

Abstract
We have developed a methodology we call ROMA (representational oligonucleotide microarray analysis), for the detection of the genomic aberrations in cancer and normal humans. By arraying oligonucleotide probes designed from the human genome sequence, and hybridizing with “representations” from cancer and normal cells, we detect regions of the genome with altered “copy number.” We achieve an average resolution of 30 kb throughout the genome, and resolutions as high as a probe every 15 kb are practical. We illustrate the characteristics of probes on the array and accuracy of measurements obtained using ROMA. Using this methodology, we identify variation between cancer and normal genomes, as well as between normal human genomes. In cancer genomes, we readily detect amplifications and large and small homozygous and hemizygous deletions. Between normal human genomes, we frequently detect large (100 kb to 1 Mb) deletions or duplications. Many of these changes encompass known genes. ROMA will assist in the discovery of genes and markers important in cancer, and the discovery of loci that may be important in inherited predispositions to disease.
Cancer is a disease caused, at least in part, by somatic and inherited mutations in genes called oncogenes and tumor suppressor genes. It is likely that we know only a minority of the critical genes that are commonly mutated in the major cancer types. The identification of these genes can lead to rational targets for chemotherapy. Moreover, in many cases, the knowledge of which genes have been mutated can predict the course of neoplasias, including their therapeutic vulnerabilities, if any. This knowledge is likely to become increasingly important as cancers, or suspected cancers, are detected at earlier and earlier stages.
Methods for finding cancer genes date back to the early 1980s, but general methods have only recently been developed. This problem is being addressed by a variety of evolving techniques, some capable of detecting the genetic losses and amplifications that often accompany the mutation of tumor suppressor genes or oncogenes, respectively. We describe here our success with ROMA (representational oligonucleotide microarray analysis), a technique that evolved from an earlier method, RDA (representational difference analysis; Lisitsyn et al. 1993). Like RDA, ROMA detects differences present in cancer genomes. ROMA also has applications to the identification of genetic variation in individuals caused by gene deletions or duplications, some of which may be related to inherited disease.
We developed RDA as one general approach to the cancer problem. RDA compares two genomes by subtractive hybridization. To apply RDA, the complexity of the two genomes must first be reduced so that hybridization can go nearly to completion. To achieve this, we use low-complexity representations, a PCR-based method (Lisitsyn et al. 1993; Lucito et al. 1998). To compare genomes, they are cleaved in parallel with a restriction endonuclease, ligated to oligonucleotide adapters, and amplified by PCR. The shorter restriction endonuclease fragments are preferentially selected after many cycles of PCR, resulting in the reduced nucleotide complexity that is the essential characteristic of representations.
RDA has been successfully used to detect deletions and amplifications in tumors, and its use has led to the discovery of several candidate tumor suppressor genes and oncogenes (Li et al. 1997; Hamaguchi et al. 2002; Mu et al. 2003). However, RDA does not lend itself to the high-throughput genomic profiling of hundreds to thousands of cancer samples that can then be analyzed in parallel. Such vast parallel analysis is likely to be needed if the majority of complex genetic causes of cancer are to be identified.
Microarray analysis is a high-throughput method that has been widely used to profile gene expression in cancers (DeRisi et al. 1996; Golub et al. 1999; Van't Veer et al. 2002), and three groups, including ours, have adapted microarrays to detect genomic deletions and amplifications in tumors. Pinkel et al. (1998) have used arrays of BAC DNAs as hybridization probes; Pollack et al. (1999) have used cDNA fragments as probes; and in our first implementation, we used microarrays of fragments from representations as probes to analyze genomic representations (Lucito et al. 2000). All three methods use the comparative “two-color” scheme, in which simultaneous array hybridization detects a “normal” genome at one fluorescent wavelength and a pathological genome at another.
We previously demonstrated that complexity reduction of samples by representation improves signal-to-noise performance, and diminishes the amount of sample required for analysis, relative to other microarray hybridization methods (Lucito et al. 2000). However, useful interpretation of genomic array hybridization data requires that the arrayed probes be mapped, and this was a daunting task when we used fragments as probes. Moreover, in our previous implementation we used random fragment libraries, and we therefore could not create arrays focused in certain regions of the genome at will.
Adopting microarrays of oligonucleotide probes solves these problems. Representations are based on amplification of short restriction endonuclease fragments, and hence are predictable from the nucleotide sequence of the genome. Therefore, with the publication of the rough draft of the human genome (Lander et al. 2001), we can now design oligonucleotide probes that will hybridize to representations, and map them computationally. We developed algorithms for choosing from each predicted short fragment a 70-mer (“long”) oligonucleotide probe with a minimal degree of sequence overlap to the rest of the genome. Through computation on the published human sequence, we can design almost any distribution of probes within the genome.
There are many other advantages to oligonucleotide-based microarrays. Based on our experience with the earlier implementation of this method using fragment arrays, the quality and reproducibility of printed oligonucleotide arrays (“print format”) are superior. Although there is a large initial capital outlay to purchase large sets of oligonucleotides, the printed arrays are very inexpensive per unit when costs are amortized, and laborious and expensive replication of an underlying collection is not required. Furthermore, “long” oligonucleotide probes can be synthesized directly on an array surface (photoprinted arrays), and we demonstrate herein the equivalence of the two formats. In the photoprint format, there is no underlying physical collection at all (Singh-Gasson et al. 1999). In either case, whether printed or photoprinted, the composition of the array can be absolutely specified and hence is completely reproducible by others.
We show results from two array formats. The printed arrays are a format that is readily achievable. The regions that are represented on the array can be changed to suit the user. A whole-genome array can be printed with the desired resolution. Smaller ROMA arrays can be designed and printed to focus on specific regions of the genome if wished, the advantage being that less capital outlay would be required for a smaller set of oligonucleotides. Results from the second format used, photoprint array, were presented to demonstrate the power of high-resolution copy number analysis.
In this paper, we demonstrate our system, illustrating results and analytical techniques, present high-resolution analysis of cancer genomes, and provide initial evidence for widespread copy number polymorphism in humans. We discuss applications of our method, compare our method to other methods for global genomic analysis, and outline likely future developments.
RESULTS
Overview
This paper describes a complex procedure, observations, and methods of analysis that are highly interactive. We therefore give here an overview of our results to guide the reader through a sensible reading of this portion of the manuscript. The first section reviews the technique of representations, and in particular “depleted” representations. Next we describe the design and selection of probes selected to hybridize well to representations. We introduce the two array formats that we use. The third section illustrates how to use hybridization to depleted representations to validate the composition of an array design, and the fourth section illustrates the use of such hybridization data to characterize probes and model overall array performance. Next we view essentially raw data of tumor and normal genomes, using two very different array formats, and show that the data from both formats are highly comparable. In the next section, we demonstrate a new statistical approach to gene copy number analysis based on segmentation analysis, and apply the method to two cancer genomes. The clonal nature of the cancers appears evident, as does the highly turbulent nature of their genomic rearrangements. The concordance between copy number analysis and our mathematical model is re-examined. Then we look more closely at several genetic lesions detected by our arrays following our statistical processing. Several distinct types of lesions are illustrated, including large regions of amplification and very narrow regions of homozygous and hemizygous deletion. Different types of inferences that can be made by the method are demonstrated. In the final section, we find a surprising abundance of “normal” variation in copy number between two individuals, and illustrate the need to coordinate data about such variation with interpretation of cancer data.
Representations
Representations reduce the complexity of samples in a reproducible way, thereby increasing signal to noise during hybridization to arrayed probes. Representations also provide a means to amplify the quantity of sample, and allow a very convenient way to validate and simulate array performance.
In our present studies, we have limited ourselves to the use of representations made with BglII, an enzyme with a typical 6-bp recognition site. BglII is one of many restriction enzymes that satisfy these useful criteria: It is a robust enzyme; its cleavage site is not affected by CpG methylation; it leaves a four-base overhang; and its cleavage sites have a reasonably uniform distribution in the human genome. After cleavage with BglII, we ligate adapters, and use the resulting product as a template for a PCR reaction. Because PCR selects small fragments, BglII representations are made up of the short BglII fragments, generally smaller than 1.2 kb, and we estimate that there are ∼200,000 of them, comprising ∼2.5% of the human genome, with an average spacing of 17 kb.
For array characterization, we use “depleted” BglII representations. These are representations made according to the usual protocol, but prior to PCR (to selectively amplify small BglII fragments), the adaptor-ligated BglII fragments are cleaved with a second restriction endonuclease. Cleavage destroys the capacity of some fragments to be exponentially amplified. For example, a BglII representation-depleted by EcoRI would consist of all small BglII fragments of the genome that do not contain within them EcoRI sites. Depleted representations are used for probe validation and modeling performance because we can remove a known subset of fragments from the representation, and observe the consequence upon hybridization to those probes complementary to the depleted fragments.
In all of the experiments described herein, we have used comparative hybridization of representations prepared in parallel. Our approach works best if the DNA from two samples being compared is prepared at the same time, from the same concentration of template, using the same protocols, reagents and thermal-cycler. This diminishes the “noise” created by variable yield upon PCR amplification.
Design and Selection of Probes, and Composition of Probes for Microarrays Formats
We describe the design (length and composition) and selection of probes using two very distinct formats for the synthesis of arrayed probes.
Our probes are derived from the short BglII restriction endonuclease fragments that we predict to exist from analysis of the human genome sequence. We initially evaluated probes of length 30 through 70, using methods described in the next section. The signal-to-noise ratio was maximal for probes of 70 nt in length, and we chose that length as our standard.
We selected our probes to be as unique as possible within the human genome, and tried to minimize short homologies to all unrelated sequences. We devised algorithms by which we could annotate any sequence of the genome with its frequency of exact matches in the genome (Healy et al. 2003). These algorithms were used to choose regions within the predicted BglII fragments that are unique for their constituent 18-mers or 21-mers, and then within these regions, choose 70-mers with the minimal arithmetic mean of their constituent 15-mer exact matches. Subsets of the 70-mers were then tested for uniqueness in the human genome by a low homology search using BLAST.
We used two formats for constructing microarrays. In the first of these, the “print” format, we purchased nearly 10,000 oligonucleotides made with solid-phase chemistry, and printed them with quills on a glass surface. In the second format, “photoprint arrays,” oligonucleotides were synthesized directly on a silica surface using laser-directed photochemistry by NimbleGen Systems Inc. The photoprint arrays were a gift of NimbleGen Systems Inc., and fabricated to our design. Many more probes can be synthesized per array with laser-directed photochemistry, and in these experiments our arrays contained 85,000 oligonucleotide probes.
The probe composition for the 85K set was determined by a combination of design and selection, as described below. Unlike oligonucleotide probes synthesized by standard phosphoramidite solid-phase chemistry, certain oligonucleotides synthesized by laser-directed photochemistry are made in poor yield. However, unlike probes synthesized by the solid-phase chemistry and then printed, the cost of testing a set of probes synthesized directly on a chip is no more than the cost of the chip itself. Therefore, we tested ∼700,000 unique 70-nt probes (see Methods) predicted to be complementary to small BglII fragments, arrayed on eight chips. These were hybridized with standard BglII and EcoRI-depleted BglII representations, and we picked the 85,000 with the most intense signal when hybridized to a single normal human DNA, “J. Doe.” These 85,000 were then arrayed on a single chip.
In both our 10K and 85K formats, probes are arrayed in a random order, to minimize the possibility that a geometric artifact during array hybridization will be incorrectly interpreted as a genomic lesion.
Validation of Printed Arrays With Depleted Representations
We should be able to observe a very clear and predictable pattern to arrays hybridized with depleted representations if and only if these conditions are met: The available human genome sequence assembly is accurate; our method of probe design and selection is valid; our hybridization conditions are sufficiently robust to give a good signal-to-noise ratio for our probe population; and we have correctly deconvoluted the probe addresses on our arrays during data processing. We put all our array designs through such tests. Moreover, the data we collect can further be used for probe calibration and to create simulations that predict the power of the array hybridization to detect various genomic lesions, as will be described in a following section.
To illustrate this process with a 10K array, we show in Figure 1 results obtained with BglII representations depleted by HindIII. In Figure 1A, we graph the ratios of hybridization intensity of each probe along the Y-axis. (See Methods for a description of how we process raw scanned data. We perform no background subtraction, as that only increases noise.) Each experiment is performed in color reversal, and the geometric mean of ratios from the separate experiments is plotted. Probes that we predict to detect fragments in both the full and depleted representations, based on the published human sequence, are grouped on the left. There are ∼8000 probes that are predicted to be present in both depleted and nondepleted representations. Probes that we predict will not detect fragments in the depleted representation are grouped on the right. There are ∼1800 probes predicted as being depleted.
Figure 1.

The predictability of informatics and accuracy of the array measurements using 10K microarrays. (A) The results, where the samples hybridized are BglII representation and BglII representation depleted of fragments with a HindIII cleavage site. The Y-axis (Mean Ratio) is the mean measured ratio from two hybridizations of depleted representation to normal representation plotted in log scale. The X-axis (Index) is a sorted index, such that those probes that derive fromfragments that do not have an internal HindIII restriction cleavage site sort first and those with an internal HindIII site sort last. This allows the separation of these two subsets for visualization of the cleavage results. (B) The reproducibility of the duplicate experiments used to generate the average ratio in A. The Y-axis (Ratio Exp1) is the measured ratio from experiment 1, and the X-axis (Ratio Exp2) is the measured ratio of experiment 2. Both axes are plotted in log scale. (C) Graph of the normalized ratio on the Y-axis as a function of intensity of the sample that was not depleted on the X-axis. Both the ratio and intensity were plotted in log scale. (D) Data generated by simulation. The X-axis (Index) is a false index. Probes, in groups of 600, detect increasing copy number, from left to right; 600 flanking probes detect normal copy number. The Y-axis (Mean Ratio) is the mean ratio calculated from two hybridizations.
From the experiment shown in Figure 1A, we can infer that the promise of the method is largely fulfilled: The restriction profile of representational fragments is correctly predicted, the probes are correctly arrayed, and the probes detect the predicted fragment with acceptable signal intensity.
To calculate the data shown in Figure 1A, each hybridization was performed in color reversal, and the geometric mean of ratios from the separate experiments was plotted. In Figure 1B, the agreement between the ratios of the color reversal experiments is graphed, as a log-log scatter plot, showing excellent correlation of the data regardless of the labeling choice.
Modeling Array Hybridization
Variation in the ratio of intensities is evident from Figure 1A. Some probes fail to exhibit the predicted elevated ratios. There are several possible explanations for this. For example, the oligonucleotide probe may not have been correctly or completely synthesized, or the respective BglII fragment may not be present in the representation as predicted. The latter can happen, for example, if the public genome sequence is in error, or if there is a polymorphism at one of the BglII sites in the sample genome resulting in a longer BglII fragment than expected.
When, as here, there is significant variation in measurements, statistical methods need to be used for the most accurate interpretation of data. It is also often useful to construct a mathematical model that can simulate measurement. Moreover, a good model can help predict the limits of detection, and be of assistance in the design of experiments. In this section, we describe a mathematical model that fits the data, and in a later section we describe statistical methods for data analysis. The mathematical model is useful for individual probe characterization, a clearer interpretation of the data, and the sharpening of statistical tools.
There is always more than one way to model data, and various enhancements can be added, but for our arrays we have found that a simple equation and sampling technique creates a model with great predictive power. This model will be described in detail in a subsequent manuscript, but it is based on an equation for the intensity of the i-th probe in a given channel, I[i]:
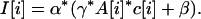 |
In this equation, c[i] is the concentration of BglII fragment complementary to the i-th probe prior to representation; and A[i] is the combined “performance character” of the probe and its complementary BglII fragment. The parameters of the equation are elements of distributions. α is a multiplicative system noise; β is an additive system noise that encompasses background hybridization; and γ is the multiplicative noise created during parallel representation and labeling. By definition, both α and γ have a mean of 1, and for a diploid genome, c[i] = 1.
A[i] can be viewed as the “brightness” of the i-th probe, and is a major determinant of the signal-to-noise ratio. In principle, A[i] should depend on at least two factors: the proportionate amplification of the fragment complementary to the probe during representation; and the purity of the probe. For example, a probe that is complementary to a poorly amplified fragment will have a low A value. Conversely, a probe complementary to a well-amplified fragment should be “bright” and have a high signal-to-noise ratio. Similarly, a probe that is synthesized with poor yield will have a low intensity and a poor signal-to-noise ratio. Other factors may influence A, such as the secondary structure of the probe and its base composition.
In the actual data, the highest ratios are observed from the most intense probes (see Fig. 1C). According to the model, this is explained by a fairly constant nonspecific signal for most probes. That is, β is independent of the probe. Thus, the “brightest” probes also have the highest specific to nonspecific signal. This observation was the basis for our selection of the probes of the 85K set in the photoprint format (see above).
The model makes additional predictions: First, actual ratios are linearly related to measured ratios, and second, the standard deviation of probe measurement is a strong function of ratio, being a minimum for ratios of unity. Using parameters derived from the experiments displayed in Figure 1, we illustrate these relationships in Figure 1D. We assume 15 sets of 600 probes with various copy numbers n/4, with n = 0-14, bracketed by 600 probes of diploid copy number (4/4) on either end, measured against a diploid genome (c[i] = 1), and measured in duplicate. Note that the mean measured ratio of a set of probes is a linear function of the “true” copy number, the number of gene copies per cell, and the mean measured ratio, RM, of a subset of probes reflects their true ratio, RT, by the following equation:
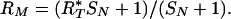 |
This is one general form of a linear equation in which RM = 1 when RT = 1. SN is an experimental character, which we think of as “specific to nonspecific” noise. We can solve for SN from any pair of nonunitary RM and RT values. We use this tool below to analyze two cancer genomes, below.
Views of Tumor Genomes at 10K and 85K Resolution
Array hybridization data can be readily viewed, after deconvolution of probes into genomic order, without any model. In particular, genomic lesions, whether deletions or amplifications, are visually obvious. We show in the matrix of panels of Figure 2 the array hybridization data for three genomic comparisons. Figure 2, A1-A3, shows breast cancer (aneuploid) versus “normal” (diploid) data from the same biopsy of a patient (CHTN159). Figure 2, B1-B3, shows a breast cancer cell line (SK-BR-3) derived from a patient of unknown ethnicity versus an unrelated normal male (“J. Doe”) of mixed European and African parentage. Figure 2, C1-C3, shows a normal male (African pygmy) versus the same J. Doe. In each case, the samples were hybridized twice, with color reversal, and the geometric mean ratio (on a log scale) is plotted versus the genome order of the probes.
Figure 2.

The genomic profiles for (A) a primary breast cancer sample (CHTN159), with aneuploid nuclei compared with diploid nuclei from the same patient; (B) a breast cancer cell line compared with a normal male reference; and (C) a normal male compared with a normal male reference, using the 10K printed array (A1,B1,C1) and the 85K photoprint array (A2,B2,C2). In each case (rows 1 and 2), the Y-axis is the mean ratio, and the X-axis (Gen Index) is an index of the probes' genomic order based on the June 2002 assembly, that is, NCBI Build 30. The probes were put into genomic order concatenating Chromosomes 1 through Y. (A3,B3,C3) The correspondence of the ratios measured from “brother” probes (see text for details) present in the 10K and the 85K microarrays. The Y-axis is the measured ratio from the 10K microarray, and the X-axis is the measured ratio from the 85K microarray.
The samples from Figure 2A were derived by flow sorting the nuclei of a surgical biopsy into aneuploid and diploid fractions, and making representations from as few as 15,000 nuclei (∼100 ng of DNA). We estimate that the aneuploid fraction has perhaps 10% contamination from diploid nuclei, whereas the diploid fraction is not expected to be completely normal. Nevertheless, highly interpretable data result.
These data are in two formats: the 10K print format (Fig. 2A1,B1,C1) and an 85K photoprint format (Fig. 2A2,B2,C2). Unlike the 10K format, probes of the 85K format were also selected for performance, as described and justified in earlier sections. This selection procedure produces a slight bias, in that no probe from the 85K set will detect a small BglII fragment that is homozygously missing in J. Doe. The consequences of this bias can be seen in comparisons of the 10K print format with the 85K photoprint format. In results from the 10K print format, there are roughly equal numbers of extreme “singlets” above and below a copy number of 1 (most apparent in Fig. 2C1). In contrast to this, using the 85K format, more extreme singlets are below rather than above a copy number of 1 (Fig. 2C2).
In Figure 2, A1, A2, B1, B2, C1, C2, increased copy number is indicated by a ratio above 1, and decreased copy number by a ratio below 1. Even at this global view, with all probes displayed, several interesting observations can be made. There are clearly profiles to the cancer genomes, large regions of amplification, some quite high, and large regions of deletion (Fig. 2A,B). The profiles of the cancer genomes are varied. In contrast, the profile of the normal-normal appears to be flat, although some features can be seen. These will be examined more closely below.
There are, in all three genomes, many stand-alone probes detecting minor losses and gains, which we attribute to heterozygous BglII polymorphism. These are manifest in the normal-normal comparison (Fig. 2C2) as a “shell” of probes that approach ratios of 0.5 and 2.0 throughout the genome.
In contrast, in the tumor-normal comparison, wherein the normal is matched, there is only one stand-alone probe detecting major gains, and the stand-alone probes detecting major losses are more or less confined to extensive regions showing minor loss. This pattern is consistent with a hypothesis of allelic polymorphism and loss of heterozygosity (LOH). For a patient with heterozygosity at a BglII fragment, with a large and a small fragment, loss of the small allele will result in the virtual loss of specific signal because the large allele will not be abundant in the representation. This will present as an apparent major loss. On the other hand, a loss of the large allele, for example, by gene conversion, would at most result in a twofold increase in ratio, appearing as a minor gain.
It is evident, looking at the results of the 10K print and the 85K photoprint formats in Figure 2, A1, A2, B1, B2, C1, C2, that the two systems capture a similar view of the larger genomic features. A correspondence between the two formats can be seen quantitatively. We call probes “brothers” if they share complementarity to the same BglII fragment. Brothers do not necessarily have overlapping sequence, or may be complementary across their entire length. In Figure 2, A3, B3, C3, we plot the ratios of brothers from one format to ratios of their brothers from the other format. There are in excess of 7000 brother probes. For all three experiments, in spite of the fact that the probe sequences differ between formats, the order of arraying is different, the hybridization conditions differ, and the surfaces of the array are different, there is remarkable concordance between the ratios of brother probes regardless of format.
Automated Segmentation and Whole-Genome Analysis
Because of the extent of the data, and its statistical nature, automated tools for feature recognition that are statistically based are extremely useful. One part of our group has developed a statistical segmentation algorithm termed circular binary segmentation (CBS) that parses the probe ratio data into segments of similar mean after taking variance into account (Olshen et al. 2002). The algorithm works by analyzing one chromosome at a time and, within that chromosome, recursively identifying the best possible segmentation. Each proposed split is accepted or rejected based on the probability that the difference in mean could have arisen by chance. This probability is determined using a randomization method. The algorithm is a novel modification of binary segmentation (Sen and Srivastava 1975). Because of its nonparametric nature, the algorithm cannot identify aberrations with fewer than three probes. We discuss detecting smaller lesions below.
Figure 3 illustrates some of the output for the analysis of the cancer cell line SK-BR-3 at 85K resolution. We show four chromosomes, the highly turbulent Chromosome 8, a somewhat less active Chromosome 17, Chromosome 5, and the X-chromosome. The segmentation profiles and segment means for the 10K and 85K sets are very similar (data not shown), but clearly are not identical. More features are seen with the 85K set. In the next section, we inspect some of the data more closely. The full data, and that for the other two genomes, can be viewed at our Web site (http://roma.cshl.org/).
Figure 3.

Several chromosomes with varying copy number fluctuations from analysis of the tumor cell line SK-BR-3 as compared with the normal reference. The Y-axis (Mean Ratio) represents the mean ratio of two hybridizations in log scale. The X-axis (Gen Index) is an index of the genomic coordinates, as described above. (A) Copy number fluctuations identified for Chromosome 5, (B) for Chromosome 8, (C) for Chromosome 17, and (D) for the X-chromosome.
Once segmented, we can assign to every probe the mean ratio of the segment to which it belongs, and then view the assigned mean ratios in sorted order. We do this for the two cancer genomes in Figure 4, A (CHTN159) and C (SK-BR-3). It is evident from the figure the segment mean ratios within each genome are quantized, with major and minor plateaus of similar value. In fact, it is likely that we can deduce the copy number by counting. As determined by flow analysis, the tumor is subtriploid, and the cell line is tetraploid. Assuming each sample is roughly monoclonal, then the two major plateaus in the tumor would be two and three copies per cell, and the major plateaus in the cell line are likely to be three and four copies per cell.
Figure 4.

The mean segmentation calculated from the analysis of SK-BR-3 compared with (A,B) the normal reference and (C,D) CHTN159. In all panels, the Y-axis is the value of the mean segment for each probe in log scale. In A and C, the X-axis (Mean Segment Index) is in ascending value of the assigned mean segment. In B and D, the X-axis (Gen Index) is the genomic index, as described above. Plotted on top of the mean segment data is a copy number lattice extrapolated from the array data using formulas within the text (horizontal lines). The calculated copy number for each horizontal line is to the right of the lattice.
We can then use the copy number assumptions of the major plateaus to solve the ploidy and SN for each experiment. Our method is to use a version of equation 2 for each plateau. We select RM, the mean measured ratio, as the average of the probes of the segments in the plateau. We first set RT to CN/P, where CN is the “true” copy number. CN is the number of gene copies per cell, assumed to be known and equal for the plateau. P is the ploidy of the tumor genome. The result is two equations and two unknowns, with the unknowns being P and SN. For the tumor biopsy experiment (Fig. 4A), we calculate the ploidy P to be 2.60, and SN to be 1.13. For the cell line experiment (Fig. 4C), we calculate that P is 3.93, and SN is 1.21. We can then use equation 2 again to calculate what mean ratios would be expected for higher and lower copy numbers. These expectations are marked on the respective graphs, from zero to a copy number of 12, with horizontal lines forming a “copy number lattice.” The assigned mean-segment values for probes are displayed in genome order, embedded with the expected copy number lattice (Fig. 4B,D).
The copy number lattice fits remarkably well the minor plateaus of the data, especially for the higher copy numbers. However, there appears to be error in the expected ratios for probes detecting loss. The assigned mean-segment ratios of probes detecting loss cluster around values somewhat below the predicted values. In other words, the array appears to perform better for deletions than predicted based on the major plateaus and our present model. This deviation might be explained if we reexamine our assumption of clonality, and will be investigated further.
Specific Illustrative Examples
There is clearly too much data to be described in a printed paper, and the reader is invited to visit our Web page (http://roma.cshl.org/). In this section, we discuss a few examples taken from the array data of SK-BR-3 that illustrate several aspects of our system.
The first example is a closer inspection of a region of a break in the X-chromosome, seen in Figure 3D. SK-BR-3, which derives from a female, has been compared to an unrelated male. The expectation is that probes in the X-chromosome will have elevated ratios. This is the case through much of the long arm of Chromosome X. In the midst of Xq13.3, over a region spanning 27 kb, there is a sharp break in copy number, and for the remainder of the chromosome, ratios near 1 are observed (Fig. 5A). This example demonstrates the boundaries that can be drawn from the array data by segmentation. In our data there are other examples of sharp copy number transitions that must break genes.
Figure 5.

In all panels, the Y-axis (Mean Ratio SK-BR-3) is the mean ratio of two hybridizations of SK-BR-3 compared with a normal reference in log scale. The X-axis (Gen Index) is the genomic index, as described. (A) A region from the X-chromosome with a region of loss. Plotted over the measured array ratio is the calculated segmentation value. (B) A region of Chromosome 8 (c-myc located to the right of the center of the graph) from results of SK-BR-3 in comparison to normal reference. Plotted on top of the data are the segmentation values for SK-BR-3 in comparison to the normal reference in red and the segmentation values for the primary tumor CHTN159 in green. (C) A lesion on Chromosome 5 demonstrating the resolving power of the 85K as compared with the 10K array. Results are from SK-BR-3 compared with a normal reference. Spots in red are from the 10K printed microarray, and spots in blue are from the 85K photoprint array. Horizontal lines are copy number estimates, based on modeling from mean-segment values. (D) Comparison of SK-BR-3 to normal reference, displaying a region of homozygous deletion on Chromosome 19. The mean-segment value is plotted as a red line, and horizontal lines are copy number estimates as described.
There are three to four narrow amplifications in SK-BR-3, each containing two or fewer genes, among which are transmembrane receptors. But broad amplifications can also be informative. The second example comes from the highly turbulent Chromosome 8 (see Fig. 3B). Despite the abundance of aberrations, we can clearly discern distinct regions of amplification. One such region is shown in Figure 5B. The rightmost peak is approximately a 1-Mb stretch, comprised of 37 probes (probe coordinates 45099-45138, June 2002 assembly, or NCBI build 30 genome coordinates 126815070-128207342). Yet it contains a single Ref-Seq gene, c-myc.
There is a second very broad peak in SK-BR-3, ascending to the left of the c-myc peak, and off the graph. This broad peak has a broad shoulder on its right (probe coordinates 44994-45051, June 2002 assembly, or NCBI build 30 genome coordinates 123976563-125564705), with a very narrow peak in its midst. We can overlay on this the segmentation data from the tumor genome, CHTN159, which has an even broader peak encompassing c-myc (probe coordinates 44996-45131, June 2002 assembly, or NCBI build 30 genomic coordinates 124073565-127828283). The peak in CHTN159 also encompasses the shoulder of the second SK-BR-3 peak (Fig. 5B). Thus, the shoulder may contain candidate oncogenes that merit attention. Within that region, at the narrow peak, we find TRC8, the target of a translocation implicated in hereditary renal carcinoma (Gemmill et al. 1998). This example illustrates the value of coordinating data from multiple genomes, and the need for automated methods for analyzing multiple data sets.
We next show an example of a narrow deletion that highlights the need for high-resolution arrays, and also raises additional questions. The lesion occurs on Chromosome 5. In Figure 5C, we show a combined 10K (red) and 85K (blue) view. We do not show segmentation, but show the copy number lattice. A deletion is evident at both 10K and 85K resolutions (probe coordinates 26496-26540, June 2002 assembly, or NCBI build 30 genomic coordinates 14231414-15591226), one we judge to be hemizygous loss, but which may represent the presence of one copy in a tetraploid genome. The boundaries are much more clearly resolved at 85K. This region contains TRIO, a protein having a GEF, SH3, and serine threonine kinase domain (Lin et al. 2000); ANKH, a transmembrane protein (Nurnberg et al. 2001); and FBXL, a component of the ubiquitin ligase mediated protein degradation pathway (Ilyin et al. 2000).
It is also clear from the data that the lesion does not appear “neat.” In the middle of the deletion are four or five probes that report ratios near 1. We can consider several explanations for this result. First, the hybridization to those probes may have failed for a variety of reasons. For example, the probes might not have been completely synthesized, or their complementary BglII fragments might not have amplified well. However, the intensities of these probes are in the middle range for all probe intensities, which diminishes the likelihood of this hypothesis. Second, the human assembly may be in error, and the outlier probes have been incorrectly posted at this location. Third, the deletion event may indeed be complex, the result of a localized genomic instability.
Our last example is a region of homozygous loss (Fig. 5D). In this example, a cluster of zinc-finger proteins on Chromosome 19 is affected (probe coordinates 77142-77198, June 2002 assembly, or NCBI build 30 genomic coordinates 21893948-24955961). These genes, having zinc-finger domains, may encode transcription factors, whose deletion may have a role in tumorigenesis.
There are an abundance of narrow hemizygous and homozygous lesions. These are seen both in the analysis of the cancer cell line and the cancer biopsy. However, as described below, we must take caution in their interpretation. Our next examples will all be in the context of normal-normal variation.
Examining Normal Genomic Variation
In this section, we demonstrate the need to coordinate cancer genome analysis with a knowledge base of normal genomic variation.
When the tumor DNA cannot be matched against normal DNA, and an unrelated normal DNA is used as a reference, the differences observed may be the result of polymorphic variation. This variation can be of two sorts, the run-of-the-mill point sequence variation, of the sort that creates or destroys a BglII fragment, SNPs for example, or actual copy number fluctuation present in the human gene pool. The former is relatively harmless, as it will produce scattered noise that can largely be filtered by statistical means.
We illustrate the application of a very mild filtration algorithm: If a ratio is the most deviant of the surrounding four, we replace it with the closer ratio of its two neighbors. In Figure 2C2, we showed a normal-normal comparison. The data look flat, with a cloud of scattered polymorphisms. In Figure 6A (combined 10K and 85K sets), we have applied filtration. The data no longer look so flat, and the cloud of scattered polymorphism is lifted, revealing nonrandom clusters of deviant probe ratios. These clusters reflect large-scale genomic differences between normal individuals, and we will say more of this presently.
Figure 6.

(A) The results of a normal genomic profile compared with a normal, identical to that displayed in Figure 2C2 with the exception that singlet probes have been filtered as described in the text. (B) The serial comparison of experiments for a small region from Chromosome 4. The Y-axis is the mean ratio in log scale. The X-axis is the genomic index, as described. The blue (85K) and red (10K) spots are from the comparison of SK-BR-3 to normal. The green is a comparison of a pygmy to the normal reference. (C) A lesion found in the normal population on Chromosome 6. The blue spots are plotted by mean ratio for analysis of the pygmy to the normal reference. The red line is the mean-segment value for the pygmy-to-normal reference comparison. The green line is the mean-segment value for the SK-BR-3-to-normal reference comparison. The blue line is the segment value from the primary tumor (CHTN159 aneuploid to diploid) comparison. (D) A region of Chromosome 2. The data shown in blue circles are from the comparison of SK-BR-3 to the normal reference. The mean-segment line for this comparison is shown in green. The mean-segment line for the comparison of a pygmy to the normal reference is shown in red and for the primary tumor CHTN159 in blue. For C and D, the calculated copy number for the horizontal lines is found to the right of the panel.
Polymorphic variation of the scattered variety can also be filtered by serial comparison of experiments. We illustrate such a process in Figure 6B. In this figure, we display data from SK-BR-3 compared with normal donor J. Doe, the 85K ratios displayed in blue circles, and the 10K in red. On the same graph we display the ratios of J. Doe compared with another normal, DNA from an African pygmy, in green triangles. This is a fairly typical field of view. We see three probes of extreme ratio in the SK-BR-3-normal hybridization that can be identified as polymorphisms by comparison to hybridization between the two normal individuals. The simplest interpretation is that J. Doe is +/+, pygmy +/-, and SK-BR-3 -/-, where + designates the presence of a small BglII fragment and designates the absence of a fragment (most likely a SNP at a BglII site). In general, pairwise comparisons of three genomes allow interpretable calls of allele status. Hence, we suggest that when a malignant genome cannot be paired to a matched normal, or perhaps even when it can, such genomes should be compared with a single reference normal donor, whose allele status can be firmly established by extensive comparisons against other normals.
Polymorphism in copy number, however, presents a different sort of problem. In this case, many probes within a region will show a deviation from a ratio of unity, and the pattern will appear coherent, not scattered. Statistical means will not suppress this signal. But do such variations commonly exist, and are they likely to be a source of misinterpretation if ignored? The perhaps surprising answer is emphatically, yes.
Figure 6A indicates that there are gross regional differences in the normal-normal comparison. Indeed, many regions that display altered copy number between the two normal individuals are revealed upon segmentation analysis. Close inspections of two such regions are displayed in Figure 6, C and D, with ratios as connected blue dots and copy number lattice values in orange. In Figure 6C, the abnormal region is 135 kb on Chromosome 6p21 (probe coordinates 32518-32524, June 2002 assembly, or NCBI build 30 genomic coordinates 35669083-35804705), and encompasses three known genes. In Figure 6D, the region is a 620-kb region from Chromosome 2p11 (probe coordinates 9927-9952, June 2002 assembly, or NCBI build 30 genomic coordinates 88787694-89385815) that contains a number of heavy chain variable regions.
We observe on the order of a dozen such regions in any normal-normal comparison. They range from 100 kb to >1 Mb in length and are more frequently observed near telomeres and centromeres, but can apparently occur anywhere. They often encompass known genes. We are presently investigating this phenomenon more fully, and will report on them subsequently. For now, we show how they impact the interpretation of cancer-normal data.
In Figure 6, C and D, we have overlain the segmentation values from the analysis of SK-BR-3 in green. The copy number lattice for SK-BR-3 is plotted as orange lines. Figure 6C illustrates a region in SK-BR-3 that would be called a deletion in comparison to the normal. In SK-BR-3 compared to normal, the flanking region occurs at a copy number that we judge to be two copies per cell, and within that region, copy number becomes reduced to one. But the same region appears in the comparison of pygmy DNA to the same normal. In Figure 6D, we observe an analogous condition on Chromosome 2p11. In this panel, we have also plotted segmentation data from the tumor. This region is evidently abnormal there as well. Hence, we are inclined to view this “lesion” as pre-existing in the normal cells of the patient.
DISCUSSION
Comparison of Methodologies for Global Genomic Analysis
We have described a method, representational oligonucleotide microarray analysis, or ROMA, that is useful for detecting amplifications and deletions and sites of breakage in cancer and normal genomes. Detection of these events can in principle be used to discover genes involved in cancer and other diseases of genetic origin, and serve as markers or guides for the diagnosis and treatment of such diseases. Because our method is sensitive to even single nucleotide polymorphisms at restriction endonuclease sites, it could in principle also be used as a high-density array for detecting SNPs.
There are other methods for global analysis of cancers. Most well known is the gene expression microarray (Chee et al. 1996; DeRisi et al. 1996). This method does not find the primary lesions in a cancer, but rather the sequels of mutation. Gene expression microarrays are based on RNA extracted from tumors, and RNA is a very unstable molecule, difficult to extract in a reliable manner. Moreover, the outcome of expression array analysis will be extremely dependent on difficult-to-control factors such as sample handling, and other complicating physiological variables such as tumor infiltration by normal stroma and inflammatory cells. Our method is based on DNA, a very stable molecule, easily extracted even from tissue that has been mishandled. The DNA is the repository of the causative molecular events, and the presence of normal infiltrating stroma and inflammatory cells dilutes the signal but does not change it. We do not intend for our method to exclude RNA analysis, and in fact the two together would be more valuable than either alone.
There are other DNA-based methods for measuring changes in copy number in cancers. The oldest of these is fluorescent in situ hybridization (FISH), which is used clinically to evaluate amplification at the ErbB-2 locus in breast cancer, for example (Tkachuk et al. 1990; Bartlett and Mallon 2003). In work in progress, we have shown that our method is essentially equivalent for evaluating amplification at ErbB-2, but, of course, our method evaluates the entire genome, not just a single locus that may be important in selecting cancer therapy. The major advantage of FISH is that it is essentially a single-cell assay that can thus be performed on very few cells, such as might be available upon needle biopsy. Our method requires perhaps ∼2000 cells, and is a mass measurement, not a single-cell assay. However, our method points to loci that may be converted into FISH-based assays, and that is a major strength.
Another DNA-based method is the BAC array, which is a method that is more commonly known, and more widely practiced, than our method (Pinkel et al. 1998; Snijders et al. 2003). Present BAC arrays suffer from much lower resolution, on the order of 3000 probes. At their maximum, 30,000 member arrays, there are still fewer probes into the genome, and the size of the BAC, 150 to 200 kb, ultimately obscures high resolution. For example, we can observe very small deletions and amplifications that would be entirely missed with even high-density BAC arrays. Additionally, because our method is based on representations, our sample size can be smaller than is needed for the standard BAC array protocol. (However, users of BAC arrays may use our representational approaches to diminish their need for large sample sizes.) Furthermore, BAC arrays cannot be fabricated to industrial standards, as can our arrays. The composition of our arrays is precisely specified, nucleotide for nucleotide, and a highly reproducible standard product can be made available for wide usage. Again, each and every one of our probes can be readily calibrated for performance, a property that cannot be readily done with BAC probes. Finally, our arrays are based on oligonucleotides derived from the human sequence assembly, the lingua franca of human genetics, and can therefore be precisely and automatically mapped into all the databases of all mapped genes and genetic disorders. This cannot be done with BACs, which can be unstable under propagation and can be chimeric. The one advantage of BAC arrays is that they are presently cheaper, but that is likely to be a short-lived advantage.
cDNA arrays have also been used for measuring copy number mutations (Pollack et al. 1999; Hyman et al. 2002), whereby whole genomic DNA is hybridized to a cDNA expression array. These are presently insensitive. Moving averaging of the measured probe ratios is used to decrease system noise, and this results in a decrease in resolution. Therefore this methodology is useful for the detection of larger amplifications and deletions. However, detecting deletions is problematic because of overall signal-to-noise issues of single fragment or oligonucleotide probes. ROMA has overcome this problem by decreasing the complexity of the genome, thereby increasing the signal-to-noise ratio for each probe.
Is Our Knowledge of Cancer Complete?
Science has identified many of the commonly mutated genes in cancer, and we know many of the cellular pathways on which they act. Some think a basic theory of cancer is comprised of only a few basic principles, sufficient to explain the nature of the disease. However, it is a poor and unnecessary gamble to act as though our theory is correct, or that our knowledge of specific facts is nearly complete. Future progress in detection, prognosis, and treatment of cancer will depend on the accuracy and completeness of our understanding of its specific molecular causes.
There are simple tests for the completeness of our understanding and knowledge of how cancers survive in and kill their hosts. If our knowledge of the genes were complete, we would see a plateau in the number of common mutant genes found in all cancers. If our understanding of the principles were complete, even advanced cancers with a large number of accumulated genetic lesions would show only a small number of commonly affected pathways. It follows from this, that if mutation in a single gene were sufficient to affect a given pathway, even advanced cancers would show only a small number of commonly affected genes, the remainder of lesions being highly sporadic.
The microarray-based method we have just described can partially address these issues. We can readily identify loci in the genome that undergo amplification, deletion, and imbalanced breaks. Although there are many other possible mechanisms that alter critical genes, such as point mutations, balanced translocations, and possibly stable epigenetic changes, many if not most oncogenes and tumor suppressor genes will eventually be found in the types of lesions that we can readily detect. Moreover, if a region is commonly found altered in cancers, that region harbors a good candidate cancer gene. Therefore, the application of our method to a large series of cancers, and the comprehensive comparative analysis of such data, should reveal the existence and number of candidate cancer genes in cancers.
Sources of Cancer Genomes
We have demonstrated the application of our method to two types of sample: a tumor and a cancer cell line. There are advantages and problems associated with each type. Cancer cell lines are “universal” reagents. They are self-replenishing, and can be passed between investigators. There is always ample material for analysis, and they tend to be monoclonal. They are suitable for further functional analysis, whether by gene expression profiling, genetic manipulation to restore or block a suspected tumor suppressor gene or oncogene, or by tumorigenicity studies. Almost always there is no matched normal to control for scattered polymorphic variation, but as we have described above, this is not a serious limitation, as long as the unmatched normal can be characterized. The significant disadvantages of cell lines are that they can drift genetically, and they have undergone selection by virtue of their survival in tissue culture. There is a limited repertoire of such cell lines, and no correlations between clinical presentation and copy number can be made.
The direct analysis of tumor material offers many opportunities. There is a virtually unlimited source of different samples, and they can often be matched to the same normal, easing somewhat the analytical burden of interpretation. It is in principle possible to determine whether there are clinical parameters, such as survival and drug responsiveness, that correlate with specific gene amplification, deletions, and breakage, or overall patterns of genomic instability. These correlations may find utility in the treatment of patients. The disadvantages of tumor material are also clear. Tumors are always contaminated with stroma, can be oligoclonal, poorly preserved, and available in limiting amounts. Fortunately, our method seems to be highly sensitive, and does not require vast amounts of starting material. We routinely start from 50 ng of sample, which corresponds to ∼10,000 nuclei, and the method can be practiced with as few as 2000 nuclei or less. Either flow sorting or microdissection can enrich tumor purity, but amplifications and many deletions will be observable even with material that is only 50% tumor (reconstruction experiments; data not shown).
Technological Critique
Our method rests on three pillars: complexity reduction by representations, the human genome assembly, and oligonucleotide microarrays.
Because of the success of the human genome sequencing project, and the reproducibility of representations, we are able to design oligonucleotide probes that are complementary to a given representation, such as the BglII representations that we have used here. Because the human genome sequence is very reliable, at least locally, we are able to experimentally validate our computationally derived designs by exploiting the known restriction endonuclease sites in our fragments (see Fig. 1). In principle, we can thus calibrate every probe's performance. The detection of these ∼1800 predicted probes validates the ability of this method to detect and identify copy number fluctuations. There are ∼10% of the probes that are poor performers in the pin printed format. By calibrating the probes, performance can be accounted for during further analysis. Performance improves with the photoprint format because of the empirical selection of the oligonucleotides.
Of the 8000 probes predicted to hybridize to fragments not cleaved by HindIII (see Fig. 1), ∼16 appear to hybridize to BglII fragments that are in fact cleaved. We estimate that these 16 detect homozygous and heterozygous HindIII sites, in equal proportion. We attribute this to a divergence of about one nucleotide in 300 between our sample and the published human sequence, which could result from either polymorphism or sequencing errors. If this number were mainly caused by polymorphism, then roughly one in 30 BglII fragments would also be polymorphic. From other experiments, we estimate that the rate of BglII polymorphism between unrelated individuals is more on the order of one in 60, corresponding to a divergence from the published human sequence of 1 in 600. Because the public human sequence is reasonably well assembled, we automatically have associated map positions for every probe that are as accurate as the genome assembly. The algorithms we use for designing these probes are in part described here, and in part in Healy et al. (2003). Our approach allows us to design probes that have minimal cross-reaction to the remainder of the genome. Microarrays for any species, for example, mouse, can be built in short order once a reliably complete and assembled genome sequence is publicly available.
There are many advantages to an oligonucleotide microarray format. The composition of the microarray is precisely formulated, and hence entirely reproducible by others. The work presented here demonstrates the equivalence of measurements achieved by the printed and light directed microarray formats. Using printed arrays we can achieve densities of 30,000 probes per slide, and using in situ light-directed synthesis, we have achieved densities of 190,000, although only 85K data are illustrated here. The latter technique has many advantages over the printed array. Besides achieving higher density, the layout of probes and the choice of probes are flexible. Although the unit cost of printed arrays is presently below the costs of light-directed microarrays, with the latter there is no need for a large initial capital expenditure for the purchase of oligonucleotides.
Our method is dependent on representations. Without complexity reduction, which increases the concentration of DNA complementary to the probes, signal intensity from specific hybridization is too weak to measure above background. Dependence on representations is a mixed blessing. Representations use PCR both for the amplification of sample and complexity reduction. As a consequence, very little sample is required. However, PCR does introduce noise, and this requires that the test sample be compared with a control sample that is prepared exactly in parallel. We find that if the starting DNAs of test and control are of comparable quantity and quality, then subsequent parallel sample preparation, from PCR to labeling, is usually sufficient to give data of the type that is illustrated in this report.
There are a finite number of repeat-free 70-mer-long oligonucleotide probes in the genome that are useful for measuring BglII representations. We estimate that there are on the order of 120,000 of these scattered about the genome in a Poisson-like distribution, and the distribution of probes does not reflect the distribution of genes. At present we only array ∼85,000 probes. Although the average distance between these 85,000 probes is ∼30 kb, there are regions of the genome that are very poorly represented. We are therefore designing other types of representations, and other formats of probes, that will give us even higher coverage of the genome. In principle, any desired density of coverage is possible.
Data Interpretation
All array-based data require interpretation using statistical tools of varying sophistication. Ours is no exception, but our system is relatively unique. First, unlike cDNA expression profiling, there are clear theoretical expectations of copy number measurements. When comparing a test sample to a normal genome, there is a clear expectation of how normals, except for polymorphisms, will behave. Moreover, if the test sample is clonal, we expect probe ratios to be clustered, reflecting discrete integral copy numbers per cell. Second, because the restriction endonuclease profile of fragments is known, virtually all probes can be calibrated, and array performance can be very accurately modeled. Third, because the probes are ordered in the genome, and lesions are expected to be regional, with defined starts and stops, the expectation is that consecutive probe ratios within these regions will share a distribution. Thus, we have developed “segmentation” algorithms that are designed to parse the data into regions with similar distributions.
Our present segmentation algorithm requires a minimum of three probes to define a lesion, but clearly this is conservative. For example, when our tumor sample is compared with a matched normal, polymorphisms are controlled, and even a single probe with an elevated copy number in the tumor is likely to be meaningful. Other approaches to data analysis should be pursued, and we are attempting to integrate polymorphism data, probe calibration data, and probe intensity data into a more comprehensive model. Our present methods are not finished, but they are clearly already useful. We expect that the borders of regions can be drawn very sharply, most often to within a single probe, and this is confirmed in modeling experiments (data not shown).
We will report on our progress in statistical methods in subsequent publications. In the end, however, no statistical interpretation of a single experiment is certain, and only the accumulation of larger data sets and molecular confirmation can increase confidence in a conclusion.
Normal Polymorphic Variation
Scattered polymorphism is evident in comparison of normal individuals, and even in the comparison of a single individual in a “depletion” experiment (see Fig. 1). Most of these likely arise from single nucleotide polymorphisms in the human population. For example, loss of a BglII site may cause a fragment to be absent in a BglII representation. Such events can interfere in data interpretation in several ways. Except for the case of increased copy number in a matched tumor-normal, the ratio from a single probe outlier cannot be considered a somatic lesion, as it may represent a genetic polymorphism, with or without loss of heterozygosity. Similarly, the boundaries of a segment may not be accurately called if the bounding probe is complementary to a polymorphic fragment. Lastly, a string of probes that by chance are all complementary to polymorphic fragments may give rise to the appearance of a consistent lesion. Fortunately, the frequency of these polymorphisms is low, less than one fragment in 30, so most boundaries are not obscured, and runs of polymorphisms with the appearance of a lesion will occur rarely. Much of the informatic “damage” caused by polymorphisms can be contained, either by filtering out scattered outliers, or by accumulating data on normal genomes used for comparisons.
There is another type of “polymorphism” that we see, which for now we call “copy number” polymorphism. This type is much more interesting, and more pernicious, than scattered polymorphism, and it is documented in Figure 6. A series of regionally clustered probes may display a consistently altered ratio in the comparison of one normal sample against another. We see these regions in every normal-normal comparison that we have made, and many of these lesions appear in cancer-normal comparisons. In fact, some of these regions may be prone to genomic instability (see Fig. 6D). They vary in size from <100 kb to in excess of 1 Mb, and in most cases encompass genes. Creating a large database of normal-normal comparisons may mitigate the misinterpretation of these lesions as somatic events occurring in cancer, and this is something we intend to do.
Our present hypothesis is that these normal-normal variations are in fact copy number polymorphisms, genetic in origin, but this is by no means proven here, nor is it the only plausible hypothesis. For example, these variant regions might be caused by locally high sequence divergence, or the consequence of highly altered chromatin structure, affecting the yield of DNA during purification from nuclei. Additional experimentation is needed to resolve these questions, and work in progress strongly indicates that the majority of these normal variations are, indeed, alterations in the gene pool. If there is, in fact, widespread copy number variation in humans, such variation might well contribute to human traits, including disease susceptibility and resistance.
METHODS
Reagents
Oligonucleotides were synthesized by Illumina Inc. Human Cot-1 DNA (15279-011) and yeast tRNA (15401-029) were supplied by Invitrogen Inc. Restriction enzymes, ligase, and Klenow fragments (M0212M) were supplied by New England Biolabs. The Megaprime labeling kit, Cy3-conjugated dCTP, and Cy5-conjugated dCTP were supplied by Amersham-Pharmacia. Taq polymerase was supplied by Eppendorf. Centricon YM-30 filters were supplied by Amicon (42410), and formamide was supplied by Amresco (0606-500). Phenol:chloroform was supplied by Sigma (P2069). NimbleGen photoprint arrays were a gift from NimbleGen Systems Inc.
Representation
BglII representations, in general, were prepared as previously described (Lucito et al. 2003b). A major change is that amplification was carried out in an MJ Research Tetrad. Sixteen 250-μL tubes were used for amplification of the representation. The cycle conditions were 95°C for 1 min, 72°C for 3 min, for 25 cycles, followed by a 10-min extension at 72°C. The contents of the tubes were pooled when completed. Representations were cleaned by phenol:chloroform extraction, precipitated, resuspended, and the concentration determined. Representations depleted of specific fragments by restriction enzyme were prepared in the same manner with the following modification. After ligation of adaptor, the mixture was cleaned by phenol:chloroform extraction, precipitated, and resuspended. The ligated fragments were then digested with the second chosen enzyme. In the text, HindIII was used. This material was then used as template in the PCR reaction.
Probe Selection
We performed an in silico BglII digestion of the human genome by locating all BglII restriction sites within the present draft assembly and storing all sequences of BglII fragments that are between 200 and 1200 bp in length. Fragments were annotated with the counts of their substituent, overlapping 15-mers and 21-mers using the “mer-engine” constructed from the same draft assembly (see accompanying manuscript by Healy et al. 2003). For each fragment, the following attributes were determined for every substituent, overlapping 70-mer: maximum 21-mer count, arithmetic mean of 15-mer counts, percent GC content, the quantity of each base, and the longest run of any single base. All 70-mer probes that possess any of the following characteristics were eliminated: maximum 21-mer count >1, GC content <30% or >70%, a run of A/Ts >6 bases, a run of G/Cs >4 bases. From the remaining set of 70-mers, the one (or more) that has a GC/AT proportionality closest to that of the genome as a whole as well as a minimal mean 15-mer count were selected. As a final check for overall uniqueness, the optimal probes for each fragment were compared with the entire genome using BLAST (default parameters were used with the exception of filtration of low complexity sequence, which was not performed). Any probe found to have any degree of homology along 50% or more of its length to any sequence other than itself was eliminated.
Printed Arrays
We used the Cartesian PixSys 5500 (Genetic Microsystems) to array our probe collection onto slides. We are presently using a 4 × 4 pin configuration. The dimension of each printed array was roughly 2 cm2. Our arrays were printed on commercially prepared silanated slides (Corning ultraGAPS #40015). Pins used for the arrayer are from Majer Precision.
Labeling
DNA was labeled as described (Lucito et al. 2003a). Briefly, place DNA template (dissolved in TE at pH 8) in a 0.2-mL PCR tube. Add 10 μL of Primers from the Amersham-Pharmacia Megaprime labeling Kit and pipette up and down several times. Bring volume up to 100 μL with dH2O, and mix. Place tubes in Tetrad at 100°C for 5 min, then place on ice for 5 min and add 20 μL of labeling buffer from the Amersham-Pharmacia Megaprime labeling Kit, 10 μL of label (Cy3-dCTP or Cy5-dCTP), and 1 μL of NEB Klenow fragment. Place the tubes in a Tetrad and incubate at 37°C for 2 h. Combine the labeled samples (Cy3 and Cy5) into one Eppendorf tube and add 50 μL of 1 μg/μL human Cot 1 DNA, 10 μL of 10 mg/mL stock yeast tRNA, and 80 μL of Low TE (3 mM Tris at pH 7.4, 0.2 mM EDTA). Load all into a Centricon Filter and centrifuge for 10 min at 12,600 rcf. Discard flowthrough and wash with 450 μL of Low TE. Centrifuge at 12,600 rcf and repeat twice. Collect the labeled sample by inverting the centricon column into a new tube and centrifuging for 2 units at 12,600 rcf. Transfer labeled sample to a 200-μL PCR tube and adjust volume to 10 μL of Low TE.
Slide Preparation
Slides were prepared as in Lucito et al. (2003a) with the following changes. Prehybridization buffer for printed microarrays consisted of the following, 25% deionized formamide, 5× SSC, and 0.1% SDS. Pour into a coplin jar or other slide processing chamber and preheat to 61°C. UV cross-link DNA to slide (using a Strategene Statalinker, set Energy to 300 mJ, rotate slide 180°, keeping the slide in the same spot in the cross-linker, and repeat). NimbleGen photoprinted arrays do not require UV cross-linking. Wash slides in the following solutions: 2 min in 0.1% SDS, 2 min in milliQ H2O, 5 min in milliQ H2O that has boiled, and finally in ice cold 95% benzene-free EtOH. Dry slides by placing in a metal rack and spin at 75 rcf for 5 min. Printed microarray slides were incubated in the 61°C prehyb solution. After 2 h, wash slides in milliQ H2O for 10 sec. Dry slides by placing in a metal slide rack and spin for 5 min at 75 rcf. NimbleGen photoprinted arrays do not require prehybridization.
Hybridization
The hybridization solution for printed slides consisted of 25% formamide, 5× SSC, and 0.1% SDS. The hybridization solution for NimbleGen photoprinted arrays consisted of 50% formamide, 5× SSC, and 0.1%SDS. For each, 25 μL of hybridization solution was added to the 10 μL of labeled sample and mixed. Samples were denatured in an MJ Research Tetrad at 95°C for 5 min, and then incubated at 37°C for 30 min. Samples were spun down and pipetted onto a slide prepared with lifter slip and incubated in a hybridization oven such as the Boekel InSlide Out oven set at 58°C for printed arrays or 42°C for NimbleGen photoprinted arrays for 14 to 16 h. After hybridization, slides were washed as follows: brief wash in 0.2% SDS/0.2× SSC to remove the cover-slip, 1 min in 0.2% SDS/0.2× SSC, 30 sec in 0.2× SSC, and 30 sec in 0.05× SSC. Slides were dried as before by placing in a rack and spinning at 75 rcf for 5 min, and then scanned immediately. An Axon GenePix 4000B scanner was used setting the pixel size to 10 μm for printed arrays and 5 μm for NimbleGen photoprinted arrays. GenePix Pro 4.0 software was used for quantitation of intensity for the arrays. Array data were imported into S-PLUS for further analysis. Measured intensities without background subtraction were used to calculate ratios. Data were normalized using an intensity-based lowest curve fitting algorithm similar to that described in Yang et al. (2002). Data obtained from color reversal experiments were averaged and displayed as presented in the figures.
Acknowledgments
We thank Emile Nuwaysir and Todd Richmond of NimbleGen Systems Inc. for providing slides and support, and Masaaki Hamaguchi for critical comments on the manuscript. We also thank Joe Derisi and Michael Eisen for technical comments on the printing of oligonucleotides. Tumor samples were supplied by the Cooperative Human Tissue Network, which is funded by the National Cancer Institute. Other investigators may have received samples from these same tissues. This work was supported by grants awards to M.W. from the National Institutes of Health and NCI (5R01-CA78544; 1R21-CA81674; 5R33-CA81674-04); Tularik Inc.; 1 in 9: The Long Island Breast Cancer Action Coalition; Lillian Goldman and the Breast Cancer Research Foundation; The Miracle Foundation; The Marks Family Foundation; Babylon Breast Cancer Coalition; Elizabeth McFarland Group; and Long Islanders Against Breast Cancer. Support was granted to R.L. from the National Institutes of Health and NCI (K01 CA93634-01). M.W. is an American Cancer Society Research Professor.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
[The photoprint arrays were a kind gift of NimbleGen Systems Inc. and were fabricated to our design.]
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.1349003. Article published online before print in September 2003.
References
- Bartlett, J. and Mallon, E.C.T. 2003. The clinical evaluation of HER-2 status: Which test to use? J. Pathology 199: 418-423. [DOI] [PubMed] [Google Scholar]
- Chee, M., Yang, R., Hubbell, E., Berno, A., Huang, X.C., Stern, D., Winkler, J., Lockhard, D.J., Morris, M.S., and Fodor, S.P. 1996. Accessing genetic information with high-density DNA arrays. Science 274: 610-614. [DOI] [PubMed] [Google Scholar]
- DeRisi, J., Penland, L., Brown, P.O., Bittner, M.L., Meltzer, P.S., Ray, M., Chen, Y., Su, Y.A., and Trent, J.M. 1996. Use of a cDNA microarray to analyse gene expression patterns in human cancer. Nat. Genet. 14: 457-460. [DOI] [PubMed] [Google Scholar]
- Gemmill, R.M., West, J.D., Boldog, F., Tanaka, N., Robinson, L.J., Smith, D.I., Li, F., and Drabkin, H.A. 1998. The hereditary renal cell carcinoma 3;8 translocation fuses FHIT to a patched-related gene, TRC8. Proc. Natl. Acad. Sci. 95: 9572-9577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golub, T.R., Slonim, D.K., Tamayo, P., Huard, C., Gaasenbeek, M., Mesirov, J.P., Coller, H., Loh, M.L., Downing, J.R., Caligiuri, M.A., et al. 1999. Molecular classification of cancer: Class discovery and class prediction by gene expression monitoring. Science 286: 531-537. [DOI] [PubMed] [Google Scholar]
- Hamaguchi, M., Meth, J.L., von Klitzing, C., Wei, W., Esposito, D., Rodgers, L., Walsh, T., Welcsh, P., King, M.-C., and Wigler, M. H. 2002. DBC2, a candidate for a tumor suppressor gene involved in breast cancer. Proc. Natl. Acad. Sci. 99: 13647-13652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Healy, J., Thomas, E.E., Schwartz, J.T., and Wigler, M.H. 2003. Annotating large genomes with exact word matches. Genome Res. (this issue). [DOI] [PMC free article] [PubMed]
- Hyman, E., Kauraniemi, P., Hautaniemi, S., Wolf, M., Mousses, S., Rozenblum, E., Ringner, M., Sauter, G., Monni, O., Elkahloun, A., et al. 2002. Impact of DNA amplification on gene expression patterns in breast cancer. Cancer Res. 62: 6240-6245. [PubMed] [Google Scholar]
- Ilyin, G.P., Rialland, M., Pigeon, C., and Guguen-Guillouzo, C. 2000. cDNA cloning and expression analysis of new members of the mammalian F-box protein family. Genomics 67: 40-47. [DOI] [PubMed] [Google Scholar]
- Lander, E.S., Linton, L.M., Birren, B., Nusbaum, C., Zody, M.C., Baldwin, J., Devon, K., Dewar, K., Doyle, M., FitzHugh, W., et al. 2001. Initial sequencing and analysis of the human genome. Nature 409: 860-921. [DOI] [PubMed] [Google Scholar]
- Li, J., Yen, C., Liaw, D., Podsypanina, K., Bose, S., Wang, S.I., Puc, J., Miliaresis, C., Rodgers, L., McCombie, R., et al. 1997. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 275: 1943-1947. [DOI] [PubMed] [Google Scholar]
- Lin, M.Z. and Greenberg, M.E. 2000. Orchestral maneuvers in the axon: Trio and the control of axon guidance. Cell 101: 230-242. [DOI] [PubMed] [Google Scholar]
- Lisitsyn, N., Lisitsyn, N., and Wigler, M. 1993. Cloning the differences between two complex genomes. Science 258: 946-951. [DOI] [PubMed] [Google Scholar]
- Lucito, R., Nakimura, M., West, J.A., Han, Y., Chin, K., Jensen, K., McCombie, R., Gray, J.W., and Wigler, M. 1998. Genetic analysis using genomic representations. Proc. Natl. Acad. Sci. 95: 4487-4492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucito, R., West, J., Reiner, A., Alexander, J., Esposito, D., Mishra, B., Powers, S., Norton, L., and Wigler, M. 2000. Genetic alterations in cancer detected by hybridization to micro-arrays of genomic representations. Genome Res. 10: 1726-1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucito, R. and Wigler, M. 2003a. Preparation of Slides and Hybridization. In Microarray-based representational analysis of DNA copy number (eds. D. Bowtell and J. Sambrook), pp. 394-399. Cold Spring Harbor Press, Cold Spring Harbor, NY.
- Lucito, R. and Wigler, M. 2003b. Preparation of Target DNA. In Microarray-based representational analysis of DNA copy number (eds. D. Bowtell and J. Sambrook), pp. 386-393. Cold Spring Harbor Press, Cold Spring Harbor, NY.
- Mu, D., Chen, L., Zhang, X., See, L.-H., Koch, C.M., Yen, C., Tong, J.J., Spiegel, L., Nguyen, K.C.Q., Servoss, A., et al. 2003. Genomic amplification and oncogenic properties of the KCNK9 potassium channel gene. Cancer Cell 3: 297-302. [DOI] [PubMed] [Google Scholar]
- Nurnberg, P., Thiele, H., Chandler, D., Hohne, W., Cunningham, M.L., Ritter, H., Leschik, G., Uhlmann, K., Mischung, C., Harroop, K., et al. 2001. Heterozygous mutations in ANKH, the human ortholog of the mouse progressive ankylosis gene, result in craniometaphyseal dysplasia. Nat. Genet. 28: 37-41. [DOI] [PubMed] [Google Scholar]
- Olshen, A.B. and Venkatraman, E.S. 2002. Change-point analysis of array-based comparative genomic hybridization data. American Statistical Association, Alexandria, VA.
- Pinkel, D., Segraves, R., Sudar, D., Clark, S., Poole, I., Kowbel, D., Collins, C., Kuo, W. L., Chen, C., Zhai, Y., et al. 1998. High resolution analysis of DNA copy number variation using comparative genomic hybridization to microarrays. Nat. Genet. 20: 207-211. [DOI] [PubMed] [Google Scholar]
- Pollack, J.R., Perou, C.M., Alizadeh, A.A., Eisen, M.B., Pergamenschikov, A., Williams, C.F., Jeffrey, S.S., Botstein, D., and Brown, P.O. 1999. Genome-wide analysis of DNA copy-number changes using cDNA microarrays. Nat. Genet. 23: 41-46. [DOI] [PubMed] [Google Scholar]
- Sen, A. and Srivastava, M.S. 1975. On tests for detecting change in mean. Ann. Stat. 3: 98-108. [Google Scholar]
- Singh-Gasson, S., Green, R.D., Yue, Y., Nelson, C., Blattner, F., Sussman, M.R., and Cerrina, F. 1999. Maskless fabrication of light-directed oligonucleotide microarrays using a digital micromirror array. Nat. Biotech. 17: 974-978. [DOI] [PubMed] [Google Scholar]
- Snijders, A.M., Nowee, M.E., Fridlyand, J., Piek, J.M., Dorsman, J.C., Jain, A.N., Pinkel, D., van Diest, P.J., Verheijen, R.H., and Albertson, D.G. 2003. Genome-wide-array-based comparative genomic hybridization reveals genetic homogeneity and frequent copy number increases encompassing CCNE1 in Fallopian tube carcinoma. Oncogene 22: 4281-4286. [DOI] [PubMed] [Google Scholar]
- Tkachuk, D.C., Westbrook, C.A., Andreeff, M., Donlon, T.A., Cleary, M.L., Suryanarayan, K., Homge, M., Redner, A., Gray, J., and Pinkel, D. 1990. Detection of bvr-abl fusion in chronic myelogeneous leukemia by in situ hybridization. Science 250: 559-562. [DOI] [PubMed] [Google Scholar]
- Van't Veer, L.J., Dai, H., van de Vijver, M.J., He, Y.D., Hart, A.A.M., Mao, M., Peterse, H.L., van der Kooy, K., Marton, M.J., Witteveen, A.T., et al. 2002. Gene expression profiling predicts clinical outcome of breast cancer. Nature 415: 530-536. [DOI] [PubMed] [Google Scholar]
- Yang, Y.H., Dudoit, S., Luu, P., Lin, D.M., Peng, V., Ngai, J., and Speed, T.P. 2002. Normalization for cDNA microarray data: A robust composite method addressing single and multiple slide systematic variation. Nucleic Acids Res. 30: e15-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
WEB SITE REFERENCES
- http://roma.cshl.org/; ROMA.