ylthio)acrylate 3 and methyl (Z)-3-(1-methyl-1H-imidazol-2-ylthio)acrylate 4
With the reaction conditions described by Fan et al., methimazole was reacted with methyl propiolate in the presence of DABCO or triethylamine.[18, 19] Methimazole (0.51 mmol, 58 mg) was dissolved in 4 mL of dichloromethane and triethylamine (0.0508 mmol, 7 μL) was added with continuous stirring. Methyl propiolate (0.51 mmol, 45.2 μL) was then added, and the reaction mixture was stirred at RT for 90 min after which time the solvent was removed under reduced pressure. The crude product was purified by reversed-phase HPLC (Ultrasphere® Si column, no. 235340, 10 mm × 25 cm, 5 μm particle size; Beckman-Coulter, Brea, CA) with a gradient of 9–17% acetonitrile 0.1% TFA over 13 min at 3 mL/min. Water, 0.1% TFA was the second mobile phase, and UV absorbance at 280 nm was used for detection. The E:Z ratio of methyl esters 3 and 4 was nearly 1:1 with triethylamine as the catalyst and 3:1 with DABCO as the catalyst. (Further attempts to optimize the reaction conditions for the formation of the E-isomer of methyl ester 3 as the major product were not pursued.) The fractions corresponding to methyl esters 3 and 4 in the eluate from the HPLC column were collected; the acetonitrile was removed under reduced pressure, and the samples were lyophilized. Methyl (E)-3-(1-methyl-1H-imidazol-2-ylthio)acrylate 3. 1H NMR (400 MHz, CDCl3) δ 3.69 (s, 3H), 3.74 (s, 3H), 5.60 (d, J = 15.1 Hz, 1H), 7.17 (d, J = 1.4 Hz, 1H), 7.33 (d, J = 1.4 Hz, 1H), 7.53 (d, J = 15.1 Hz, 1H); 13C NMR (400 MHz, CDCl3): δ 34.85, 52.03 120.43, 125.30, 126.10, 134.99, 139.15, 164.28; HREI-MS: m/z 198.0472 [M•]+. Methyl (Z)-3-(1-methyl-1H-imidazol-2-ylthio)acrylate 4. 1H NMR (400 MHz, CDCl3): δ 3.84 (s, 3H), 3.85 (s, 3H), 6.27 (d, J = 9.6 Hz, 1H), 7.16 (d, J = 1.8 Hz, 1H), 7.49 (s, 1H), 7.61 (d, J = 9.6 Hz, 1H); 13C NMR (400 MHz, CDCl3): δ 35.33, 52.32, 118.30, 123.61, 124.04, 139.26, 141.28, 167.00; HREI-MS: m/z 198.0471 [M•]+.
3-(1-Methyl-1H-imidazol-2-ylthio)propanoic acid 5
A 100-mL round-bottom flask was charged with methimazole (0.01 mol, 1.14 g), 3-bromopropanoic acid (0.01 mol; 1.53 g), and 50 mL of dichloromethane. The mixture was heated at reflux for 48 h, during which time a white, crystalline solid formed. The solvent was removed under reduced pressure to afford 1.42 g (54%) of 2-(2-carboxyethylthio)-1-methyl-1H-imidazol-1-ium bromide. For analytical purposes, a sample (50 mg) of the bromide salt was dissolved in water, and the pH was brought to 5.5 by addition of 1 M Na2CO3. The mixture was extracted with dichloromethane (3 × 10 mL). A colorless residue was obtained after removal of the solvent. 1H NMR (400 MHz, CDCl3): δ 2.72–2.75 (t, 2H, J = 7.2 Hz), 3.25–3.29 (t, 2H, J = 7.2), 3.62 (s, 3H), 6.941–6.944 (d, 1H, J = 1.2 Hz), 7.119–7.122 (d, 1H, J = 1.2 Hz), 10.8 (br, s, 1H); 13C NMR (400 MHz, CDCl3): δ 30.3, 34.6, 35.1, 120.6, 125.4, 139.5, 175.1 HRESI-MS: m/z 187.0533 [M + H]+
Acknowledgments
This work was supported by the P30 ES006694 Southwest Environmental Health Sciences Center (SSL), T32 ES007091 and T32 ES016652 (both to CMH) at the University of Arizona. We would like to acknowledge Dr. Richard T. Miller for critical review of the manuscript.
References
- 1.Sheu SS, Nauduri D, Anders MW. Targeting antioxidants to mitochondria: A new therapeutic direction. Biochim Biophys Acta. 2006;1762:256–265. doi: 10.1016/j.bbadis.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 2.Anders MW, Robotham JL, Sheu SS. Mitochondria: new drug targets for oxidative stress-induced diseases. Expert Opin Drug Metab Toxicol. 2006;2:71–79. doi: 10.1517/17425255.2.1.71. [DOI] [PubMed] [Google Scholar]
- 3.Szeto HH. Mitochondria-targeted peptide antioxidants: Novel neuroprotective agents. AAPS J. 2006;8:E521–E531. doi: 10.1208/aapsj080362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Murphy MP, Smith RAJ. Targeting antioxidants to mitochondria by conjugation to lipophilic cations. Annu Rev Pharmacol Toxicol. 2007;47:629–656. doi: 10.1146/annurev.pharmtox.47.120505.105110. [DOI] [PubMed] [Google Scholar]
- 5.Ripcke J, Zarse K, Ristow M, Birringer M. Small-molecule targeting of the mitochondrial compartment with an endogenously cleaved reversible tag. ChemBiochem. 2009;10:1689–1696. doi: 10.1002/cbic.200900159. [DOI] [PubMed] [Google Scholar]
- 6.Roser KS, Brookes PS, Wojtovich AP, Olson LP, Shojaie J, Parton RL, Anders MW. Mitochondrial biotransformation of ω-(phenoxy) alkanoic acids, 3-(phenoxy) acrylic acids, and ω-(1-methyl-1H-imidazol-2-ylthio)alkanoic acids: A prodrug strategy for targeting cytoprotective antioxidants to mitochondria. Bioorg Med Chem. 2010;18:1441–1448. doi: 10.1016/j.bmc.2010.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Joshi GD, Deshmukh ARAS, Gore KG, Kulkarni GH. Michael addition-reaction on propynoic acid and its esters - synthesis of insecticidally active aralkyl-3-substituted-2(E)-propenoates. Chem Ind (London) 1991:281–282. [Google Scholar]
- 8.Fujinami A, Mine A. Effect of substitutedβ-phenoxyacrylates on shoot elongation of barnyard grass. Agric Biol Chem. 1970;34:1157–1161. [Google Scholar]
- 9.Heindel ND, Reid JR. 7H-[1,2,4]Triazolo[5,1-B][1,3]thiazin-7-ones by deamination cyclization of S-acrylates of “4-amino-3(3H)-1,2,4-triazolethiones. J Org Chem. 1980;45:2479–2482. [Google Scholar]
- 10.Montanari F. The stereochemistry of nucleophilic addition of thiophenols to propiolic acid and ethyl propiolate. Tetrahedron Lett. 1960:18–22. [Google Scholar]
- 11.Itoh T, Honma M, Ogura H. Studies on heterocyclic-compounds.17. reaction of 6-mercapto-1,3-dimethyluracil with electrophilic reagents. Chem Pharm Bull. 1976;24:1390–1393. [Google Scholar]
- 12.Di Santo R, Costi R, Massa S, Artico M. Arylsulfonylpyrroles from reaction of tosylmethyl isocyanide (TosMIC) with 3-arylsulfonyl acrylates as Michael acceptors. Synth Commun. 1998;28:1801–1815. [Google Scholar]
- 13.Di Santo R, Costi R, Massa S, Artico M. Pyrrole-annulated heterocyclic systems. Synthesis of 2H-pyrrolo[3,4-b][1,5]benzothiazepine 4, 4-dioxide derivatives. Synth Commun. 1998;28:2517–2530. [Google Scholar]
- 14.Undheim K, Riege LA. Adduct formation between pyridine-2-thiones and acetylenic carbonyl derivatives. J Chem Soc, Perkin Trans. 1975;1:1493–1496. [Google Scholar]
- 15.Truce WE, Heine RF. The stereochemistry of base-catalyzed additions of p-toluenethiol to several negatively-substituted acetylenes -an exception to the rule of trans-nucleophilic addition. J Am Chem Soc. 1957;79:5311–5313. [Google Scholar]
- 16.Wade JJ. Reaction of 2H-Benzimidazole-2-Thione with Dimethyl Acetylenedicarboxylate. J Org Chem. 1979;44:1816–1819. [Google Scholar]
- 17.Haefliger W, Petrzilk T. Synthesen substituierter Butenolide. Helv Chim Acta. 1966;49:1937–1950. [Google Scholar]
- 18.Fan MJ, Li GQ, Yang SD, Liang YM. DABCO-catalyzed reaction of phenols or 1,2-diphenols with activated alkynes leading to the formation of alkenoic acid esters or 1,3-dioxole derivatives. Synthesis. 2006:2286–2292. [Google Scholar]
- 19.Fan MJ, Li GQ, Liang YM. DABCO catalyzed reaction of various nucleophiles with activated alkynes leading to the formation of alkenoic acid esters, 1,4-dioxane, morpholine, and piperazinone derivatives. Tetrahedron. 2006;62:6782–6791. [Google Scholar]
- 20.Wu WJ, Feng YG, He X, Hofstein HA, Raleigh DP, Tonge PJ. Stereospecificity of the reaction catalyzed by enoyl-CoA hydratase. J Am Chem Soc. 2000;122:3987–3994. [Google Scholar]
- 21.Yavari I, Souri S, Sirouspour M, Djahaniani H, Nasiri F. Synthesis of alkyl 3-aryloxypropenoates and alkyl 2-arylacrylates through nucleophilic addition to alkyl propiolates. Synthesis. 2005:1761–1764. [Google Scholar]
- 22.Tanaka H, Taniguchi M, Kameyama Y, Torii S, Sasaoka M, Shiroi T, Kikuchi R, Kawahara I, Shimabayashi A, Nagao S. Deprotection of carboxylic esters assisted by hydrogen-bonding network in phenolic media - its application to β-lactam antibiotic syntheses. Tetrahedron Lett. 1990;31:6661–6662. [Google Scholar]
- 23.Tweit RC, Kreider EM, Muir RD. Synthesis of antimicrobial nitroimidazolyl 2-sulfides, 2-sulfoxides, and 2-sulfones. J Med Chem. 1973;16:1161–1169. doi: 10.1021/jm00268a021. [DOI] [PubMed] [Google Scholar]
- 24.Hutton AT, Irving HMNH. 3-carboxymethylthio-1,5-diphenylformazan - potential terdentate ligand with unusual properties. J Chem Soc, Perkin Trans. 1980;2:139–145. [Google Scholar]
- 25.Sharaf MAF, Ezat EHM, Hammouda HAA. Reactions with 4,5-di(p-chlorophenyl) imidazoline-2-thione.2. Synthesis of fused imidazolines and some N-substituted derivatives. J Chem Res, Synop. 1996:322–323. [Google Scholar]