

Abstract
Redundancy of the genetic code dictates that a given protein can be encoded by a large collection of distinct mRNA species, potentially allowing mRNAs to simultaneously optimize desirable RNA structural features in addition to their protein-coding function. To determine whether natural mRNAs exhibit biases related to local RNA secondary structure, a new randomization procedure was developed, DicodonShuffle, which randomizes mRNA sequences while preserving the same encoded protein sequence, the same codon usage, and the same dinucleotide composition as the native message. Genes from 10 of 14 eubacterial species studied and one eukaryote, the yeast Saccharomyces cerevisiae, exhibited statistically significant biases in favor of local RNA structure as measured by folding free energy. Several significant associations suggest functional roles for mRNA structure, including stronger secondary structure bias in the coding regions of intron-containing yeast genes than in intronless genes, and significantly higher folding potential in polycistronic messages than in monocistronic messages in Escherichia coli. Potential secondary structure generally increased in genes from the 5′ to the 3′ end of E. coli operons, and secondary structure potential was conserved in homologous Salmonella typhi operons. These results are interpreted in terms of possible roles of RNA structures in RNA processing, regulation of mRNA stability, and translational control.
Single-stranded RNA molecules can form local secondary structures through the interactions of complementary segments. These secondary structure elements may influence many cellular processes, including mRNA stability and localization, transcription, RNA processing, and translation. Functionally important RNA secondary structures can be found in untranslated regions (UTRs), introns, and coding sequences. For example, in eukaryotes, stem-loop structures in 5′ UTRs may prevent association of the 40S ribosomal subunit with the mRNA, inhibiting translation initiation (Gray and Wickens 1998; Meijer and Thomas 2002). Similarly, secondary structure elements in bacterial 5′ UTRs can reduce the rate of mRNA degradation through the inhibition of nuclease activity (Diwa et al. 2000). Introns and coding regions may also contain important structural elements. For example, mutations predicted to disrupt a stem-loop structure in intron 10 of the human tau gene cause higher levels of inclusion of exon 10 and are linked to debilitating neurodegenerative conditions (Grover et al. 1999). Additionally, a stem-loop in the coding sequence of the yeast ASH1 gene can localize ASH1 mRNA to the bud tip (Chartrand et al. 1999).
Most previous computational analyses of mRNA secondary structures have focused on the 5′ and 3′ UTRs, excluding coding regions. It is widely believed that secondary structure in ORFs can interfere with translation (Klionsky et al. 1986; Guisez et al. 1993; Schmittgen et al. 1994), giving rise to the expectation that RNA structure should generally be avoided in coding regions. However, very few studies have addressed this issue. Rocha and colleagues noted that the coding regions of Bacillus subtilis contain stable secondary structures (Rocha et al. 1999). Similarly, examination of 51 random mRNAs from GenBank revealed a bias toward more negative folding energies in native sequences, controlling for encoded amino acid sequence and codon usage (Seffens and Digby 1999). This study was later challenged by another group (Workman and Krogh 1999), who re-examined this set of mRNAs and concluded that the apparent higher stability of RNA structures in native sequences could be explained by differences in dinucleotide composition between the native and randomized mRNAs, since RNA folding thermodynamics is strongly dependent on dinucleotide stacking interactions.
We sought to resolve this contradiction by developing a new protocol, called DicodonShuffle, which randomizes mRNAs preserving dinucleotide composition (as in Workman and Krogh 1999), while at the same time preserving amino acid sequence and codon usage (as in Seffens and Digby 1999). The key issue is that, whereas swaps between synonymous codons preserve transcript dinucleotide composition at the (1,2) and (2,3) codon positions, they often change the dinucleotide composition at the (3,1) positions of the mRNA, that is, dinucleotides formed by the last base of a codon followed by the first base of the next. The essential idea of this new algorithm is to make only those synonymous codon swaps which either (1)preserve (3,1) dinucleotide composition by themselves; or (2)which can be paired with another reciprocal synonymous codon swap, such that simultaneous swapping of both codon pairs results in no net change in (3,1) dinucleotide composition.
In contrast to previous analyses, we studied local RNA structures—structures formed within short sequence regions of 50 bases—rather than the structures predicted for folding of an entire mRNA. Such local structures are more likely to form in vivo in actively translating mRNAs. Finally, the sets of mRNAs used by the two previous groups contained a random collection of sequences from organisms as diverse as Escherichia coli and Homo sapiens, potentially blurring any effects that might be present in prokaryotes but absent from eukaryotes, for example. Therefore, we analyzed each organism separately.
We took advantage of the recent availability of numerous complete genomes and analyzed thousands of coding regions from 28 different organisms with several representatives each from Archaea, Eukaryota, and Eubacteria. Overall, coding regions were found to contain significantly more local RNA secondary structure than expected in most eubacterial species studied, but this phenomenon was observed only sporadically in archaeal and eukaryotic organisms. To investigate the possible roles of secondary structures in coding regions, we systematically studied the folding potential of mRNAs in relation to their expression levels, half-lives, positions in bacterial operons, and other properties.
RESULTS
Secondary Structure Bias Exists in Coding Regions of Many Organisms
To determine whether coding regions of genes in E. coli are biased against (or for)local RNA secondary structure potential, the folding free energies of native mRNAs were compared with folding free energies computed from sequences randomized by three different methods.
The first randomization procedure, which we call CodonShuffle, preserves the same encoded protein sequence and the same codon usage of the native mRNA, as in Seffens and Digby (1999; see Methods). The resulting shuffled sequences are, on average, ∼80% identical to the corresponding native mRNAs at the nucleotide level (data not shown). Native and randomized sequences were then folded in a sliding window of 50 bases, and the average free energy over all windows was compared between the two datasets. The average free energy for native mRNAs, (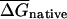 was –218.6 kcal/mole per kilobase for native E. coli mRNAs, compared with a value of
was –218.6 kcal/mole per kilobase for native E. coli mRNAs, compared with a value of 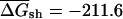 kcal/mole/kb for the corresponding CodonShuffled mRNAs; data not shown). These values are significantly different (P < 2.2e-16; Wilcoxon test, see Methods). However, as pointed out by Workman and Krogh (1999), this result by itself does not imply selection for RNA structure, because the CodonShuffle protocol does not preserve dinucleotide composition at the (3,1) positions of codons (i.e., the dinucleotide formed by the third base of one codon with the first base of the next), and dinucleotide composition is a primary contributor to folding free energy (Zuker and Stiegler 1981).
kcal/mole/kb for the corresponding CodonShuffled mRNAs; data not shown). These values are significantly different (P < 2.2e-16; Wilcoxon test, see Methods). However, as pointed out by Workman and Krogh (1999), this result by itself does not imply selection for RNA structure, because the CodonShuffle protocol does not preserve dinucleotide composition at the (3,1) positions of codons (i.e., the dinucleotide formed by the third base of one codon with the first base of the next), and dinucleotide composition is a primary contributor to folding free energy (Zuker and Stiegler 1981).
Next, the comparison between native and shuffled mRNAs was repeated with sequences randomized preserving the same dinucleotide frequencies, encoded protein, and codon usage as the native mRNA using a newly developed method that we call DicodonShuffle (see Methods). DicodonShuffle generates sequences that are, on average, more similar to the corresponding native coding regions than CodonShuffle (85% versus 80% identity; data not shown) because of the additional constraint that must be satisfied. Nevertheless, native sequences again had significantly different (more negative)average free energies than DicodonShuffled mRNA controls (P < 2.2e-16; Tables 1 and 2).
Table 1.
Effect of Window Size on Folding Potential in E. coli Coding Regions
| Window size (bp) | 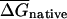 |
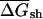 |
P-value |
|---|---|---|---|
| 25 | - 128.4 | - 126.4 | E-05 |
| 50 | - 218.6 | - 213.8 | <2.2E-16 |
| 100 | - 278.3 | - 272.5 | <2.2E-16 |
| 200 | - 318.3 | - 312.0 | <2.2E-16 |
Native and randomized coding regions of 4290 E. coli genes were divided into windows of the indicated size and the minimum folding free energies were computed for each window using the RNA fold program. Mean free energies over all windows are listed for native and randomized sequences (scaled in per kilobase units). These mean values were compared using Wilcoxin t-test.
Table 2.
Folding Potential of Coding Regions From Diverse Organisms
| Empire | Species | No. genes | 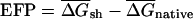 |
EFP | Wilcoxon t-test | %G + C |
|---|---|---|---|---|---|---|
| Archaea | Thermoplasma volcanium | 1525 | -143.2 | +1.0 | NS | 43.14 |
| Methanobacterium thermoautotropicum | 1873 | -204.0 | +4.0* | E-12 | 52.27 | |
| Sulfolobus solfataricus | 2977 | -121.8 | -0.2 | NS | 39.01 | |
| Sulfolobus tokodaii | 2826 | -110.6 | -0.2 | NS | 36.49 | |
| Pyrococcus abyssi | 1769 | -165.4 | +1.4 | NS | 47.22 | |
| Aeropyrum pernix | 1840 | -244.6 | +0.2 | NS | 58.86 | |
| Archaeoglobus fulgidus | 2407 | -187.6 | -0.6 | NS | 51.17 | |
| Halobacterium sp. NRC-1 | 2058 | -328.4 | +5.2* | <E-16 | 69.43 | |
| Eubacteria | Escherichia coli | 4290 | -218.6 | +4.8* | <E-16 | 52.90 |
| Salmonella typhi | 4600 | -227.6 | +6.0* | <E-16 | 54.30 | |
| Bacillus subtilis | 4100 | -165.6 | +2.8* | E-13 | 45.92 | |
| Mycoplasma pneumoniae | 689 | -147.6 | +6.6* | <E-16 | 42.58 | |
| Vibrio cholerae | 2752 | -193.2 | +4.4* | <E-16 | 50.13 | |
| Aquifex aeolicus | 1522 | -152.2 | +1.4 | NS | 45.52 | |
| Borellia burgdorferi | 850 | -98.4 | +1.6 | NS | 31.47 | |
| Thermotoga maritima | 1846 | -173.4 | +2.2* | E-05 | 48.33 | |
| Haemophilus influenzae | 1709 | -142.0 | +2.2* | E-05 | 40.79 | |
| Synechocystis PCC6803 | 3169 | -185.4 | -1.8* | E-07 | 50.35 | |
| Treponema pallidum | 1031 | -219.0 | +2.8* | E-03 | 54.85 | |
| Clostridium acetobutylicum | 3672 | -106.0 | +1.8* | <E-16 | 34.05 | |
| Rickettsia prowazekii | 834 | -102.6 | +1.2 | NS | 32.75 | |
| Helicobacter pylori | 1491 | -139.2 | -0.6 | NS | 41.96 | |
| Eukaryota | Plasmodium falciparum | 422 | -70.8 | +0.8 | NS | 26.60 |
| Saccharomyces cerevisiae | 6314 | -129.0 | +1.2* | <E-16 | 42.20 | |
| Arabidopsis thaliana | 2676 | -162.2 | -0.1 | NS | 47.80 | |
| Caenorhabditis elegans | 762 | -149.0 | -2.8* | E-07 | 46.40 | |
| Homo sapiens | 1855 | -207.8 | -0.8 | NS | 54.10 | |
| Drosophila melanogaster | 1956 | -210.2 | +0.01 | NS | 55.90 |
All sequences were randomized using the Dicodon Shuffle program. EFP as defined in Methods. (NS) Not significant.
Statistically significant bias in favor (positive EFP) or against (negative EFP) RNA structure.
For completeness, the sequences were also randomized preserving only dinucleotide composition (but not encoded amino acid sequence or codon usage)using a protocol called DiShuffle (Methods). DiShuffled sequences have essentially no detectable sequence similarity to the native mRNA (just slightly above 25% identity). A significant bias toward secondary structure in native sequences was still present (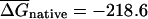 ,
, 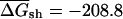 , P < 2.2e-16).
, P < 2.2e-16).
Applying the three shuffling protocols to human mRNAs revealed a significant bias toward RNA structure in native mRNAs when compared with CodonShuffled sequences (data not shown), but this effect disappeared when DiShuffle or DicodonShuffle was used (data not shown; Table 2, respectively), indicating that it is an artifact resulting from the failure to control for dinucleotide composition. These results are similar to those obtained previously (Seffens and Digby 1999; Workman and Krogh 1999) on the basis of analysis of a dataset comprised of mostly human and mouse mRNAs. To avoid this potential artifact, all of the remaining analyses described here were performed using DicodonShuffled sequences as controls.
The general result reported above for E. coli mRNAs was invariant over a range of window sizes tested (Table 1), with native mRNAs always showing a highly significant bias toward more negative folding free energy (higher secondary structure potential). For most subsequent calculations, we chose a window size of 50 bases (with step size of 10 bases), as most known functionally important secondary structures are small and local, and because RNA folding algorithms are most accurate for short sequences (Mathews et al. 1999). Moreover, a recent global analysis of translation in Saccharomyces cerevisiae suggests that the typical density of ribosomes on mRNAs is about 1 every 150 bases (D. Herschlag, pers. comm.), so it is probable that local secondary structures on the order of 50 bases could form during translation, but that substantially larger structures would often not have an opportunity to form in actively translating mRNAs.
To investigate the generality of the bias toward local secondary structures observed in E. coli mRNAs, the folding potential was computed for native and randomized mRNAs in the genomes of 27 additional species chosen to represent evolutionarily diverse taxa (Table 2). For each organism, the average folding energies were determined for a set of native and DicodonShuffled mRNAs, and the excess folding potential, 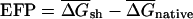 per kilobase of sequence was calculated. A positive EFP value for a given organism indicates that native mRNAs have, on average, more potential secondary structure than the corresponding randomized controls, whereas a negative EFP indicates that less secondary structure is present than expected. Strikingly, the genes in 10 of 14 eubacterial genomes studied exhibited a statistically significant bias in favor of RNA structure, whereas 3 of 14 had no significant bias, and only 1 showed significant avoidance of RNA structure (Table 2). Thus, the presence of excess secondary structure in coding regions of mRNAs appears to be a general feature of most Eubacteria. These results are not inconsistent with the findings of Workman and Krogh (1999), as only 2 of 46 sequences in their analyses came from Eubacteria. Two of eight Archaea studied, the thermophiles M. thermoautotropicum and Halobacterium sp. NRC-1, exhibited significantly positive EFP values, whereas the other six had no significant bias. In eukaryotes, only the single-celled S. cerevisiae exhibited a significant bias in favor of RNA structure, whereas the others had no bias, or, in one case (Caenorhabditis elegans), exhibited a bias against secondary structure (Table 2).
per kilobase of sequence was calculated. A positive EFP value for a given organism indicates that native mRNAs have, on average, more potential secondary structure than the corresponding randomized controls, whereas a negative EFP indicates that less secondary structure is present than expected. Strikingly, the genes in 10 of 14 eubacterial genomes studied exhibited a statistically significant bias in favor of RNA structure, whereas 3 of 14 had no significant bias, and only 1 showed significant avoidance of RNA structure (Table 2). Thus, the presence of excess secondary structure in coding regions of mRNAs appears to be a general feature of most Eubacteria. These results are not inconsistent with the findings of Workman and Krogh (1999), as only 2 of 46 sequences in their analyses came from Eubacteria. Two of eight Archaea studied, the thermophiles M. thermoautotropicum and Halobacterium sp. NRC-1, exhibited significantly positive EFP values, whereas the other six had no significant bias. In eukaryotes, only the single-celled S. cerevisiae exhibited a significant bias in favor of RNA structure, whereas the others had no bias, or, in one case (Caenorhabditis elegans), exhibited a bias against secondary structure (Table 2).
Secondary Structure Bias at the Gene Level
To determine whether a few genes with extremely negative free energies of folding account for the overall bias toward structure potential, or whether it is an additive effect of many genes, the genome of E. coli was analyzed on a per gene basis. For each E. coli mRNA, 20 different DicodonShuffled sequences were generated, and the average folding free energy was computed as described previously. The mean and standard deviations of these values were then calculated and used to determine a Z score for each of the native coding regions, indicating the number of standard deviations from the mean of the shuffled sequences (see Methods). A negative Z score indicates that the native mRNA sequence has a more negative folding energy (greater secondary structure potential) than the average of the random sequences. For example, Figure 1 demonstrates that the entire distribution of Z scores in the genome of E. coli is shifted to the left (mean Z-score, Z̄ = –0.7), consistent with the previously observed bias toward structure in mRNAs from this organism (Table 2). This result indicates that many genes contribute to the overall excess folding potential.
Figure 1.

Distribution of Z scores for E. coli genes. Each gene in the E. coli genome was shuffled 20 times, and a Z score was calculated for each as described in the text. A histogram of these Z scores (solid) and a standard normal distribution (dashed) are shown.
Calculating Z scores is more computationally intensive by an order of magnitude than the simpler EFP statistic, so we applied this type of analysis to only a few representative organisms. In all cases tested, there was good agreement between these two measures of secondary structure bias. Specifically, mean Z-score values of –0.45, –1.04, –0.44, and –0.25 were determined for Bacillus subtilis, Mycoplasma pneumoniae, Treponema pallidum, and S. cerevisiae, respectively, all organisms that have significant bias toward secondary structure according to the EFP statistic (Table 2). On the other hand, Helicobacter pylori had a mean Z-score of –0.01, consistent with the absence of significant bias for secondary structure observed using the EFP statistic (Table 2).
Distribution of Secondary Structure Along the Coding Regions
To further characterize the nature of this bias for structure, EFP was measured for different portions of coding regions separately. For each native and DicodonShuffled coding region in the E. coli genome, 12 subsets of nucleotide sequence segments were constructed corresponding to 140 base regions spanning the length of a typical ORF, and S. cerevisiae coding regions were treated similarly (Fig. 2). The average free energy of folding was then determined for each native and shuffled subset, and the EFP was calculated for each such 140-base segment. As shown in Figure 2, the bias toward secondary structure (positive EFP)occurs across all parts of the coding region in both E. coli and S. cerevisiae. In yeast, the bias is stronger at the 5′ ends of ORFs than at the 3′ ends, whereas in E. coli, secondary structure potential is fairly evenly distributed across ORFs, except for the first 140 bases, which have less bias.
Figure 2.

Distribution of EFP along coding regions in (A) E. coli and (B) S. cerevisiae. For native and shuffled mRNAs, six subsets of 50-bp sequence windows from each end of the coding region (corresponding to the overlapping windows 1–10, 11–20, 21–30, 31–40, 41–50, 51–60 relative to the 5′ or 3′ end) were folded, average folding free energies were calculated for native (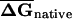 ) and CodonShuffled (
) and CodonShuffled (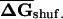 ) sequences, and the
) sequences, and the 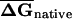 was determined for each segment. Because the step size between successive windows is 10 bp, each bin corresponds to 140 bases of sequence.
was determined for each segment. Because the step size between successive windows is 10 bp, each bin corresponds to 140 bases of sequence.
Can One Region Per Gene Account for the Bias in Structure?
Next, we asked whether a single 50-bp window per gene could potentially account for the observed EFP in coding regions, or whether it must result from an additive effect of several nonoverlapping sequence windows. For every coding sequence in S. cerevisiae and E. coli, the 50-bp window with most negative folding free energy was removed, as well as all overlapping 50-base windows. As controls, the corresponding sequence segments were removed from the DicodonShuffled version of each mRNA.
Comparing the average folding free energy over all remaining windows in native sequences to the corresponding regions of randomized controls showed that in yeast, the EFP was reduced by two-thirds (EFP = 0.4 kcal/mole/kb, versus 1.2 kcal/mole/kb in Table 2), whereas in E. coli, it was reduced by one-half (EFP = 2.4 kcal/mole/kb, versus 4.8 in Table 2). This result indicates that the secondary structure bias observed in yeast and bacteria can potentially be attributed to as few as one or two short (∼50 bp) segments per gene.
To identify highly folded subregions in individual genes, we used a local Z statistic, in which the free energy of folding of a 50-base window is compared with the mean and standard deviations of folding free energies for the corresponding window in DicodonShuffled sequences (see Methods). Such local Z scores were calculated for every 50-base window in ORFs from the E. coli, S. cerevisiae, and B. subtilis genomes. Some examples of genes/windows with extremely low local Z scores from these organisms are listed in Supplemental Tables 1–3 (available online at www.genome.org). These genes and regions provide potential targets for the experimental investigation of the roles of local mRNA structures in gene expression.
Secondary Structure in Bacterial Operons
In bacterial genomes, functionally related genes are frequently clustered into operons and transcribed as a single unit. Comparison of the Z scores for 857 polycistronic genes from E. coli (Huerta et al. 1998)with the Z scores for the 3427 independently transcribed genes revealed a significantly stronger bias toward secondary structure in operons Z̄operon = –0.95 than in independently transcribed genes, Z̄non-operon = –0.65 (Z̄operon < Z̄non-operon; P = 3.1e-10). Furthermore, the average secondary structure potential, as measured by the Z score, increased in the second genes of operons relative to the first, in the third relative to the second, and so forth, up to the fifth gene (Fig. 3). Too few sixth, seventh, and higher genes were available to determine reliable average Z scores (e.g., only 27 sixth genes; Fig. 3A).
Figure 3.

Bias for secondary structure in bacterial operons. Operons were classified on the basis of their locations in annotated polycistronic messages, and the average Z score was calculated for each subset. Numbers 1–6 correspond to the location of genes within operons relative to the 5′ end of the transcript. Genes in position 7or higher within operons are not shown. The number of genes in each data set is indicated.
Similar results were obtained for the genome of Salmonella typhi, using operon annotation inferred by homology to the better-annotated E. coli genome (see Methods). Specifically, secondary structure potential also tended to increase from the first gene in operons to the sixth (Fig. 3B). Given these results, it was of obvious interest to ask whether there was evidence for conservation of secondary structure potential between homologous E. coli/S. typhi genes, above and beyond that resulting from conservation of the encoded protein sequence. For this purpose a program, EvolveGene, was developed to generate synthetic S. typhi mRNAs, designated S. typhiS, from alignments of homologous E. coli/S. typhi genes. In brief, EvolveGene generates a random synthetic S. typhi homolog of a given E. coli gene, which has exactly the same degree of sequence similarity to the E. coli gene as the true S. typhi homolog, and the same amino acid and codon usage as the S. typhi homolog (see Methods for details). For this experiment, we focused on the subset of 147 polycistronic E. coli genes that had high-folding potential (Z score < –2)and also had clear S. typhi orthologs. The result was that the mean Z score for the native S. typhi homologs of these E. coli genes was significantly more negative than for the synthetic S. typhiS genes (Z̄s.typhi = –2.06 < Z̄s.typhiS = –1.75, P < 0.01). This result provides evidence that natural selection is acting to preserve RNA secondary structure in the coding regions of polycistronic genes in these two bacteria.
Six operons contained multiple genes with significant conserved folding potential (Z score < –2 for both E. coli and S. typhi homologs), the atp operon (genes atpH, atpG, atpD), the lpx operon (lpxB, dnaE), the hyb operon (hybB, hybE), the nuo operon (nuoF, nuoH), the S10 operon (rplD, rplB), and the spc operon (rplX, rplR). Interestingly, at least three of these operons—atp, S10 and spc—are known to be regulated at the level of translation rate and/or differential mRNA stability (Freedman et al. 1987; Lindahl et al. 1989; Mattheakis et al. 1989; McCarthy 1990; McCarthy et al. 1991). As systematic annotation of operons becomes available for other bacteria, it will be of interest to determine how widespread this association between secondary structure potential and location within operons may be.
High Secondary Structure Potential in Mitochondrial-Encoded mRNAs
Mitochondria are believed to have arisen through a symbiotic fusion between an ancestral α-Proteobacterium with the capacity for oxidative phosphorylation and a nucleus-containing organism (Fridovich 1974; Gray 1992; Gray et al. 1999). Because most of the organisms that have a bias for secondary structure in coding regions are Eubacteria (Table 2), we asked whether mitochondrial (mt) genomes also have excess folding potential. The mt genome of S. cerevisiae was chosen for this analysis because of its relatively large size. It contains 17 protein-coding genes, of which most are required for self-maintenance and production of ATP. Each mt gene was randomized 20 times using DicodonShuffle, and a Z score was calculated for each gene as described previously. The distribution of Z scores was found to be shifted substantially to the left (Z̄ = –1.5), to an even greater extent than for E. coli genes (Fig. 1), indicating that the coding regions of mtencoded genes have very strong folding potential (Fig. 4A). Similar results were obtained for another large fungal mt genome, that of Podospora anserina (data not shown). The possibility that some of the secondary structures in mt mRNAs might relate to group I or group II intron splicing (Tanner and Sargueil 1995; Rho and Martinis 2000; Bonen and Vogel 2001) is illustrated in Figure 4B, in which regions of high folding potential appear commonly just before introns or clusters of introns in the S. cerevisiae mt-encoded COX1 gene. Although too few mt genes/introns are available to evaluate this effect statistically, a similar association was observed in the COX1 gene of P. anserina (data not shown).
Figure 4.

Genes in the yeast mitochondrial genome have unusually high folding potential. (A) Z scores were calculated for every gene in the mitochondrial genome of S. cerevisiae. For alternatively spliced genes, only the longest isoform was chosen for analysis. One gene was excluded because of its location inside the intron of another gene. (B) Positions of the group I (blue) and group II (green) introns relative to the start site are shown superimposed on the minimum free energy plot for the mitochondrial gene COX1 (magenta). Coordinates and identity of introns were obtained from GenBank (accession no. AJ011856).
Folding Potential of Yeast mRNAs Correlates With Presence of Introns, But Not With Measures of Expression Level or Half-Life
To explore the biological significance of mRNA secondary structure in eukaryotes, we focused on S. cerevisiae, as this organism exhibits a bias toward RNA structure (Table 2) and has been the subject of a number of large-scale studies of gene expression. Two publicly available data sets were used to study the relationship between expression level, half-life, and folding potential of mRNAs. First, the transcriptional profile of 1003 genes measured under different conditions by Northern analysis (Brown et al. 2001) were compared with their folding free energies. Folding potential was found to be uncorrelated with the level of expression at steady state or after heat shock (Supplemental Fig. 1A,B). Using RNA polymerase II mutants and microarrays (Holstege et al. 1998), Wang and colleagues measured the rate of mRNA decay for thousands of genes in yeast (Wang et al. 2002). Again, we found no correlation between mRNA half-life and folding potential (Supplemental Fig. 1C). Codon Adaptation Index (CAI) is a measure of codon usage bias in a gene relative to a reference set of highly expressed genes that was originally developed as a predictor of the robustness of gene expression (Sharp and Li 1987). Folding potential was found to be uncorrelated with CAI (Supplemental Fig. 1D). Finally, we examined whether secondary structure in coding regions is associated with RNA splicing in the yeast nuclear genome by comparing the Z scores of coding regions derived from intron-containing and intron-less genes. We found that the mean Z score was significantly lower for the 201 annotated intron-containing genes than for the 6113 annotated intron-less genes (Z̄ = –0.50 versus –0.24, respectively; P = 0.004), suggesting that secondary structure in yeast exons might play a role in nuclear RNA processing. Although the set of S. cerevisiae genes that contain introns is heavily biased toward ribosomal protein genes, the mean Z-score for ribosomal proteins was –0.20, comparable with that observed for intronless genes, implying that the secondary structure bias is associated with presence of an intron rather than with this particular functional group of genes.
DISCUSSION
In a large-scale study using thousands of mRNA sequences derived from 28 different species, we found evidence of selection for local RNA structure in the coding regions of a number of different organisms (Table 2). In 10 of 14 eubacterial species, mRNA coding regions had a small, but significant bias toward more local secondary structure potential than expected (Table 2), and yeast nuclear and mitochondrial genes share this bias (Results). Our method for measuring secondary structure bias randomizes mRNAs, while preserving the same encoded protein sequence (thereby controlling for selection acting at the protein sequence or protein structure level), as well as the same codon usage and dinucleotide composition as in the native mRNA. Codon usage may be under selection related to translation rate and is, therefore, a variable that should be controlled for in this context. Dinucleotide composition was not controlled for in a previous study (Seffens and Digby 1999), leading to justified criticism (Workman and Krogh 1999). The issue is not that dinucleotide composition is likely to be a selected property of mRNAs, but rather that dinucleotide composition is impacted by other, largely nonselective factors such as DNA mutation and repair processes (Campbell et al. 1999). Because RNA folding free energies depend on dinucleotide composition, it is a factor that must be controlled for in order to confidently assess whether there is evidence of selection for or against RNA structure in any set of sequences. This was the consideration that motivated us to develop the DicodonShuffle method, which is described in the Methods section.
What aspects of mRNA biogenesis or function could account for the observed biases toward local secondary structure potential in many yeast and bacterial ORFs? Formally, secondary structure might play a role at any step in the lifetime of an mRNA molecule, for example, transcription, processing/modification, subcellular localization, translation, or degradation. We consider these possibilities in turn, omitting mRNA localization, for which no large-scale data sources are currently available.
RNA Secondary Structure and Transcription
In prokaryotes, stable secondary structures are associated with transcription termination, but the secondary structures we observe in coding regions presumably do not have this function. RNA secondary structure is known to play a role in regulating transcription elongation in certain retroviruses (Karn 1999; Weik et al. 2002). However, we see no significant bias for secondary structure in the ORFs of most eukaryotes, with the exception of S. cerevisiae. If there were a significant general effect of secondary structure on transcription elongation, one might expect to see a correlation between secondary structure and expression level, as presumably genes that need to be expressed at high levels would be under stronger selection for increased rates of transcription elongation. In yeast, however, where a bias toward secondary structure is observed, we saw no correlation between expression level and secondary structure potential (Supplemental Fig. 1). Thus, our data do not provide support for a general role of mRNA structure in transcriptional regulation.
RNA Secondary Structure and RNA Processing
The secondary structures of group I and group II introns play crucial roles in their mechanisms of excision, and intronic secondary structures also play a role in processing of some spliceosomal introns in yeast (Howe and Ares Jr. 1997; Libri et al. 2000). In higher eukaryotes, it is well established that exonic sequences can play important roles in splicing (e.g., Fairbrother et al. 2002 and references therein). However, it is not known whether exonic secondary structures play any general role in splicing. We observed a significantly stronger bias toward local secondary structure in the mRNAs of intron-containing nuclear genes in the yeast S. cerevisiae than in intron-less genes. The increased representation of ribosomal protein genes among yeast intron-containing genes does not appear to explain this difference (see Results). Furthermore, the distribution of secondary structures in the mt-encoded COX1 gene appeared to correlate with the locations of group I and group II introns (Fig. 4B). Therefore, we suggest that local RNA secondary structures in exons may play a role in splicing of both nuclear spliceosomal introns and mitochondrial group I/II introns in yeast. For example, local secondary structures in exons might facilitate splicing by competing with unproductive exon–intron duplexes, thereby increasing the rate of formation of proper intra-intronic secondary structures involved in splicing. Unfortunately, the limited numbers of introns present in the yeast nuclear and mt genomes prevent a more detailed computational analysis. It is important to note that intronless genes in the yeast nuclear and mitochondrial genomes also exhibit significantly more secondary structure potential than expected, suggesting that mRNA secondary structure also has other important functions in yeast in addition to a possible role in splicing, and clearly, the widespread bias for secondary structure observed in eubacterial genomes (Table 2)is related to factors other than splicing.
RNA Secondary Structure and Translation
The secondary structures of 5′ UTRs are known to play important roles in regulation of translation initiation in both prokaryotic and eukaryotic systems (Rocha et al. 1999), whereas secondary structure in coding regions is known to reduce the rate of translation (Klionsky et al. 1986; Guisez et al. 1993; Schmittgen et al. 1994). One might expect that highly expressed genes would be under the strongest pressure to increase translation rates. However, we observed no correlation between mRNA secondary structure potential and the steady-state expression levels of 1000 yeast genes as determined by Northern analysis (Supplemental Fig. 1A). One possible explanation for this observation is that maximization of translation rate (by elimination of mRNA secondary structure) may not necessarily maximize production of active protein. For example, synonymous mutations have been identified in the E. coli chloramphenicol acetyltransferase (CAT) gene, which increase translation rate, but lead to reductions in enzyme activity, presumably by leading to increased levels of protein misfolding (Komar et al. 1999). Previous computational analyses have identified translationally slow regions in mRNAs at the boundaries of protein domains (Thanaraj and Argos 1996). Therefore, coding region secondary structures might generally be used to regulate the rate of translation of messages, either to control the total level of protein produced or to facilitate protein folding by temporarily pausing translation to allow a protein domain to fold without interference from more carboxy-terminal portions of the peptide. Our data suggests that such a mechanism, if it occurs, is more prominent in prokaryotic than eukaryotic systems. Such a mechanism could be generalized to operons as well. Because many operons encode multi-subunit protein complexes, coding-region secondary structures might be used to temporarily pause translation to allow more 5′-encoded peptides to fold/assemble to provide a proper scaffold on which to fold more 3′-encoded peptides. Such a mechanism might explain the increased levels of RNA secondary structure observed toward the 3′ ends of operons (Fig. 3).
RNA Secondary Structure and mRNA Stability/Decay
In E. coli, two 3′–5′ exonucleases, RNase II and polynucleotide phosphorylase (PNPase), are responsible for the bulk of mRNA degradation to mononucleotides, whereas three endonuclease activities, RNase III, RNase E, and RNase K, carry out mRNA cleavage/inactivation and processing (Deutscher and Li 2001). RNA hairpins in both 5′ and 3′ UTRs of prokaryotic messages are known to control mRNA stability, with 3′ hairpins apparently protecting against degradation by the exonucleases RNase II and PNPase, and 5′ hairpins protecting the mRNA from inactivation by RNase E (Carrier and Keasling 1997). Such structures appear to function optimally in protecting mRNA when positioned at the extreme 5′ or 3′ ends of the message. Internal UTR hairpins may often be sites for transcript processing by RNase III; however, coding-region hairpins have not been well studied in bacteria. In polycistronic mRNA transcribed from some prokaryotic operons, hairpin structures internal to the mRNA can play a role in stabilizing secondary mRNA products (Regnier and Hajnsdorf 1991). Such products may be generated by partial transcript processing by RNase E or an exonuclease, leaving the internal hairpin structure at the terminus of the resulting product. Our data show stronger selection for secondary structure in genes in operons than in singly transcribed genes from E. coli and S. typhi, and we found evidence for conservation of RNA secondary structure in polycistronic genes with high folding potential (Results). The secondary structure bias increases from the 5′-most gene to successively more 3′ genes in operons in both of these organisms (Fig. 3). On the basis of these observations, we suggest that coding-region secondary structures may be selected for in some bacterial operons to protect secondary mRNA products containing a subset of the ORFs in the original operon transcript from further exonucleolytic degradation. Such a mechanism could be used to fine-tune the relative levels of the different protein products of ORFs in an operon. Because exonucleolytic mRNA degradation proceeds 3′–5′ in bacteria, the most 5′ ORF could be protected by hairpins in any of the downstream ORFs, whereas more 3′ ORFs could be protected only by hairpins in the subset of even more 3′ ORFs. As a consequence, the 3′-most ORFs might be under greatest selective pressure to conserve hairpins, as such hairpins could be used to protect the greatest number of ORFs.
In eukaryotes, sequences in the coding regions of certain genes such as FOS, MYC, and tubulin are known to regulate mRNA half-life (Ross 1995), but it is not known whether the secondary structures of coding regions play a general role in determining mRNA stability. We observed no correlation between the excess folding potential of mRNA coding regions and mRNA half-lives estimated by microarray analysis for >4000 yeast genes (Supplemental Fig. 1C). This observation argues against coding-region secondary structure as a primary determinant of mRNA stability in yeast, but does not rule out a role in regulating the half-lives of specific messages or under specific conditions not studied in the microarray experiments (Holstege et al. 1998; Wang et al. 2002).
Conclusions
We have documented a small, but highly significant and widespread bias toward local secondary structure potential in the coding regions of many eubacteria. This bias is stronger in polycistronic genes than in monocistronic genes, and increases toward the 3′ ends of operons. Evidence was also found that, in addition to strong conservation of protein-coding function, operon genes in E. coli/S. typhi are also under selection to conserve RNA secondary structure. These results suggest widespread regulation of translation and/or mRNA decay in prokaryotes by mechanisms involving coding-region hairpins. Experimental studies will be required to determine the details of these regulatory mechanisms. To facilitate such studies, supplemental tables are provided listing specific regions of specific mRNAs that contain significant local RNA structure. The tools developed in the course of this study, DicodonShuffle and EvolveGene (available on request) should prove useful in future studies of mRNA function and evolution.
METHODS
RNA Folding
Standard methods for RNA secondary structure prediction compute the folding free energy for the most favorable conformation from a vast number of possible structures. The Vienna RNAfold program (Hofacker et al. 1994), a widely used prediction program that implements Zuker's energy minimization algorithm, was used in this study (Zuker and Stiegler 1981). Calculations were performed with a temperature parameter setting of 37°C for all organisms. Similar results were obtained when the S. cerevisiae genome was folded at the more physiological temperature of 25°C (data not shown).
Definitions
The average folding potential, 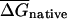 , of an mRNA or set of mRNAs, is defined as the mean of the folding free energies of all 50-bp sequence windows in the native mRNA coding region (starting with the first 50 bases, with a step size between windows of 10 bp), multiplied by 20 to generate per kilobase units (comparable to or somewhat smaller than the average ORF length in the organisms studied), and
, of an mRNA or set of mRNAs, is defined as the mean of the folding free energies of all 50-bp sequence windows in the native mRNA coding region (starting with the first 50 bases, with a step size between windows of 10 bp), multiplied by 20 to generate per kilobase units (comparable to or somewhat smaller than the average ORF length in the organisms studied), and 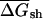 is the corresponding value for the shuffled (randomized)mRNA(s)(see below). EFP is defined as
is the corresponding value for the shuffled (randomized)mRNA(s)(see below). EFP is defined as 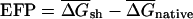 , with higher positive values indicating stronger bias toward secondary structure in the native mRNA, and negative values indicating bias against secondary structure in the native mRNA.
, with higher positive values indicating stronger bias toward secondary structure in the native mRNA, and negative values indicating bias against secondary structure in the native mRNA.
Codon Adaptation Index
Sharp and Lee assigned a parameter, relative adaptiveness, to each of the 61 codons (stop codons excluded). In their model, the relative adaptiveness of a codon was defined as its frequency relative to the most often used synonymous codon and was computed from a set of highly expressed genes (Sharp and Lee 1987).
Sequences
All sequences were obtained from GenBank (http://ncbi.nlm.nih.gov). The genomes of D. melanogaster, H. sapiens, A. thaliana, and C. elegans were annotated using the GENOA script (L.P. Lim and C.B. Burge, unpubl.), which uses spliced alignment of cDNAs to annotate gene structures. Nonredundant single hits were filtered for incomplete or incorrectly annotated coding sequences by removing entries that did not maintain an ORF. All of the remaining sequences for D. melanogaster, A. thaliana, and C. elegans, and 1855 randomly chosen cDNAs for H. sapiens were kept for further analysis.
mRNA Randomization Procedures
The three randomization protocols used in this study, CodonShuffle, DicodonShuffle, and DiShuffle are described below.
CodonShuffle randomly permutes the set of codons used in a transcript to encode each amino acid, preserving the exact count of each codon and the precise order of encoded amino acids as in the original transcript. This algorithm is identical to that used previously (Seffens and Digby 1999). For example, if a short native peptide MPFRYPRLR is encoded by the nucleotide sequence AUGCCGUUUCGAUACCCACGGCUGCGU, 12 possible shuffled sequences with the same encoded amino acids could be generated by the program CodonShuffle, including the following:
AUGCCGUUUCGGUACCCACGACUGCGU;
AUGCCAUUU CGGUACCCGCGACUGCGU; and
AUGCCGUUUCGAUACCCACGUCUGCGG.
(Changed codons are in boldface).
The CodonShuffle protocol preserves dinucleotide composition at the (1,2) and (2,3) positions of codons (first/second bases and second/third codon bases, respectively) of the native sequence, because it preserves codon usage. However, it does not, in general, preserve the dinucleotide composition at (3,1) positions, that is, dinucleotides formed by the last base of one codon and the first base of the next.
The DicodonShuffle algorithm, described below, remedies this limitation, preserving the dinucleotide composition at (3,1), (1,2), and (2,3) positions, as well as the same encoded amino acid sequence and codon usage of the native mRNA. The essential idea of this new algorithm is to make only those synonymous codon swaps which either (1)preserve (3,1) dinucleotide composition by themselves, or (2) which can be paired with another reciprocal synonymous codon swap, such that simultaneous swapping of both codon pairs results in no net change in (3,1) dinucleotide composition. The steps in the DicodonShuffle algorithm are as follows: (1)set the sequence to be randomized equal to the native mRNA; (2) pick an amino acid at random, indicated by the letter i, from the list of amino acids not yet considered (initially, all 20 amino acids); (3) generate a random number j between 1 and Ni, the number of codons for amino acid i that occur in the native mRNA, and consider the consequences of making a swap between the first and jth codons for this amino acid that occur in the native mRNA; (4) if the swap chosen in step 3 would not change the (3,1) dinucleotide composition of the sequence, make the swap; however, if the swap would alter the (3,1) dinucleotide composition, make a list of reciprocal swaps (from among all synonymous codon pairs in the sequence—see below), pick one such swap at random from this list, then make both swaps in the sequence, and mark the codons in the reciprocal pair as having been swapped and therefore unavailable for future swaps (if the list of reciprocal swaps is empty, then do not make any swaps); (5) generate a random number k between 2 and Ni, and consider the swap between codon 2 and codon k, as in steps 3 and 4, and repeat until all codons for amino acid i have been considered; then go to step 2 and repeat the procedure until all 20 amino acids have been considered.
By way of example, in the sample mRNA sequence above, AUGCCGUUUCGAUACCCACGGCUGCGU, the net effect of a swap between the two proline codons, CCG and CCA, is to eliminate one AU dinucleotide and one GC dinucleotide and to create one GU dinucleotide and one AC dinucleotide at (3,1) positions. A swap between the first (CGG) and second (CGA) arginine codons in this sequence is reciprocal to this swap, as it eliminates one GU and one AC dinucleotide, while creating one AU and one GC dinucleotide. Therefore, the following sequence could be generated by the DicodonShuffle program:
AUGCCAUUUCGGUACCCGCGACUGCGU.
(Changed codons are in bold face.) Note that this randomized mRNA has identical encoded amino acid sequence, codon usage, and dinucleotide composition as the native mRNA, as desired. Tests on short mRNAs show that DicodonShuffle samples almost all of the possible sequences that have these properties. For example, 94% of the possible randomized sequences with these properties were generated in 1000 shuffles of a 12-codon mRNA.
The DiShuffle program simply derives first-order Markov transition probabilities (conditional probability of nucleotide j at a given position given nucleotide i at the previous position) from the conditional frequencies in the input sequence, and generates a random sequence from this model.
Source code for all three of these randomization programs—CodonShuffle, DicodonShuffle, and DiShuffle—is available upon request to the authors.
Analysis of E. coli and S. typhi operons
The gene annotation of the S. typhi genome was searched for syntenic blocks corresponding to annotated E. coli operons to infer candidate S. typhi operons. Next, amino acid and nucleotide global alignments of orthologous pairs of E. coli and S. typhi operon genes were constructed using the ALIGN program (Myers and Miller 1989). These alignments are taken as input files to the program EvolveGene, which then generates a synthetic S. typhi sequence, S.typhiS, with the same amino acid and codon usage as the native S. typhi sequence, and the same degree of similarity to the E. coli homolog. For example, consider the following nucleotide alignment:
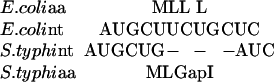 |
On the basis of this alignment, EvolveGene might generate the following synthetic S. typhi sequence:
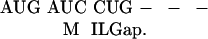 |
Statistical Analyses
The statistical package R was used for data analysis and display (http://www.r-project.org/). Statistical significance for the biases observed in computed free energies between native and randomized sequences were measured for nonoverlapping windows using the Wilcoxon test. Z-scores were calculated as:
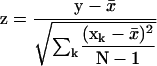 |
y = average folding free energy for windows in native sequence; x̄ = mean of average folding free energies for N DicodonShuffled sequences;
xk = average folding free energy for kth shuffled sequence; N = total number of randomizations (at least 20 for all studies).
Acknowledgments
We thank Alan P. Jasanoff for suggesting the idea to analyze RNA secondary structure in coding regions and for many helpful discussions about this work. We also thank Phil Green, Dirk Holste, and Uttam RajBhandary for helpful suggestions on the manuscript, and Daniel Herschlag for communication of results prior to publication. This work was supported by a Functional Genomics Innovation Award (C.B.B. and Phillip A. Sharp). L.K. is supported by an NIH postdoctoral fellowship.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
[Supplementary material available online at www.genome.org.]
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.1257503.
References
- Bonen, L. and Vogel, J. 2001. The ins and outs of group II introns. Trends Genet. 17: 322–331. [DOI] [PubMed] [Google Scholar]
- Brown, A.J., Planta, R.J., Restuhadi, F., Bailey, D.A., Butler, P.R., Cadahia, J.L., Cerdan, M.E., De Jonge, M., Gardner, D.C., Gent, M.E., et al. 2001. Transcript analysis of 1003 novel yeast genes using high-throughput northern hybridizations. EMBO J. 20: 3177–3186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell, A., Mrazek, J., and Karlin, S. 1999. Genome signature comparisons among prokaryote, plasmid, and mitochondrial DNA. Proc. Natl. Acad. Sci. 96: 9184–9189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrier, T.A. and Keasling, J.D. 1997. Controlling messenger RNA stability in bacteria: Strategies for engineering gene expression. Biotechnol. Prog. 13: 699–708. [DOI] [PubMed] [Google Scholar]
- Chartrand, P., Meng, X.H., Singer, R.H., and Long, R.M. 1999. Structural elements required for the localization of ASH1 mRNA and of a green fluorescent protein reporter particle in vivo. Curr. Biol. 9: 333–336. [DOI] [PubMed] [Google Scholar]
- Deutscher, M.P. and Li, Z. 2001. Exoribonucleases and their multiple roles in RNA metabolism. Prog. Nucleic Acid Res. Mol. Biol. 66: 67–105. [DOI] [PubMed] [Google Scholar]
- Diwa, A., Bricker, A.L., Jain, C., and Belasco, J.G. 2000. An evolutionarily conserved RNA stem-loop functions as a sensor that directs feedback regulation of RNase E gene expression. Genes & Dev. 14: 1249–1260. [PMC free article] [PubMed] [Google Scholar]
- Fairbrother, W.G., Yeh, R.F., Sharp, P.A., and Burge, C.B. 2002. Predictive identification of exonic splicing enhancers in human genes. Science 297: 1007–1013. [DOI] [PubMed] [Google Scholar]
- Freedman, L.P., Zengel, J.M., Archer, R.H., and Lindahl, L. 1987. Autogenous control of the S10 ribosomal protein operon of Escherichia coli: Genetic dissection of transcriptional and posttranscriptional regulation. Proc. Natl. Acad. Sci. 84: 6516–6520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fridovich, I. 1974. Evidence for the symbiotic origin of mitochondria. Life Sci. 14: 819–826. [DOI] [PubMed] [Google Scholar]
- Gray, M.W. 1992. The endosymbiont hypothesis revisited. Int. Rev. Cytol. 141: 233–357. [DOI] [PubMed] [Google Scholar]
- Gray, M.W., Burger, G., and Lang, B.F. 1999. Mitochondrial evolution. Science 283: 1476–1481. [DOI] [PubMed] [Google Scholar]
- Gray, N.K. and Wickens, M. 1998. Control of translation initiation in animals. Annu. Rev. Dev. Biol. 14: 399–458. [DOI] [PubMed] [Google Scholar]
- Grover, A., Houlden, H., Baker, M., Adamson, J., Lewis, J., Prihar, G., Pickering-Brown, S., Duff, K., and Hutton, M. 1999. 5′ splice site mutations in tau associated with the inherited dementia FTDP-17 affect a stem-loop structure that regulates alternative splicing of exon 10. J. Biol. Chem. 274: 15134–15143. [DOI] [PubMed] [Google Scholar]
- Guisez, Y., Robbens, J., Remaut, E., and Fiers, W. 1993. Folding of the MS2 coat protein in Escherichia coli is modulated by translational pauses resulting from mRNA secondary structure and codon usage: A hypothesis. J. Theor. Biol. 162: 243–252. [DOI] [PubMed] [Google Scholar]
- Hofacker, I.L., Fontana, W., Stadler, P.F., Bonhoeffer, L.S., Tacker, M., and Schuster, P. 1994. Fast folding and comparison of RNA secondary structures. Monatshefte f. Chemie 125: 167–188. [Google Scholar]
- Holstege, F.C., Jennings, E.G., Wyrick, J.J., Lee, T.I., Hengartner, C.J., Green, M.R., Golub, T.R., Lander, E.S., and Young, R.A. 1998. Dissecting the regulatory circuitry of a eukaryotic genome. Cell 95: 717–728. [DOI] [PubMed] [Google Scholar]
- Howe, K.J. and Ares Jr., M. 1997. Intron self-complementarity enforces exon inclusion in a yeast pre-mRNA. Proc. Natl. Acad. Sci. 94: 12467–12472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huerta, A.M., Salgado, H., Thieffry, D., and Collado-Vides, J. 1998. RegulonDB: A database on transcriptional regulation in Escherichia coli. Nucleic Acids Res. 26: 55–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karn, J. 1999. Tackling Tat. J. Mol. Biol. 293: 235–254. [DOI] [PubMed] [Google Scholar]
- Klionsky, D.J., Skalnik, D.G., and Simoni, R.D. 1986. Differential translation of the genes encoding the proton-translocating ATPase of Escherichia coli. J. Biol. Chem. 261: 8096–8099. [PubMed] [Google Scholar]
- Komar, A.A., Lesnik, T., and Reiss, C. 1999. Synonymous codon substitutions affect ribosome traffic and protein folding during in vitro translation. FEBS Lett. 462: 387–391. [DOI] [PubMed] [Google Scholar]
- Libri, D., Lescure, A., and Rosbash, M. 2000. Splicing enhancement in the yeast rp51b intron. RNA 6: 352–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindahl, L., Archer, R.H., McCormick, J.R., Freedman, L.P., and Zengel, J.M. 1989. Translational coupling of the two proximal genes in the S10 ribosomal protein operon of Escherichia coli. J. Bacteriol. 171: 2639–2645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathews, D.H., Sabina, J., Zuker, M., and Turner, D.H. 1999. Expanded sequence dependence of thermodynamic parameters improves prediction of RNA secondary structure. J. Mol. Biol. 288: 911–940. [DOI] [PubMed] [Google Scholar]
- Mattheakis, L., Vu, L., Sor, F., and Nomura, M. 1989. Retroregulation of the synthesis of ribosomal proteins L14 and L24 by feedback repressor S8 in Escherichia coli. Proc. Natl. Acad. Sci. 86: 448–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy, J.E. 1990. Post-transcriptional control in the polycistronic operon environment: Studies of the atp operon of Escherichia coli. Mol. Microbiol. 4: 1233–1240. [DOI] [PubMed] [Google Scholar]
- McCarthy, J.E., Gerstel, B., Surin, B., Wiedemann, U., and Ziemke, P. 1991. Differential gene expression from the Escherichia coli atp operon mediated by segmental differences in mRNA stability. Mol. Microbiol. 5: 2447–2458. [DOI] [PubMed] [Google Scholar]
- Meijer, H.A. and Thomas, A.M. 2002. Control of eukaryotic protein synthesis by upstream open reading frames in the 5′-untranslated region of an mRNA. Biochem. J. 367: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers, E.W. and Miller, W. 1989. Optimal alignments in linear space. Comput. Appl. Biosci. 4: 11–17. [DOI] [PubMed] [Google Scholar]
- Regnier, P. and Hajnsdorf, E. 1991. Decay of mRNA encoding ribosomal protein S15 of Escherichia coli is initiated by an RNase E-dependent endonucleolytic cleavage that removes the 3′ stabilizing stem and loop structure. J. Mol. Biol. 217: 283–292. [DOI] [PubMed] [Google Scholar]
- Rho, S.B. and Martinis, S.A. 2000. The bI4 group I intron binds directly to both its protein splicing partners, a tRNA synthetase and maturase, to facilitate RNA splicing activity. RNA 6: 1882–1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocha, E.P., Danchin, A., and Viari, A. 1999. Translation in Bacillus subtilis: Roles and trends of initiation and termination, insights from a genome analysis. Nucleic Acids Res. 27: 3567–3576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross, J. 1995. mRNA stability in mammalian cells. Microbiol Rev. 59: 423–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmittgen, T.D., Danenberg, K.D., Horikoshi, T., Lenz, H.J., and Danenberg, P.V. 1994. Effect of 5-fluoro- and 5-bromouracil substitution on the translation of human thymidylate synthase mRNA. J. Biol. Chem. 269: 16269–16275. [PubMed] [Google Scholar]
- Seffens, W. and Digby, D. 1999. mRNAs have greater negative folding free energies than shuffled or codon choice randomized sequences. Nucleic Acids Res. 27: 1578–1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp, P.M. and Li, W.H. 1987. The codon Adaptation Index—A measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 15: 1281–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanner, N.K. and Sargueil, B. 1995. Dissecting and analyzing the secondary structure domains of group I introns through the use of chimeric intron constructs. J. Mol. Biol. 252: 583–595. [DOI] [PubMed] [Google Scholar]
- Thanaraj, T.A. and Argos, P. 1996. Ribosome-mediated translational pause and protein domain organization. Protein Sci. 5: 1594–1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y., Liu, C.L., Storey, J.D., Tibshirani, R.J., Herschlag, D., and Brown, P.O. 2002. Precision and functional specificity in mRNA decay. Proc. Natl. Acad. Sci. 99: 5860–5865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weik, M., Modrof, J., Klenk, H.D., Becker, S., and Muhlberger, E. 2002. Ebola virus VP30-mediated transcription is regulated by RNA secondary structure formation. J. Virol. 76: 8532–8539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Workman, C. and Krogh, A. 1999. No evidence that mRNAs have lower folding free energies than random sequences with the same dinucleotide distribution. Nucleic Acids Res. 27: 4816–4822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuker, M. and Stiegler, P. 1981. Optimal computer folding of large RNA sequences using thermodynamics and auxiliary information. Nucleic Acids Res. 9: 133–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
WEB SITE REFERENCES
- http://ncbi.nlm.nih.gov; National Center for Biotechnology Information Web site, home of GenBank database.
- http://www.r-project.org/; home page of the R Project—free version of the S+ statistical software package.