

Abstract
BACKGROUND
Whole-genome sequencing may revolutionize medical diagnostics through rapid identification of alleles that cause disease. However, even in cases with simple patterns of inheritance and unambiguous diagnoses, the relationship between disease phenotypes and their corresponding genetic changes can be complicated. Comprehensive diagnostic assays must therefore identify all possible DNA changes in each haplotype and determine which are responsible for the underlying disorder. The high number of rare, heterogeneous mutations present in all humans and the paucity of known functional variants in more than 90% of annotated genes make this challenge particularly difficult. Thus, the identification of the molecular basis of a genetic disease by means of whole-genome sequencing has remained elusive. We therefore aimed to assess the usefulness of human whole-genome sequencing for genetic diagnosis in a patient with Charcot–Marie–Tooth disease.
METHODS
We identified a family with a recessive form of Charcot–Marie–Tooth disease for which the genetic basis had not been identified. We sequenced the whole genome of the proband, identified all potential functional variants in genes likely to be related to the disease, and genotyped these variants in the affected family members.
RESULTS
We identified and validated compound, heterozygous, causative alleles in SH3TC2 (the SH3 domain and tetratricopeptide repeats 2 gene), involving two mutations, in the proband and in family members affected by Charcot–Marie–Tooth disease. Separate subclinical phenotypes segregated independently with each of the two mutations; heterozygous mutations confer susceptibility to neuropathy, including the carpal tunnel syndrome.
CONCLUSIONS
As shown in this study of a family with Charcot–Marie–Tooth disease, whole-genome sequencing can identify clinically relevant variants and provide diagnostic information to inform the care of patients.
The practice of medical genetics requires gene-specific analyses of DNA sequences and mutations to definitively diagnose disease, provide prognostic information, and guide genetic counseling regarding the risk of recurrence. Studies of autosomal recessive traits such as cystic fibrosis1 and some dominant traits such as neurofibromatosis type 12 revealed the role of single “disease genes” in conveying traits. However, many phenotypes of mendelian diseases (see the Glossary) are genetically heterogeneous: causative mutations have been identified in more than 100 genes for deafness and retinitis pigmentosa, for instance. Moreover, specific mutations may confer phenotypes that segregate as dominant, recessive, or even digenic3 or triallelic4 traits. There is also ample evidence of modifying loci in mendelian disorders.5,6 Thus, even when there are simple patterns of inheritance in syndromes with a well-characterized pathologic course, the underlying mutational events, which need to be resolved for precise molecular diagnosis, within individual families may be complex.
Charcot–Marie–Tooth disease is an inherited peripheral neuropathy with two forms: a demyelinating form (type 1) affecting the glia-derived myelin and an axonal form (type 2) affecting the nerve axon. The two forms can be distinguished by means of electrophysiological or neuropathological studies. Charcot–Marie–Tooth disease has been used as a model disease to describe genetic heterogeneity, posit the relation of hereditary pattern to clinical severity, and investigate the relative importance of principal and modifying genes in determining human diseases.7,8 Mutant alleles underlying Charcot–Marie–Tooth disease can segregate in an autosomal dominant, recessive, or X-linked manner (Fig. 1). Both single-base variants (single-nucleotide polymorphisms [SNPs]) and copy-number variants,10 at 39 separate loci, confer susceptibility to Charcot–Marie–Tooth disease. Most of these susceptibility variants cause dominant forms of the disease, although mutations in genes at 14 of the loci cause recessive disease.
Figure 1. Charcot–Marie–Tooth (CMT) Disease Phenotypes, Their Genetic Forms of Inheritance, and Their Mapped Genes and Loci.
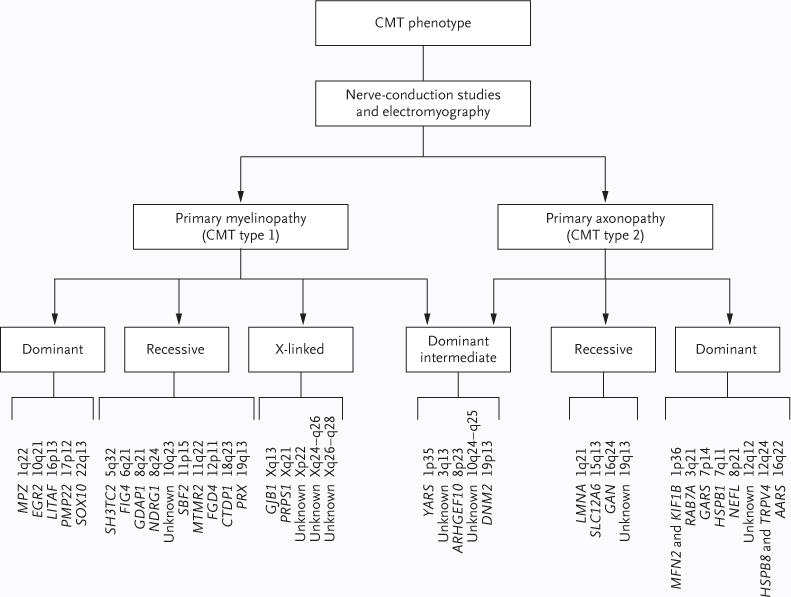
CMT is divided in two major phenotypic types — glial myelinopathy (CMT type 1) and neuronal axonopathy (CMT type 2) — according to electrophysiological, clinical, and nerve-biopsy evaluations. Each type can be inherited in a dominant, recessive, or X-linked fashion. There are also autosomal dominant intermediate forms of CMT that can have features of both axonal and demyelinating neuropathies. Several genes have been associated with CMT disease to date, and other loci have been associated and mapped but their genes not yet identified. MPZ, GDAP1, and GJB1 are known to be associated with CMT type 1, but select mutations in these genes can also cause CMT type 2; NEFL is known to be associated with CMT type 2, but select mutations convey a CMT type 1 phenotype. Dominant intermediate forms of CMT have been reported to be associated with MPZ mutations. Specific recessive alleles related to CMT have also been reported for EGR2 and PMP22. Of the 31 genes in 39 known CMT loci, only 15 genes are currently available for clinical testing. Current evidence-based clinical guidelines for distal symmetric polyneuropathy recommend genetic testing consisting of screening for common mutations, including the CMT1A duplication copynumber variant and point mutations of the X-linked GJB1 gene.9
Adult-onset Charcot–Marie–Tooth disease is highly variable in presentation but is characterized by distal symmetric polyneuropathy,9 with slowly progressive distal muscle weakness and atrophy (particularly peroneal muscular atrophy) resulting in foot dorsiflexor weakness, foot drop, and secondary steppage gait. Pes cavus (highly arched feet) or pes planus (flat feet) occurs in most patients.
We applied next-generation-sequencing methods to identify the cause of disease in a family with inherited neuropathy that had been previously screened, with negative results, for alterations of some common Charcot–Marie–Tooth genes, including PMP22,11 MPZ, PRX, GDAP1, and EGR2.
METHODS
STUDY PARTICIPANTS
The study family consisted of four affected siblings, four unaffected siblings, and an unaffected mother and father, all of whom provided written informed consent for participation in the study. The study was approved by the institutional review board at Baylor College of Medicine. The diagnosis of Charcot–Marie–Tooth type 1 disease in the proband and the three affected siblings was based on the results of physical examination (distal muscle weakness and wasting, pes cavus, and absence of deep-tendon reflexes) and electrophysiological studies.
NEUROPHYSIOLOGICAL ASSESSMENTS
Neurophysiological studies consisted of a standard battery of nerve-conduction studies, including motor responses of the median, ulnar, tibial, and peroneal nerves with F-wave latencies; orthodromic median-, ulnar-, and sural-nerve sensory potentials; and bilateral tibial H-reflexes. When these studies revealed demyelinating features, tests of blink reflexes were generally performed. Limbs were warmed to a temperature of at least 32°C in all instances. Demyelination was judged to be present if conduction velocities were significantly slowed and the late-response latencies were substantially delayed. Median-nerve mononeuropathy at the wrist was judged to be present when there was prolonged motor terminal latency or slowed median-nerve sensory velocity with disproportionate slowing in the palm-to-wrist segment, or both. The four affected subjects, all of whom had diffuse slowing of conduction, were also thought to have a median-nerve mononeuropathy at the wrist, since the median-nerve motor terminal latency was much more prolonged than the ulnar-nerve motor terminal latency (14.9 vs. 8.1, 10.2 vs. 7.5, 11.6 vs. 6.2, and 9.2 vs. 6.2 msec) (Table 1).
Table 1.
Neurophysiological Findings in the Study Family.*
| Subject No. | Age yr |
Sex | Demyelination | Axonopathy | Median-Nerve Entrapment | Diagnosis |
|---|---|---|---|---|---|---|
| I-1 | 80 | M | No | Motor: peroneal, 2.2 mV | Yes | |
| I-2 | 77 | F | No | Sural: absent; motor: peroneal, 0.5 mV; tibial, 2.8 mV | Yes | Axonal neuropathy |
| Maternal grandmother of proband | 90 | F | No | Normal | Yes; SCV, 43 m/sec | MMM |
| II-I | 58 | F | No | Normal | Yes; SCV, 46 m/sec | MMM |
| II-2 | 57 | M | No | Sural: absent; motor: peroneal, 0.2 mV; tibial, 1.4 mV; H-reflexes: 38 msec | Yes | Axonal neuropathy |
| III-1 | 37 | M | No | Normal | No | |
| III-2 | 35 | M | Yes | No | Yes; median-nerve motor TL, 14.9 msec; ulnar-nerve motor TL, 8.1 msec | CMT |
| III-3 | 34 | F | No | No | Yes; SCV, 42 m/sec; median-nerve motor TL, 4.4 msec | MMM |
| III-4 (proband) | 32 | M | Yes | No | Probably; median-nerve motor TL, 10.2 msec; ulnar-nerve motor TL, 7.5 msec | CMT |
| III-5 | 31 | F | No | No | No | |
| III-6 | 29 | F | Yes | No | Yes; median-nerve motor TL, 11.6 msec; ulnar-nerve motor TL, 6.2 msec | CMT |
| III-7 | 26 | F | No | Motor: peroneal, 36 m/sec; H-reflexes: 35 msec | Yes; SCV, 36 m/sec; median-nerve motor TL, 4.8 msec | MMM |
| III-8 | 25 | M | Yes | No | Yes; median-nerve motor TL, 9.2 msec; ulnar-nerve motor TL, 6.2 msec | CMT |
Subject I-1 was a carpenter for more than 50 years. The maternal grandmother of the proband is not included in the pedigree. The ages listed are the ages at the time at which the subjects were evaluated. The normal value for median-nerve motor terminal latency (TL) is less than 4.0 msec and for sensory conduction velocity (SCV) is 48 m/sec or more. The diagnosis column lists the conclusion based on the aggregate findings: severe, widespread slowing of conduction was interpreted as evidence of demyelination, and low-potential amplitudes in multiple nerves with relative preservation of conduction velocities were interpreted as evidence of axonal damage. CMT denotes Charcot–Marie– Tooth disease, CTS the carpal tunnel syndrome, and MMM mild mononeuropathy of the median nerve.
DNA SEQUENCE ANALYSIS
DNA sequencing was performed with the use of the SOLiD (Sequencing by Oligonucleotide Ligation and Detection) system (Applied Biosystems), a next-generation-sequencing platform that involves ligation-based sequencing and a two-base encoding method in which four fluorescent dyes are used to tag various combinations of dinucleotides. Its accuracy in sequencing 50-base reads is estimated at approximately 99.94%.12 Multiple sequences can be read simultaneously, and when the sequence reads overlap, the overall accuracy increases further, reducing the risk of false positive determinations and the need for additional data validation. We determined bases from the primary sequencing data, using the standard SOLiD analysis software. (For details, see the Supplementary Appendix, available with the full text of this article at NEJM.org.)
ARRAY-BASED COMPARATIVE GENOMIC HYBRIDIZATION
For array-based comparative genomic hybridization and analysis of the copy-number variants in the proband as compared with those in a male control, we used a 1-million-probe high-resolution oligonucleotide whole-genome array (Agilent), a 2.1-million-oligonucleotide whole-genome array (NimbleGen), and a 44,000-oligonucleotide array (Agilent) that was custom-designed to assay genes previously implicated in inherited neuropathy. Analysis of the copy-number variants was performed according to the manufacturer’s instructions and software.
BIOINFORMATIC ANALYSIS OF SNP VARIANTS
Analysis of SNP variants and cross-referencing of them with the Human Gene Mutation Database (www.hgmd.cf.ac.uk), the Online Mendelian Inheritance in Man database (www.ncbi.nlm.nih.gov/omim), and the PolyPhen database (http://genetics.bwh.harvard.edu/pph/data, based on the National Center for Biotechnology Information [NCBI] dbSNP, build 126) were performed with the use of Perl scripts. Alignment of the orthologous SH3TC2 (SH3 domain and tetratricopeptide repeats 2) proteins was performed with the use of the ClustalW program and reference SH3TC2 proteins from the following organisms: human (accession number, NP_078853), chimpanzee (XP_527069), macaque (XP_001104761), dog (XP_546315), horse (XP_001501607), cow (XP_616288), mouse (NP_766216), rat (XP_225887), opossum (XP_ 001380773), and chicken (XP_424256).
SEGREGATION ANALYSIS
Exons 5 and 11 of the SH3TC2 gene were amplified by means of a polymerase-chain-reaction (PCR) assay and directly sequenced in all members of the study family. To verify the Arg954ter amino acid mutation (R954X), corresponding to a G→A mutation in the genomic DNA in exon 11 of SH3TC2 on chromosome 5 at nucleotide 148,386,628, we also generated a 312-bp PCR fragment and incubated it with the restriction enzyme TaqI; the nucleotide mutation results in elimination of the restriction site for TaqI.
RESULTS
NERVE-CONDUCTION STUDIES
In addition to the Charcot–Marie–Tooth type 1 phenotype that segregates as a recessive trait, we identified through electrophysiological means an axonal neuropathy in one parent and one grandparent of the proband. Further evidence of a subtle phenotype evidenced by, at a minimum, median-nerve mononeuropathy at the wrist was also observed among all the proband’s grandparents and both parents but had an unclear pattern of inheritance. Its variable presentation (Table 1) included three neurophysiologically defined phenotypes: a normal phenotype with superimposed severe median-nerve mononeuropathy at the wrist, thought to be an incidental finding in an 80-year-old man who had been a carpenter for more than 50 years (Subject I-1), a mild median-nerve mononeuropathy at the wrist (the proband’s maternal grandmother and mother [Subject II-1]), and a more severe median-nerve mononeuropathy at the wrist associated with evidence of a more widespread axonal polyneuropathy (Subjects I-2 and II-2). The latter phenotype is similar to that of patients with hereditary neuropathy with liability to pressure palsies (Online Mendelian Inheritance in Man number, 162500), a disorder pathologically characterized by patchy myelin abnormalities and attributed to haploinsufficiency of PMP22 (as a consequence of genomic deletion)13; duplication of PMP22 causes Charcot–Marie–Tooth type 1A disease, the most common form.14
GENOME VARIATION
The sequencing of DNA samples obtained from the proband produced a mappable yield of 89.6 Gb of sequence data, representing an average depth of coverage of approximately 30 times per base. The data from sequential machine runs consisted of 8.3 Gb of 35-bp fragment sequence reads (one run), 30.3 Gb of 25-bp mate-pair sequence reads (two runs), and 51.0 Gb of 50-bp mate-pair sequence reads (one run).
We identified the differences between the consensus sequence of the proband and the human genome reference sequence. These were used to produce a list of putative single-base DNA substitutions, small insertions, and deletions and potential changes in DNA copy number. This list of variants included 3,420,306 SNPs. A total of 2,255,102 of the SNPs were in extragenic regions and 1,165,204 SNPs were within gene regions, including introns, promoters, 3′ and 5′ untranslated regions, and splice sites (Table 2). Of the intragenic SNPs, 9069 were nonredundant SNPs predicted to result in nonsynonymous codon changes, and 121 of the 9069 were nonsense mutations. The approximately 3.4 million SNPs identified represent about 0.1% of the reference haploid human genome,15 and both the total number of SNPs and the number of novel SNPs are similar to those discovered in other diploid genome sequences for individual subjects (Table 3).12,16–21 Of the more than 3.4 million SNPs, 2,858,587 were present in public databases and 561,719 were novel (Table 3). Data on the sequence reads, quality, and mapping have been deposited in the NCBI Sequence Read Archive (www.ncbi.nlm.nih.gov/Traces/sra/sra.cgi?) (accession number, SRP001734); variant data have been deposited in the dbSNP database.
Table 2.
SNPs Identified through Whole-Genome Sequencing of DNA from the Proband.*
| SNP Type | No. of SNPs |
|---|---|
| Nongene | 2,255,102 |
| Gene | 1,165,204 |
| Intron | 1,064,655 |
| Promoter | 60,075 |
| 3′ UTR | 16,350 |
| 5′ UTR | 3,517 |
| Splice regulatory site | 2,089 |
| Splice site | 112 |
| Synonymous | 9,337 |
| Stop→stop | 17 |
| Nonsynonymous | 9,069 |
| Stop→gain | 121 |
| Stop→loss | 27 |
| Total | 3,420,306 |
Stop→stop refers to synonymous substitutions within a stop codon that maintain the stop codon, stop→gain refers to nonsense mutations, and stop→loss refers to nonsynonymous substitutions that change a stop codon to any other codon. SNP denotes single-nucleotide polymorphism, and UTR untranslated region.
Table 3.
Individual Human Genomes Sequenced to Date.*
| Genome† | Technology Used | Average Depth of Coverage | SNPs | ||
|---|---|---|---|---|---|
| Total | Known | Novel | |||
|
| |||||
| ×10−6 | |||||
|
| |||||
| Venter | Sanger method | 7.5 | 3.21 | 2.80 | 0.74 |
|
| |||||
| Watson | 454 Sequencing System (Roche) | 7.4 | 3.32 | 2.71 | 0.61 |
|
| |||||
| Chinese (YH) | Genome Analyzer (Illumina) | 36 | 3.07 | 2.67 | 0.39 |
|
| |||||
| African (NA18507) | Genome Analyzer (Illumina) | 40.6 | 3.61 | 2.72 | 0.88 |
|
| |||||
| African (NA18507) | SOLiD system (Applied Biosystems) | 17.9 | 3.86 | 3.13 | 0.73 |
|
| |||||
| Korean (SJK) | Genome Analyzer (Illumina) | 28.95 | 3.43 | 3.01 | 0.42 |
|
| |||||
| Korean (AK1) | Genome Analyzer (Illumina) | 27.8 | 3.45 | 2.88 | 0.57 |
|
| |||||
| Proband in this study | SOLiD system (Applied Biosystems) | 29.9 | 3.42 | 2.85 | 0.56 |
All genomes listed have a ploidy of 2n. SNP denotes single-nucleotide polymorphism.
The surname of the individual person or the ethnic group (and HapMap sample name, in parentheses) is given. The same African (Yoruban) sample NA18507 was sequenced twice, once with the use of the Genome Analyzer and once with the use of the SOLiD (Sequencing by Oligonucleotide Ligation and Detection) system.
We used two approaches to identifying copy-number variation: array-based comparative genomic hybridization and mate-pair sequencing. We identified 234 copy-number variants ranging in size from 1690 bp to 1,627,813 bp. Of these 234 variants, 132 were confirmed by at least one other method (Table 1 in the Supplementary Appendix); 220 of the 234 (94%) overlap with reported regions of copy-number variants in the Database of Genomic Variants (http://projects.tcag.ca/variation). We found no copy-number variants affecting genes known to be involved in Charcot–Marie–Tooth disease or other neuropathies.
We cross-referenced the nonsynonymous SNPs that we detected by using whole-genome sequencing with a database of previously observed mutations implicated in human disease (the Human Gene Mutation database) (Table 4, and Table 2 in the Supplementary Appendix). Of the 174 nonsynonymous database SNPs identified in the proband, 159 had a clear association with a heritable trait (i.e., the database entry was not annotated with a question mark). Of these, 21 (13%) were described as causing mendelian disease; 16 were heterozygous in the proband, a finding that is consistent with the expected load of autosomal recessive mutations. The other five SNPs might have been erroneously assigned as disease mutations, which would explain why four of them were homozygous in the proband and have been found to be homozygous in unaffected persons. It would also explain why the sequence for the proband, who did not have adrenoleukodystrophy, contained a SNP in ABCD1 previously described as a dominant mutation that causes the X-linked disorder adrenoleukodystrophy.22 An alternative to the interpretation that the five SNPs might have been erroneously assigned as disease mutations is that these alleles might have reduced penetrance.
Table 4.
Disease and Trait Associations of Nonsynonymous SNPs Identified in the Proband, According to the Human Gene Mutation Database.*
| Disease or Trait Associated with Mutation | SNPs no. (%) |
|---|---|
| Total | 159 (100) |
| Behavioral disorder | 6 (4) |
| Cancer | 33 (21) |
| Association | 7 |
| Increased risk | 9 |
| Reduced risk | 3 |
| Susceptibility | 14 |
| Complex disease | 48 (30) |
| Mendelian disease | 21 (13) |
| Metabolic trait | 17 (11) |
| Pharmacogenetic trait | 14 (9) |
| Other traits | 20 (13) |
SNP denotes single-nucleotide polymorphism.
We examined the putative mutations in 40 genes known to cause or be linked to neuropathic or related conditions (Table 3 in the Supplementary Appendix). This exercise led to closer examination of 3148 putative SNPs, including 54 coding SNPs. Of these 54, 2 were at the SH3TC2 locus — 1 missense mutation (identified at 7.7 average depth coverage) and 1 nonsense mutation (identified at 29.9 average depth coverage) (Fig. 1 in the Supplementary Appendix). Mutations in this locus have previously been found to be associated with Charcot–Marie–Tooth type 4C disease, described in families of eastern European, Turkish or Spanish Gypsy origin.23–25 The R954X nonsense mutation has previously been implicated in Charcot–Marie–Tooth disease; the missense mutation (A→G, occurring on chromosome 5 at nucleotide 148,402,474 and corresponding to the amino acid mutation Tyr169His [Y169H]) is novel.
CORRELATION BETWEEN GENOTYPE AND PHENOTYPE
Segregation analyses verified independent maternal and paternal origins of the mutations (Fig. 2). The nonsense mutation (R954X) appeared in one parent of the proband and in two siblings who did not have Charcot–Marie–Tooth type 1 disease. The missense mutation (Y169H) was found in one parent and one grandparent, neither of whom had Charcot–Marie–Tooth disease. Only the proband (Subject III-4) and three of his siblings (Subjects III-2, III-6, and III-8) who had inherited both mutant alleles had the Charcot–Marie–Tooth type 1 phenotype (Fig. 2).
Figure 2. Pedigree of the Study Family and Segregation and Conservation of SH3TC2 Mutations.
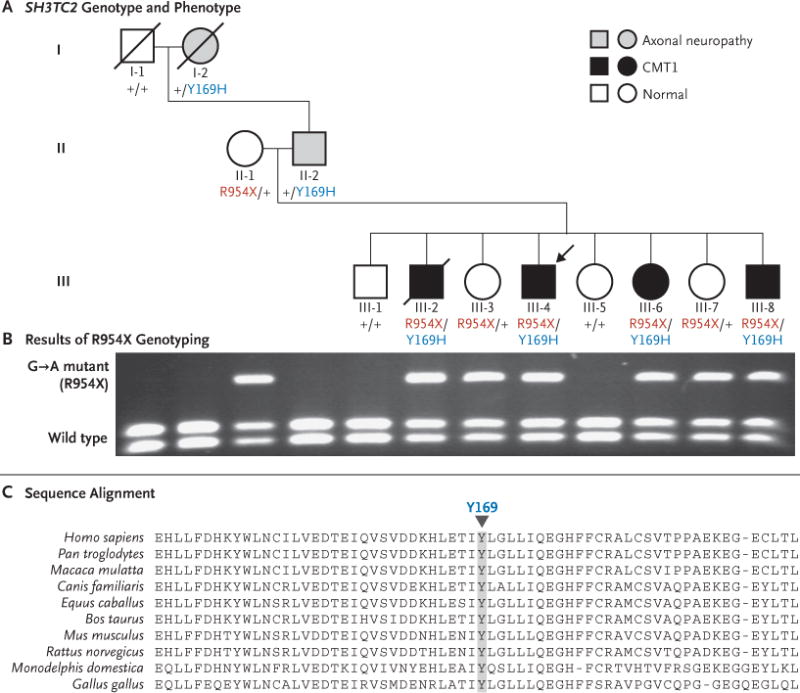
Panel A shows the pedigree of the proband (arrow) and his family and their SH3TC2 genotypes: plus signs indicate the wild-type allele; Y169H indicates the A→G mutation on chromosome 5 at nucleotide 148,402,474 and corresponding to the amino acid missense mutation Tyr169His, and R954X indicates the G→A mutation in the genomic DNA in exon 11 of SH3TC2 on chromosome 5 at nucleotide 148,386,628, leading to the amino acid nonsense mutation Arg954ter. (Genomic coordinates for the mutations in the proband are based on the human genome reference sequence, build 36.2.) Squares indicate male subjects, and circles female subjects slashes indicate deceased subjects. Subjects in generations I and II had three phenotypes. The paternal grandfather (Subject I-1) was studied 20 years ago, at 80 years of age, and had normal results, with the sole exception of a median-nerve mononeuropathy at the wrist, thought to be caused by his occupation as a carpenter. The paternal grandmother (Subject I-2, 77 years of age at the time of evaluation) and the father (Subject II-2) had evidence of a patchy axonal polyneuropathy, with definite median-nerve mononeuropathy at the wrist. The maternal grandmother (evaluated at 90 years of age; data not shown) and the mother (Subject II-1) had normal findings except for very mild median-nerve mononeuropathy at the wrist. Two of the proband’s sisters (Subjects III-3 and III-7) had this same phenotype. Two members of this generation had completely normal findings (Subjects III-1 and III-5). The other four siblings had diffuse, disproportionate conduction slowing in the distal median nerve, without evidence of conduction block, findings that are suggestive of a superimposed median mononeuropathy at the wrist. Subjects III-2, III-4, III-6, and III-8 had Charcot–Marie–Tooth type 1 (CMT1) disease. Panel B shows the results of TaqI restriction digestion of the SH3TC2 exon 11 polymerase-chain-reaction product on which the G→A mutation, corresponding to the R954X allele, occurs. This mutation was present in the proband’s mother and six of the eight siblings, as well as in the maternal grandmother (not shown). The mutation destroys the restriction site for TaqI; the wild type yields two small bands and the heterozygous mutant yields three bands, the upper of which is the uncut DNA. Panel C shows sequence alignment of the SH3TC2 protein among various species. The downward arrowhead indicates the location of the highly conserved Tyr169 amino acid that, in persons with the novel missense mutation Y169H, is changed to His. Sequences were obtained from the National Center for Biotechnology Information.
The subjects with the heterozygous missense mutation (Y169H) (Subjects I-2 and II-2) (Fig. 2) also had the apparently dominant axonal neuropathy phenotype, as detected by electrophysiological studies. These findings of axonal neuropathy (Table 1) suggest a gain of function (i.e., a toxic effect) of this mutation. In contrast, the presumed loss-of-function nonsense variant (R954X) was associated with electrophysiological evidence of the carpal tunnel syndrome, regardless of whether it was the sole mutation present (i.e., heterozygous genotype) or was accompanied by the missense variant (Y169H) (i.e., compound heterozygous genotype) (Table 1 and Fig. 2).
Discussion
We ascertained the molecular basis of an inherited disease by using next-generation-sequencing methods. We chose whole-genome sequencing over targeted, exon-capture approaches26,27 because we did not know whether the “causative” mutations would reside in known coding elements, and targeted approaches are ill suited to capturing copy-number variants. The heterogeneity of our sequence data is emblematic of the current rapid progress of sequencing technology: over the 6-month course of this study, sequence read lengths doubled (from 25 bp to 50 bp), the density of samples on the sequencing slide increased, and mapping technology improved. Overall, the sequence yield increased by a factor of three, with no appreciable increase in expense. This rapid pace of technological improvement makes it difficult to accurately determine the expense of repeating this experiment, but given that the expense of sequencing reagents for a single run on the SOLiD instrument was $25,000 in April 2009, we estimate that the entire effort would currently cost less than $50,000.
The whole-genome sequencing approach used in this proband enabled us to identify the cause of his disease as compound heterozygous mutations in the SH3TC2 gene and thus to delineate the specific biologic basis of disease in his family. The SH3TC2 protein contains both SH3 and TPR motifs; SH3 motifs mediate the assembly of protein complexes binding to proline-rich proteins, and TPR motifs are involved in protein– protein interactions.
The mouse orthologue of SH3TC2 is specifically expressed in Schwann cells, and the SH3TC2 protein localizes to the plasma membrane and to the perinuclear endocytic recycling compartment, which is consistent with a role in myelination or in axon–glia interactions.28 Mice lacking Sh3tc2 have abnormal organization of the node of Ranvier.28 Consistent with a role of SH3TC2 in endocytic processes29 is the finding that SH3TC2 mutations result in disruption of the endocytic and membrane recycling pathways.30
We observed that both of the SH3TC2 mutations, when heterozygous, have phenotypic consequences that can be detected by electrophysiological means. The Y169H missense variant segregates with an axonal neuropathy, whereas the nonsense R954X mutation is associated with subclinical evidence of the carpal tunnel syndrome; therefore, haploinsufficiency of SH3TC2 may cause susceptibility to the carpal tunnel syndrome. This susceptibility may also result from mutations in other genes related to Charcot– Marie–Tooth disease in addition to PMP22 and SH3TC2. Whole-genome sequencing of other members of the proband’s family might help clarify whether the additional 69 SNPs at the SH3TC2 locus and 3146 SNPs at the other 39 neuropathy-associated gene loci examined (including many rare variants, 466 of which have not previously been described [Table 3 in the Supplementary Appendix]) can modify the highly penetrant Y169H and R954X mutations and thereby influence the neuropathy phenotype.
The whole-genome sequencing approach that we describe here contrasts with other diagnostic approaches. A clinical-testing panel that screens for a copy-number variant that commonly causes Charcot–Marie–Tooth disease14 and nucleotide-sequence variants in 15 of the genes known to be mutated in patients with the disease can cost more than $15,000.31 Mutations in two or more genes related to Charcot–Marie–Tooth disease have been described as causing a phenotype more severe than that of our proband or other patients affected by the disease.32–34 Such groups of mutations include a combination of two SNPs at the ACBD1 locus and a copy-number variant affecting PMP22, as well as the combination of a SNP and a copy-number variant at the same locus.35,36 There is also a report of mutations in two genes related to Charcot–Marie–Tooth disease segregating in the same family as either a recessive trait or a sporadic trait, the latter of which was attributed to a de novo copy-number variant.37 Given this locus heterogeneity, with evidence of a mutational load that has clinical consequences, as well as the ease of use and accuracy of the whole-genome sequencing methods we applied, clinical and genetics experts struggling to explain poorly understood high-penetrance genetic diseases can now seriously consider this approach for illuminating the molecular causes of these diseases. The approach may ultimately contribute to the care of patients and families living with such diseases.
Our results suggest that haploinsufficiency of SH3TC2 confers predisposition to a mild polyneuropathy with particular susceptibility to the carpal tunnel syndrome. More generally, they demonstrate the diagnostic power of whole-genome sequencing in the context of genetically heterogeneous mendelian disease and inform efforts to decipher the genetic bases of complex traits. As new, rare alleles at other gene loci are implicated in conditions such as diabetes, obesity, heart disease, and cancer and as the patterns of interaction of the alleles with a patient’s phenotype are delineated, genetic susceptibility to such diseases may become clearer. As a practical matter, the identification of rare, heterogeneous alleles by means of whole-genome sequencing may be the only way to definitively determine genetic contributions to the associated clinical phenotypes.
Supplementary Material
Acknowledgments
Supported in part by grants from the National Human Genome Research Institute (5 U54 HG003273, to Dr. Gibbs) and the National Institute of Neurological Disorders and Stroke (R01 NS058529, to Dr. Lupski).
We thank Kevin McKernan, Michael Rhodes, Francisco de la Vega, Quynh Doan, and Fiona Hyland for extensive discussion and support and Cristian Coarfa for structural-variation analysis and insights.
Glossary
- Array-based comparative genomic hybridization
A hybridization method for detecting copy-number variations in DNA samples from a patient as compared with a control sample. The method provides higher resolution than cytogenetic methods but lower resolution than sequencing methods
- Average depth of coverage
The average number of times each base in the genome was sequenced, as a function of the distribution and number of sequence reads that map to the reference genome
- Coding single-nucleotide polymorphisms
Single DNA-base changes that occur in the coding regions of genes
- Copy-number variation
DNA changes that involve sequences of more than 100 bp, larger than single-nucleotide changes or microsatellites, and that vary in their number of copies among individual persons. These variants can be benign and polymorphic, but some can cause disease
- DNA template
An individual fragment of DNA that is available for sequencing
- Exon capture
Methods for isolating and sequencing gene exons, to the exclusion of the remainder of the genome. The DNA templates from exons are “captured” with the use of probes complementary to the targeted exon sequences. After capture, the targeted DNA is eluted and sequenced. The cost of exon capture can be 10 to 50% lower than that of whole-genome sequencing, although the method is insensitive to copy-number variations and mutations that are outside the targeted regions
- Fragment-sequence read
The contiguous nucleotide sequence from one end of a DNA template (as opposed to a mate-pair read)
- Haploinsufficiency
The state that occurs when a diploid organism has only a single functional copy of a gene, which does not produce enough protein to support normal function
- Mapping
The computational process of identifying the specific region of a reference genome from which an individual sequenced DNA template originated
- Mappable yield
The number of bases generated by a DNA-sequencing instrument that can be mapped to the reference genome
- Mate-pair sequencing
A sequencing strategy that permits the inference of structural changes in a genome by sequencing at both the 5′ and 3′ ends of each DNA template (as opposed to the fragment-sequencing approach)
- Mendelian disease
Human disease caused by mutations in a single gene
- Missense mutations
Single DNA-base changes that occur in the coding regions of genes and alter the resulting encoded amino acid sequence
- Next-generation sequencing
DNA-sequencing methods that involve chemical assays other than the traditional Sanger dideoxy-chain-termination method. Next-generation-sequencing methods produce much larger quantities of data at less expense, but the individual raw sequence reads that are generated from individual amplified DNA-template sequences are shorter and have lower quality
- Nonsense mutations
DNA-base changes that introduce termination codons in the coding sequences of genes, resulting in truncated proteins
- Sequence read
The sequence generated from a single DNA template
- Single-base error rate
The total number of mismatched bases found in mapped sequence reads from a sequencing run, divided by the mappable yield. This rate estimates the probability that any given mappable base is an error
- Two-base encoding
A method used in the SOLiD (Sequencing by Oligonucleotide Ligation and Detection) DNA-sequencing platform that represents a DNA sequence as a chain of overlapping dimers encoded as single-base “colors.” This allows for sequencing of the 16 unique sequence dimers with the use of only four unique dye colors and provides a method for improving the overall accuracy of the sequence reads (reducing the single-base error rate)
Footnotes
Disclosure forms provided by the authors are available with the full text of this article at NEJM.org.
References
- 1.Rommens JM, Iannuzzi MC, Kerem B, et al. Identification of the cystic fibrosis gene: chromosome walking and jumping. Science. 1989;245:1059–65. doi: 10.1126/science.2772657. [DOI] [PubMed] [Google Scholar]
- 2.Wallace MR, Marchuk DA, Andersen LB, et al. Type 1 neurofibromatosis gene: identification of a large transcript disrupted in three NF1 patients. Science. 1990;249:181–6. doi: 10.1126/science.2134734. [DOI] [PubMed] [Google Scholar]
- 3.Kajiwara K, Berson EL, Dryja TP. Digenic retinitis pigmentosa due to mutations at the unlinked peripherin/RDS and ROM1 loci. Science. 1994;264:1604–8. doi: 10.1126/science.8202715. [DOI] [PubMed] [Google Scholar]
- 4.Katsanis N, Ansley SJ, Badano JL, et al. Triallelic inheritance in Bardet-Biedl syndrome, a Mendelian recessive disorder. Science. 2001;293:2256–9. doi: 10.1126/science.1063525. [DOI] [PubMed] [Google Scholar]
- 5.Dipple KM, McCabe ERB. Phenotypes of patients with “simple” Mendelian disorders are complex traits: thresholds, modifiers, and systems dynamics. Am J Hum Genet. 2000;66:1729–35. doi: 10.1086/302938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Badano JL, Katsanis N. Beyond Mendel: an evolving view of human genetic disease transmission. Nat Rev Genet. 2002;3:779–89. doi: 10.1038/nrg910. [DOI] [PubMed] [Google Scholar]
- 7.Allan W. Relation of hereditary pattern to clinical severity as illustrated by peroneal atrophy. Arch Intern Med. 1939;63:1123–31. [Google Scholar]
- 8.Haldane JBS. The relative importance of principal and modifying genes in determining some human diseases. J Genet. 1941;41:149–57. [Google Scholar]
- 9.England JD, Gronseth GS, Franklin G, et al. Practice parameter: evaluation of distal symmetric polyneuropathy: role of laboratory and genetic testing (an evidence-based review): report of the American Academy of Neurology, American Association of Neuromuscular and Electrodiag-nostic Medicine, and American Academy of Physical Medicine and Rehabilitation. Neurology. 2009;72:185–92. doi: 10.1212/01.wnl.0000336370.51010.a1. [DOI] [PubMed] [Google Scholar]
- 10.Lupski JR. Structural variation in the human genome. N Engl J Med. 2007;356:1169–71. doi: 10.1056/NEJMcibr067658. [DOI] [PubMed] [Google Scholar]
- 11.Roa BB, Garcia CA, Suter U, et al. Charcot–Marie–Tooth disease type 1A: association with a spontaneous point mutation in the PMP22 gene. N Engl J Med. 1993;329:96–101. doi: 10.1056/NEJM199307083290205. [DOI] [PubMed] [Google Scholar]
- 12.McKernan KJ, Peckham HE, Costa GL, et al. Sequence and structural variation in a human genome uncovered by short-read, massively parallel ligation sequencing using two-base encoding. Genome Res. 2009;19:1527–41. doi: 10.1101/gr.091868.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Del Colle R, Fabrizi GM, Turazzini M, Cavallaro T, Silvestri M, Rizzuto N. Hereditary neuropathy with liability to pressure palsies: electrophysiological and genetic study of a family with carpal tunnel syndrome as only clinical manifestation. Neurol Sci. 2003;24:57–60. doi: 10.1007/s100720300072. [DOI] [PubMed] [Google Scholar]
- 14.Lupski JR, de Oca-Luna RM, Slaugen-haupt S, et al. DNA duplication associated with Charcot-Marie-Tooth disease type 1A. Cell. 1991;66:219–32. doi: 10.1016/0092-8674(91)90613-4. [DOI] [PubMed] [Google Scholar]
- 15.International Human Genome Sequencing Consortium. Finishing the euchromatic sequence of the human genome. Nature. 2004;431:931–45. doi: 10.1038/nature03001. [DOI] [PubMed] [Google Scholar]
- 16.Levy S, Sutton G, Ng PC, et al. The diploid genome sequence of an individual human. PLoS Biol. 2007;5(10):e254. doi: 10.1371/journal.pbio.0050254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wheeler DA, Srinivasan M, Egholm M, et al. The complete genome of an individual by massively parallel DNA sequencing. Nature. 2008;452:872–6. doi: 10.1038/nature06884. [DOI] [PubMed] [Google Scholar]
- 18.Bentley DR, Balasubramanian S, Swerdlow HP, et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature. 2008;456:53–9. doi: 10.1038/nature07517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang J, Wang W, Li R, et al. The diploid genome sequence of an Asian individual. Nature. 2008;456:60–5. doi: 10.1038/nature07484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ahn SM, Kim TH, Lee S, et al. The first Korean genome sequence and analysis: full genome sequencing for a socioethnic group. Genome Res. 2009;19:1622–9. doi: 10.1101/gr.092197.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim JI, Ju YS, Park H, et al. A highly annotated whole-genome sequence of a Korean individual. Nature. 2009;460:1011–5. doi: 10.1038/nature08211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dvoráková L, Storkánová G, Unterrainer G, et al. Eight novel ABCD1 gene mutations and three polymorphisms in patients with X-linked adrenoleukodystrophy: the first polymorphism causing an amino acid exchange. Hum Mutat. 2001;18:52–60. doi: 10.1002/humu.1149. [DOI] [PubMed] [Google Scholar]
- 23.Senderek J, Bergmann C, Stendel C, et al. Mutations in a gene encoding a novel SH3/TPR domain protein cause autosomal recessive Charcot-Marie-Tooth type 4C neuropathy. Am J Hum Genet. 2003;73:1106–19. doi: 10.1086/379525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Azzedine H, Ravisé N, Verny C, et al. Spine deformities in Charcot-Marie-Tooth 4C caused by SH3TC2 gene mutations. Neurology. 2006;67:602–6. doi: 10.1212/01.wnl.0000230225.19797.93. [DOI] [PubMed] [Google Scholar]
- 25.Gooding R, Colomer J, King R, et al. A novel Gypsy founder mutation, p.Arg1109X in the CMT4C gene, causes variable peripheral neuropathy phenotypes. J Med Genet. 2005;42(12):e69. doi: 10.1136/jmg.2005.034132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Albert TJ, Molla MN, Muzny DM, et al. Direct selection of human genomic loci by microarray hybridization. Nat Methods. 2007;4:903–5. doi: 10.1038/nmeth1111. [DOI] [PubMed] [Google Scholar]
- 27.Ng SB, Buckingham KJ, Lee C, et al. Exome sequencing identifies the cause of a mendelian disorder. Nat Genet. 2010;42:30–5. doi: 10.1038/ng.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Arnaud E, Zenker J, de Preux Charles A-S, et al. SH3TC2/KIAA1985 protein is required for proper myelination and the integrity of the node of Ranvier in the peripheral nervous system. Proc Natl Acad Sci U S A. 2009;106:17528–33. doi: 10.1073/pnas.0905523106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roberts RC, Peden AA, Buss F, et al. Mistargeting of SH3TC2 away from the recycling endosome causes Charcot-Marie-Tooth disease type 4C. Hum Mol Genet. 2010;19:1009–18. doi: 10.1093/hmg/ddp565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lupo V, Galindo M, Martínez-Rubio D, et al. Mutations in the SH3TC2 protein causing Charcot-Marie-Tooth disease type 4C affect its localization in the plasma membrane and endocytic pathway. Hum Mol Genet. 2009;18:4603–14. doi: 10.1093/hmg/ddp427. [DOI] [PubMed] [Google Scholar]
- 31.Athena Diagnostics home page. (Accessed March 5, 2010, at http://www.athenadiagnostics.com.)
- 32.Chung KW, Sunwoo IN, Kim SM, et al. Two missense mutations of EGR2 R359W and GJB1 V136A in a Charcot-Marie- Tooth disease family. Neurogenetics. 2005;6:159–63. doi: 10.1007/s10048-005-0217-4. [DOI] [PubMed] [Google Scholar]
- 33.Auer-Grumbach M, Fischer C, Papić L, et al. Two novel mutations in the GDAP1 and PRX genes in early onset Charcot-Marie-Tooth syndrome. Neuropediatrics. 2008;39:33–8. doi: 10.1055/s-2008-1077085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hodapp JA, Carter GT, Lipe HP, Michelson SJ, Kraft GH, Bird TD. Double trouble in hereditary neuropathy: concomitant mutations in the PMP-22 gene and another gene produce novel phenotypes. Arch Neurol. 2006;63:112–7. doi: 10.1001/archneur.63.1.112. [DOI] [PubMed] [Google Scholar]
- 35.Roa BB, Garcia CA, Pentao L, et al. Evidence for a recessive PMP22 point mutation in Charcot-Marie-Tooth disease type 1A. Nat Genet. 1993;5:189–94. doi: 10.1038/ng1093-189. [DOI] [PubMed] [Google Scholar]
- 36.Shy ME, Scavina MT, Clark A, et al. T118M PMP22 mutation causes partial loss of function and HNPP-like neuropathy. Ann Neurol. 2006;59:358–64. doi: 10.1002/ana.20777. [DOI] [PubMed] [Google Scholar]
- 37.Verny C, Ravisé N, Leutenegger AL, et al. Coincidence of two genetic forms of Charcot-Marie-Tooth disease in a single family. Neurology. 2004;63:1527–9. doi: 10.1212/01.wnl.0000142082.65144.ee. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.