

Abstract
Metabolic syndrome (MetS) affects more than 47 million people in the U.S. Even more alarming, MetS, once regarded as an “adult problem”, has become increasingly common in children. To date, most related research and intervention efforts have occurred in the adult medicine arena, with limited understanding of the root causes and lengthy latency of MetS. This review highlights new science on the early life origins of MetS, with a particular focus on exposure to two groups of environmental toxicants: endocrine disrupting chemicals (EDCs) and metals during the prenatal and early postnatal periods, and their specific effects and important differences in the development of MetS. It also summarizes available data on epigenetic effects, including the role of EDCs in the androgen/estrogen pathways. Emerging evidence supports the link between exposures to environmental toxicants during early life and the development of MetS later in life. Additional research is needed to address important research gaps in this area, including prospective birth cohort studies to delineate temporal and dose-response relationships, important differences in the effects of various environmental toxicants and their joint effects on MetS, as well as epigenetic mechanisms underlying the effects of specific toxicants such as EDCs and metals.
Keywords: diabetes, environmental toxicants, metabolic syndrome, obesity, prenatal exposure
Introduction
Metabolic syndrome (MetS) consists of a cluster of clinical conditions, including elevations in blood glucose levels and/or insulin resistance, lower levels of high density lipoprotein cholesterol (HDL-C), hypertriglyceridemia and hypertension, and abdominal obesity.1 MetS affects more than 47 million people in the U.S.2 Adults with MetS have a threefold higher risk of coronary heart disease and stroke and a five to six times higher risk of mortality.3 People with MetS are also at high-risk of type 2 diabetes.4 Even more alarming, MetS and its components, including obesity, diabetes, dyslipidemia and hypertension, once regarded as “adult problems”, have become increasingly common in children at younger and younger ages.
Additional research and public health efforts are needed to improve prevention of MetS. Most related research and intervention efforts have primarily occurred in the adult medicine arena, where there is a limited understanding of the root causes and lengthy latency of MetS. Emerging evidence supports the ‘fetal programming’ hypothesis, which highlights the importance of exposures that occur during the prenatal period in the development of metabolic disorders later in life (Fig 1).5,6 Observations that maternal malnutrition during pregnancy is associated with type 2 diabetes, cardiovascular disease and MetS in later life7,8 have led to the hypothesis that in utero determinants of birthweight may program the fetus for elevated risk of chronic disease in adulthood. Given the widespread production and use of synthetic chemicals and metals, in parallel with growing epidemics of obesity and type 2 diabetes, recent studies have also examined the potential detrimental effects of these chemicals on fetal development and health outcomes later in life. For instance, epidemiological studies found that offspring of mothers exposed to endocrine-disrupting chemicals (EDCs) had a higher body mass index (BMI) in adulthood compared with offspring whose mothers had no exposure at all.9 These findings clearly suggest that, in addition to any genetic transmission, exposure to environmental toxicants in utero and early life may lead to obese or diabetic offspring and contribute to the development of MetS. The purpose of this review is to discuss the early life origins of MetS, and in particular the role of maternal exposure to environmental toxicants including EDCs and metals during pregnancy and early childhood. Furthermore, this review also summarizes available data on the epigenetic effects of exposure to EDCs and metals, and the possible role of epigenetic mechanisms in offspring metabolic risk.
Figure 1.
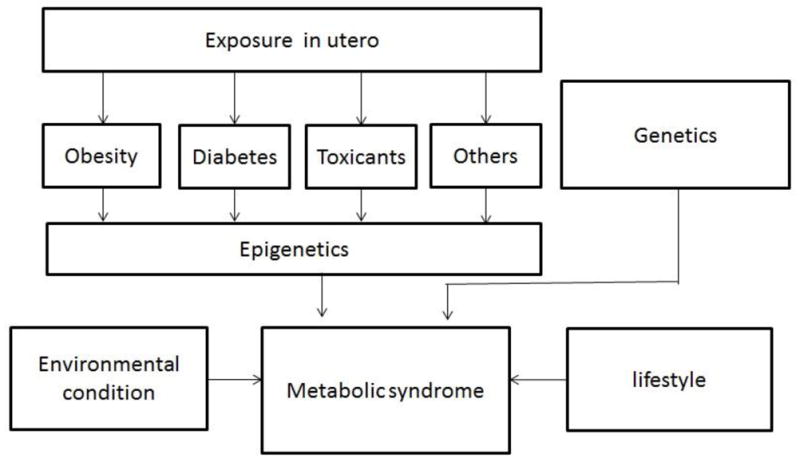
The potential mechanisms underlying the development of metabolic syndrome (MetS).
Exposure to Endocrine-Disrupting Chemicals
Endocrine-disrupting chemicals (EDCs) are defined as chemicals that act on the body’s hormonal systems, such as dichlorodiphenyltrichloroethane (DDT), dichlorodiphenyl dichloroethylene (DDE), polychlorinated biphenyls (PCBs), hexachlorobenzene (HCB), and bisphenol-A (BPA). Emerging evidence suggests that EDCs can affect human fetal development.10,11 A common characteristic of DDT, DDE, PCBs and HCB is their persistence in the environment longer than that required for their intended use. For this reason they have been grouped into the category of persistent organic pollutants.11 In contrast, BPA is categorized as a non-persistent compound.11 Humans may respond differently to EDCs exposure, particularly depending on exposure time, dose and sex. For example, high-dose exposure to EDCs can result in weight loss or growth restriction, whereas normal human environmental exposures at low-moderate levels can result in weight gain.12
Persistent organic pollutants
DDT was widely used throughout the U.S. and Europe from the 1940s to the 1970s to control insects on agricultural crops and those that carry diseases like malaria and typhus.13 Although DDT was banned in the U.S. in 1972, it is still used in some areas of the world primarily in malaria control programs. Animal studies have shown that a low dose of DDT readily passes through the placental barrier to the developing fetus where it induces embryotoxicity and fetotoxicity, but no teratogenicity.14 In humans, DDT and its metabolites (DDE) have been found in placental tissue and umbilical cord blood. Furthermore, maternal and newborn blood levels are generally similar,15 suggesting that DDT and DDE may also be able to cross the human blood-placenta barrier. PCBs have been used as lubricants and coolants in electrical appliances.16 HCB is a synthesized chlorinated monocyclic aromatic compound. HCB was widely used as a pesticide until 1965, but its use is no longer allowed in the U.S.17 PCBs and HCB have also been detected in cord blood, again suggesting their ability to transfer across the placenta to the fetus.18,19
Results of the impact of prenatal exposure to DDE/DDT, PCBs and HCB on offspring MetS have been inconsistent (Table 1). In the INMA-Sabadell study, a birth cohort recruited in 2004–2005 from Sabadell (Catalonia, Spain), prenatal DDE exposure was associated with an increased risk of rapid growth in the first 6 months of life in 518 infants born to normal weight mothers, as well as with elevated BMI at 14 months.20 The INMA-Menorca study, a birth cohort of 344 children recruited in 1997–1998 from the island of Menorca (Spain), evaluated the association between prenatal exposure to DDT, DDE and PCBs measured in cord blood with weight during childhood. Compared with the first tertile, the third tertile of PCBs exposure increased the risk of overweight in childhood by 1.7-fold, and the second tertile of DDE by 1.67-fold, but DDT was associated with overweight only in boys.21 However, in a study of male children from Chiapas, Mexico, there was no apparent association between high prenatal exposure to DDE and height or BMI in the first years of life.22 In a North Carolina cohort study, 594 children with prenatal and lactational exposures to PCBs or DDE, prenatal and early life PCBs exposures were associated with increased weight in girls at puberty.23 In terms of the relationship between early life exposures and obesity in adulthood, a study of DDE and PCBs levels in maternal blood during pregnancy in a US cohort from Michigan found that offspring who were exposed to higher prenatal levels of DDE had higher BMI at age 20–50 years, but found no association between prenatal exposure to PCBs and BMI in adulthood (Table 1).9 In terms of exposures to HCB, analysis of cord blood from 482 children from the INMA-Menorca (Spain) study showed that those in the higher HCB exposure group had a 2.5- and 3.0-fold increased risk of being overweight and obese, respectively.24
Table 1.
Some epidemiologic studies on the association between exposure to endocrine disrupting chemicals and metabolic syndrome components*
| Exposure | Population | Sample size |
Age at evaluation |
Exposure window |
Exposure Measure |
Exposure categories |
Outcome and definition |
Estimation of association |
Covariates | Reference |
|---|---|---|---|---|---|---|---|---|---|---|
| DDE | U.S. | 259 | 20–50y | In utero | Maternal blood DDE (μg/l) | BMI | mean±SD | none | Karmaus, 20089 | |
| 1st quintile | 25.8±4.9 | |||||||||
| 2nd | 26.0±5.8 | |||||||||
| 3rd | 28.1±7.7 | |||||||||
| 4th | 28.0±5.4 | |||||||||
| 5th | 28.8±8.1 | |||||||||
| Maternal blood PCBs (μg/l) | 1st quintile | 28.0±7.1 | ||||||||
| 2nd | 25.1±4.5 | |||||||||
| 3rd | 26.8±5.7 | |||||||||
| 4th | 30.0±8.6 | |||||||||
| 5th | 26.4±4.7 | |||||||||
| HCB | Spanish | 482 | 6.5y | In utero | Cord blood HCB (ng/ml) | Per log HCB ng/ml | Overweight: BMI>85th percentile | OR (95%CI): 1.69 (1.05–2.27) |
maternal age, height, pre-pregnancy overweight/obesity, education, parity, child’s sex, age | Smink, 200824 |
| DDE | Spanish | 657 | 6mo, 14mo | In utero | Maternal blood DDE (ng/g lipid) | per log ng/g lipid | Elevated BMI: >85th percentile, among normal-weight mother | RR (95%CI) 1.64 (1.24–2.18) |
age, maternal country of birth, maternal age, smoking, parental overweight; rapid growth | Mendez, 201120 |
| Maternal blood DDE (ng/g lipid) | Quartile | Rapid weight gain: change of BMI z-score >0.67, among normal-weight mother | ||||||||
| 2nd vs. 1st | 2.42 (1.25–4.67) | |||||||||
| 3rd vs. 1st | 2.47 (1.24–4.92) | |||||||||
| 4th vs. 1st | 2.47 (1.22–5.00) | |||||||||
| DDE, DDT, PCBs | Spanish | 344 | 6.5y | In utero | Cord blood DDE (ng/ml) | Tertile | Overweight: BMI>85th percentile | RR (95%CI) | birth weight, parity, maternal BMI, education, smoking, maternal age, breast-feeding | Valvi, 201221 |
| 2nd vs. 1st | 1.67 (1.10–2.55) | |||||||||
| 3rd vs. 1st | 1.28 (0.81–2.03) | |||||||||
| Cord blood DDT (ng/ml) | 2nd vs. 1st | 1.19 (0.76–1.87) | ||||||||
| 3rd vs. 1st | 1.17 (0.73–1.88) | |||||||||
| Cord blood PCBs (ng/ml) | 2nd vs. 1st | 0.97 (0.58–1.62) | ||||||||
| 3rd vs. 1st | 1.70 (1.09–2.64) | |||||||||
| DDE | Mexico | 788 | 14–22mo | In utero | Maternal serum DDE (μg/g lipid) | ≤3.0 (reference) | BMI SDS | β (95%CI) | Maternal BMI, smoking, education, rural residence, gestational age | Cupul-Uicab, 201022 |
| 3.01–6.00 | −0.11 (−0.29,0.07) | |||||||||
| 6.01–9.00 | −0.18 (−0.45, 0.08) | |||||||||
| >9.00 | 0.07 (−0.15,0.30) | |||||||||
| PCBs, DDE | Denmark | 656 | 5y and 7y | In utero, early life | Maternal blood PCBs (μg/g lipid) | PCBs quartile | BMI at 7y girl with maternal obesity | β (95%CI) | Maternal age, parity | Tang-Peronard, 201325 |
| 2nd vs. 1st | 0.76 (−0.63,2.15) | |||||||||
| 3rd vs. 1st | 0.66 (−0.98,2.29) | |||||||||
| 4th vs. 1st | 2.07 (0.59,3.55) | |||||||||
| 2nd vs. 1st | Change in BMI from 5y to 7y | 0.28 (−0.49,1.04) | ||||||||
| 3rd vs. 1st | 0.56 (−0.34,1.46) | |||||||||
| 4th vs. 1st | 1.23 (0.42,2.05) | |||||||||
| Maternal blood DDE (μg/g lipid) | DDE quartile | BMI at 7y girl with maternal obesity | Maternal age, parity | |||||||
| 2nd vs. 1st | −0.06 (−1.49,2.38) | |||||||||
| 3rd vs. 1st | −0.09 (−1.91,1.74) | |||||||||
| 4th vs. 1st | 1.29 (−0.24,2.81) | |||||||||
| 2nd vs. 1st | Change in BMI from 5y to 7y | 0.51 (−0.26,1.29) | ||||||||
| 3rd vs. 1st | 0.14 (0.82,1.10) | |||||||||
| 4th vs. 1st | 1.11 (0.30,1.92) | |||||||||
| PCBs | U.S. | 399 | At birth and 5 y | In utero | Maternal serum PCBs (ng/g lipid) | PCB percentile: 90th vs. 10th |
Birthweight: | β (se) | maternal age, BMI, height, parity, smoking, race, hypertension or preeclampsia, prenatal care, medications associated with intrauterine growth restriction, child’s sex. | Hertz-Picciotto, 200593 |
| Boy | −268 (101) | |||||||||
| Girl | −26 (73) | |||||||||
| Birth length: | ||||||||||
| Boy | −0.81 (0.55) | |||||||||
| girl | −0.48 (0.52) | |||||||||
| Standardized height at 5y: | ||||||||||
| Boy | 2.6 (1.7) | |||||||||
| Girl | 4.1 (2.1) | |||||||||
| DDE, PCBs | Belgium | 138 | 1–3y | In utero | Cord blood DDE (ng/g lipid) | Per ng/g lipid | BMI SDS | β (se) | Birth weight | Verhulst, 200927 |
| DDE | −0.002 (0.001) | |||||||||
| DDE×maternal smoking ever | −0.003 (0.001) | |||||||||
| DDE×maternal smoking×child’s age | 0.001 (0.001) | |||||||||
| Cord blood PCBs (ng/g lipid) | Per ng/g lipid PCBs | 0.003 (0.001) | Birth weigth, maternal smoking, maternal BMI height | |||||||
| PCBs, DDE | U.S. | 594 | 14y | In utero, early life | maternal blood PCB ppm fat | 0–1ppm | Self-reported weight (kg) | 51.6±1.9 | Gladen, 200023 | |
| 1–2 | 53.0±0.6 | |||||||||
| 2–3 | Girl | 52.9±0.9 | ||||||||
| >3 | 57.0±1.6 | |||||||||
| 0–1ppm | Boy | 59.6±2.2 | ||||||||
| 1–2 | 56.9±0.7 | |||||||||
| 2–3 | 56.8±1.1 | |||||||||
| >3 | 56.1±1.6 | |||||||||
| Maternal blood DDE (ppm fat) | 0–1ppm | Self-reported weight (kg) | 54.2±2.1 | |||||||
| 1–2 | 52.5±0.8 | |||||||||
| 2–3 | Girl | 54.0±0.8 | ||||||||
| 3–4 | 53.3±1.1 | |||||||||
| >4 | 51.5±1.4 | |||||||||
| 0–1ppm | Boy | 53.7±2.0 | ||||||||
| 1–2 | 5.61±1.0 | |||||||||
| 2–3 | 57.4±1.0 | |||||||||
| 3–4 | 55.6±1.2 | |||||||||
| >4 | 60.6±1.4 |
BMI, body mass index; DDT, dichlorodiphenyltrichloroethane; DDE, dichlorodiphenyl dichloroethylene; PCBs, polychlorinated biphenyls; HCB, hexachlorobenzene
Some, although limited, studies have evaluated the role of factors such as gender, maternal obesity and maternal smoking in modifying the association between EDCs and later obesity. To evaluate the modification effects of sex and maternal obesity on the association between prenatal exposure to EDCs and later obesity, Tang-Peronard et al. recruited 656 pregnant women from Denmark and measured PCBs and DDE levels in maternal serum and breast milk.25 Researchers found a significant gender difference between exposure to DDE and PCBs and BMI at age 5 and 7 years. In girls with overweight mothers, PCBs levels were associated with an increased BMI at age 7 years, and PCBs and DDE levels were associated with an increased change in BMI from 5 to 7 years of age and waist circumference at age 7. No association was found for girls with normal-weight mothers or in boys, suggesting that maternal obesity and sex are important mediators. In addition, substantial epidemiologic and toxicologic evidence, and more than 80 studies in human populations and almost 20 animal studies also support a positive association between maternal smoking and increased risk of obesity or overweight later in life.26 In a birth cohort from Belgium, maternal smoking enhanced the relationship between prenatal exposure to DDE and BMI at age 3 years, while prenatal exposure to PCBs was associated with an increase in BMI in early childhood between 1 and 3 years of age independent of maternal smoking.27 To date, most such studies have been conducted outside of the U.S. It will be important to confirm if the associations found in other populations are relevant to a U.S. population, who is typically exposed to lower levels of EDCs, and to what degree infant gender and maternal characteristics (such as BMI and smoking) modify the association between EDCs and offspring obesity, diabetes and MetS.
Bisphenol A
BPA is an endocrine-disrupting chemical used in the production of plastic food and beverage containers, leading to ubiquitous low-dose human exposure. Available data suggest widespread human exposure to BPA.28–33 Both U.S. children and pregnant women have shown nearly universal exposure to BPA.32,33 The 2005–2006 National Health and Nutrition Examination Survey (NHANES) urinary BPA data showed that median daily intake for the overall population is approximately 34 ng/kg-day.34 Importantly, BPA is rapidly absorbed and can easily cross the placental barrier to the fetus.35–38 BPA may also accumulate in the fetus37 and is measurable in cord blood,36,37,39,40 amniotic fluid,35,41 meconium and placental tissues.37 BPA is usually metabolized via the Uridine 5-diphospho-glucuronosyltransferase (UGT) system to BPA glucuronide, which does not bind to the estrogen receptor.42 However, the role of BPA glucuronidation in protecting the fetus and infant from BPA toxicity during early life is unclear.43 Relatively few epidemiological studies have examined the relationship between prenatal exposure to BPA and MetS in later life. However, animal studies provide important evidence. In a mice model, pregnant animals were treated with either vehicle or BPA during days 9–16 of gestation. At 6 months of age, male offspring exposed to BPA in utero had reduced glucose tolerance and increased insulin resistance.44 In another animal study, pregnant Wistar rats were exposed to different levels of BPA, and offspring were fed a normal diet or a high-fat diet after weaning. On a normal diet, perinatal exposure to a modest dose of BPA resulted in increased body weight, elevated serum insulin, and impaired glucose tolerance in adult offspring. Meanwhile, on a high-fat diet, such detrimental effects were accelerated and exacerbated. Furthermore, severe MetS, including obesity, dyslipidemia, hyperleptindemia, hyperglycemia, hyperinsulinemia, and glucose intolerance, was observed in high-fat-fed offspring perinatally exposed to BPA.45 Taken together, these results support the hypothesis that the fetus is highly vulnerable to exposure to environmental chemicals, resulting in a metabolic profile that shows increased risk for and susceptibility to type 2 diabetes and MetS later in life. Most studies to date were conducted in animals. Human research is needed to evaluate the role of prenatal BPA exposure and MetS in offspring.
Exposure to Metals
Metals have drawn great attention because of their widespread exposure worldwide, trans-placental and trans-breast milk passage, evidence of fetotoxicity, multi-organ adverse effects, and ability to interact with the genome and the epigenome.46,47 Our results in the Boston Birth Cohort showed a high degree of maternal to fetal transfer for mercury, lead and selenium, and that their transfer was a major source of early life exposure to these metals.48 To date, most human studies on the association of metals with obesity, diabetes and MetS have been conducted in adults. A recent systematic review of the epidemiologic evidence concluded that the available evidence is suggestive of but not sufficient for a relationship between arsenic and diabetes, and is insufficient for mercury.49
The primary source of exposure to inorganic arsenic is through drinking water contaminated by natural mineral deposits or improperly disposed chemicals. In addition, rice represents a major dietary source of inorganic arsenic both for adults and children.50 In a cross-sectional study of 788 adults aged 20 years or older who participated in the 2003–2004 National Health and Nutrition Examination Survey (NHANES) and had urine arsenic determinations, the odds ratios for type 2 diabetes was 3.58-fold (95% CI, 1.18–10.83) higher in participants within the 80th percentile of total arsenic than subjects within the 20th percentile.51 In a cross-sectional study from Wisconsin, subjects who drank water with high levels of arsenic had higher risk of hypertension than those with low levels.52 In NHANES 2003–2008, however, total arsenic and dimethylarsinate (DMA) were not associated with hypertension or blood pressure levels, although a weak association between DMA and hypertension could not be ruled out.53 In a study of 660 Taiwan residents, hair arsenic levels were associated with the risk of MetS, and with increased levels of plasma glucose, lipids, and blood pressure.54 Exposure to arsenic in utero has been related to an increased risk of cancer and cardiovascular disease both experimentally55 and in human populations.56 Arsenic also has been associated with epigenetic effects.57 Thus, it is important to evaluate the effect of early life arsenic exposures in the development of MetS including its individual components.
Regarding mercury, recently conducted research is evaluating its relationship with diabetes in adult populations. A large prospective study in young-middle aged men and women from different communities in the US found that toenail mercury levels were associated with increased diabetes risk.58 In a large cross-sectional study including 6213 adults in Korea, results showed a significant correlation between serum mercury concentrations and hypertension in both men (r=0.193, P < 0.001) and women (r =0.145, P <0.001).59 In two recent prospective cohort studies of health professionals in the U.S., however, toenail mercury level was not associated with diabetes risk in either men or women.60 While selenium has been used as an antioxidant supplement, large cross-sectional studies and interventional trials revealed that high selenium status was associated with an increased risk of diabetes.61,62 In a large prospective study in Italy, where the women were followed up a median of 16 years, dietary selenium intake was measured using a semi-quantitative food-frequency questionnaire at the baseline examination (1987–1992) and categorized into quintiles.63 The risk of type 2 diabetes in the highest quintile of selenium intake increased 2.39-fold (95% CI: 1.32, 4.32) (Table 2). Lead levels are an established risk factor of hypertension and may be related to additional metabolic abnormalities.64 A large study in Korea evaluated the joint effect of exposure to lead, mercury and cadmium as measured in blood. Serum lead levels, but not mercury and cadmium, were associated with a higher prevalence of MetS. Moreover, the sum of heavy metals exposures was associated with hypertension and elevated triglycerides.65 For cadmium, the epidemiologic evidence does not seem to support an association with diabetes.49
Table 2.
Some epidemiologic studies on the association between heavy metals and metabolic syndrome*
| Exposure | Study Design | Study Population |
Sample Size |
Age Range (years) |
Outcome definition |
Exposure Assessment |
Exposure categories |
Estimation of Associations |
Covariates | References |
|---|---|---|---|---|---|---|---|---|---|---|
| Arsenic | Cross sectional | Serbia | 746,202 | Around 60 | Type 2 diabetes: FPG≥7.8mmol.l, OGTT≥11.1mmol/l, National Registry of Type 2 Diabetes | Drinking water arsenic (μg/l) | Unexposed vs. exposed | OR (95% CI)) | none | Jovanovic, 201394 |
| Men: 1.52 (1.30–1.77) | ||||||||||
| Women:1.71 (1.49–1.97 | ||||||||||
| Arsenic | Cross sectional | U.S. | 788 | >20 | Type 2 diabetes: FPG≥126mg/dl, self- reported physician diagnosis or use of insulin or oral hypoglycemic medication | Urine arsenic (μg/l) | 80th vs 20th percentiles | OR (95% CI) | sex, age, race and ethnicity, urine creatinine, education, body mass index, serum cotinine, use of antihypertensive medication, urine arsenobetaine, blood mercury | Navas-Acien, 200851 |
| 3.58 (1.18–10.83) | ||||||||||
| Arsenic | Cross sectional | U.S. | 1185 | >20 | Hypertension: Self-reposted | Drinking water arsenic (μg/l) | >10μg/l vs | OR (95%CI) | gender, age, smoking status, and body mass index | Zierold, 200452 |
| <2μg/l | 1.68 (1.13, 2.49) | |||||||||
| Arsenic | Cross sectional | Taiwan | 660 | 35–64 | MetS: with 3 or more following factors: FPG>110mg/dl, TG>150mg/dl, HDL>40mg/dl (men) 50mg/dl (women) SBP>130mmHg, DBP>85mmHg, BMI>27kg/m2 (men) 25 (women) | Hair arsenic (μg/g) | Tertile | OR (95% CI) | Gender, work, alcohol, betel nut, smoking, and groundwater use | Wang, 200754 |
| 2nd vs. 1st | 2.54 (1.20–5.39) | |||||||||
| 3rd vs. 1st | 2.35 (1.02–5.43) | |||||||||
| Mercury | Longitudinal | U.S. | 3,875 | 20–32 | Type 2 diabetes: FPG≥7.8mmol.l, OGTT≥11.1mmol/l, HbA1C≥6.5%, use of antidiabetes medication | Toenail mercury (μg/g) | Quintile | HR (95%CI) | age, sex, ethnicity, study center, BMI, education, family history of diabetes, smoking, alcohol, physical activity, LCn3PUFAs, magnesium, toenail selenium | He, 201395 |
| 2nd vs. 1st | 1.05 (0.72–1.53) | |||||||||
| 3rd vs. 1st | 1.23 (0.85–1.78) | |||||||||
| 4th vs. 1st | 1.18 (0.79–1.77) | |||||||||
| 5th vs. 1st | 1.65 (1.07–2.56) | |||||||||
| Mercury | Cross sectional | Korea | 6213 | >20 | Hypertension: self-reported or use of hypertension medication | Serum mercury (nmol/l) | Tertile | p for trend<0.01 | none | Chio, 201359 |
| Selenium | Longitudinal | Italy | 7,182 | 34–70 | Type 2 diabetes: self-reported | Food frequency questionnaire | Quintile | OR (95%CI) | age, education, menopausal, BMI, smoking, alcohol, energy intake, saturated/polyunsaturated fatty acids ratio, animal proteins, total carbohydrates, weight change | Stranges, 201063 |
| 2nd vs. 1st | 1.42 (0.87–2.34) | |||||||||
| 3rd vs. 1st | 1.43 (0.86–2.38) | |||||||||
| 4th vs. 1st | 1.65 (0.98–2.78) | |||||||||
| 5th vs. 1st | 2.39 (1.32–4.32) | |||||||||
| Lead, mercury, cadmium | Cross sectional | Korea | 3850 | >20 | Mets: at least three of the following: (1) SBP> 130 mmHg, DBP> 85 mmHg, use of blood pressure medication; (2) HDL<40mg/dL (men) and 50 mg/dL (women); (3) TG>150 mmHg or use of antidyslipidemia medication; (4) FPG>100 mg/dL or use of antidiabetic medication; (5) WC>90 cm (men) and >85 cm (women). | Serum lead (μg/dl) | Quartile | OR (95%CI) | age, sex, region, smoking, alcohol, regular exercise, BMI | Moon, 201365 |
| 2nd vs. 1st | 0.84 (0.62–1.13) | |||||||||
| 3rd vs. 1st | 1.21 (0.90–1.62) | |||||||||
| 4th vs. 1st | 1.07 (0.79–1.45) | |||||||||
| Serum mercury (μg/l) | Quartile | |||||||||
| 2nd vs. 1st | 1.24 (0.94–1.64) | |||||||||
| 3rd vs. 1st | 1.08 (0.82–1.42) | |||||||||
| 4th vs. 1st | 1.13 (0.86–1.49) | |||||||||
| Serum cadmium (μg/l) | Quartile | |||||||||
| 2nd vs. 1st | 0.71 (0.52–0.97) | |||||||||
| 3rd vs. 1st | 0.82 (0.64–1.05) | |||||||||
| 4th vs. 1st | 0.92 (0.73–1.14) |
OR, odds ratio; HR, hazard ratio; CI, confidence interval; FPG, fasting plasma glucose; TG, triglycerides; HDL, high density lipoprotein; SBP, systolic blood pressure; DBP, diastolic blood pressure; BMI, body mass index; LCn3PUFAs, long-chain n-3 polyunsaturated fatty acids; WC, waist circumference; OGTT, oral glucose tolerance test; MetS, metabolic syndrome
There is a particular lack of prospective birth cohort studies on the metabolic consequences of exposure to metals, including arsenic, mercury, selenium and lead in utero and in early childhood. Given the available evidence on the role of some metals in diabetes and MetS in adults, the recognition of early life as a vulnerable period for environmental exposures and epigenetic alterations and for adipose tissue development and metabolic programming, it is important to evaluate the role of early metal exposure and subsequent risk of MetS.
Epigenetic Mechanisms
Epigenetic mechanisms may be the critical biological links between genetic vulnerability, in utero exposures and the development of MetS via the regulation of gene expression through DNA methylation, histone modifications, chromatin remodeling, and/or regulatory feedback by way of microRNAs. Epigenetic alterations are heritable and reversible changes in gene expression with no change in the underlying DNA code, and are believed to regulate cell differentiation and organogenesis and be a key molecular process underlying human responses to environment and gene -environment interactions.66 Some studies have shown that prenatal conditions, such as fetal growth restriction as well as nutrient restriction, can epigenetically modify gene expression by altering the DNA methylation level in promoter regions.67,68 Our work in the Boston Birth Cohort demonstrated considerable individual variation in DNA methylation, which is detectable at birth and dynamic in the first 2 years, and offered an epigenetic insight into this trans-generational effect.69 We also found that maternal obesity can alter offspring DNA methylation in multiple genes that may have important implications for future chronic diseases.70 These findings highlight the presence of in utero mechanisms that are available to respond to nutritional, hormonal, or other metabolic cues by possibly altering the timing and direction of methylation events during fetal development.
Epigenetic changes provide a ‘memory’ of developmental plastic responses to the early environment and are central to the development of phenotypes and their stability throughout the life course. There is a growing recognition that epigenetic mechanisms may contribute to MetS.71–73 Some studies have tested epigenetic changes as a result of exposure to unfavorable in utero environments. For example, Ruchat et al. measured DNA methylation at > 485,000 CpG sites in placental tissue and cord blood and found that 3,271 and 3,758 genes in the placenta and cord blood, respectively, were differentially methylated between samples exposed or not to gestational diabetes. Among the differentially methylated genes, 326 in placental tissue and 117 in cord blood were also associated with newborn weight.74 Another similar study showed that increased methylation of the Pygopus Homolog 1(PYGO1) and Ceroid-lipofuscinosis, neuronal 8 (epilepsy, progressive with mental retardation) (CLN8) genes had a relative mediation effect on the impact of exposure to maternal diabetes in utero on vascular adhesion molecule-1 (VCAM-1) levels in the offspring.75
Increasing evidence provides support for the epigenetic effects of environmental chemicals in the development of MetS. For instance, a recent study found that PCBs activate androgen receptor transcriptional activity by inducing Jarid1b, a histone demethylase that catalyzes the removal of trimethylation of lysine 4 on histone H3.76 A similar in vitro experiment showed that BPA, HCB and PCBs induce a modest decrease in global DNA methylation.77 Animal study has also provided evidence that early-life exposure to PCBs modifies the epigenome in the offspring liver, via inducing androgen receptor transcriptional activity.78 Animal studies also have supported that exposure to EDCs induce epigenetic alteration. For example, in utero exposure to BPA is associated with decreased methylation in the promoter and intron of Hoxa10 in CD-1 mice, which has been shown to persist after birth. BPA may exert its function partly through epigenetic alteration of Estrogen Response Element (ERE) sensitivity to estrogen.79 In addition, plasma DDT and DDE were found to be inversely associated with global methylation levels.80,81
One in vitro study showed gene-specific hypermethylation in a mouse cell culture with mercury levels equal to human maternal and cord blood samples.82 A study in women undergoing oocyte retrieval showed a relationship between blood mercury concentration and gene-specific hypermethylation.83 Specifically, methylation of the Glutathione S-Transferase Mu 1(GSTM)1/5 promoter was increased among women with higher mercury exposure. In a cohort of Bangladesh residents, serum selenium levels were inversely related to methylation of leukocyte DNA.84 In another study, maternal lead exposure was found to correlate with diminished Alu and long interspersed nuclear element-1(LINE-1) methylation in umbilical cord blood.85 Furthermore, the Hanna et al. study also showed a relationship between blood lead and gene-specific hypomethylation.83 They detected reduced methylation in the Collagen, Type I, Alpha 2 (COL1A2) promoter in women with higher exposure to lead. Interestingly, a mouse study found that pre- and post-natal lead exposure resulted in a shift in the expression of hundreds of genes.86 Based on this expanding list of studies, there is growing critical recognition that epigenetic mechanisms may contribute to complex human diseases87 including MetS.88 Taken together, these findings suggest that specific epigenetic modifications during key developmental periods may influence metabolic phenotypes in later life.
The Need for Future Research
Though we have learned to treat MetS using increasingly sophisticated technologies and agents, we have not been able to cure or prevent MetS. Previous research has been confined by disciplinary boundaries and specific age groups. Often, there is a disjoint that exists between our understanding of what comprises women’s health, child health and adult health. To date, while most related research and intervention efforts continue to primarily occur within the adult medicine arena, there is still a limited understanding of the root causes and lengthy latency of MetS. The future direction for medicine and public health is to use precise molecular knowledge to detect disease at the preclinical stage, and intervene before clinical disease occurs. By doing so, we will not only achieve preemptive prevention, but we will also reduce disease burden, improve population health and quality of life, and minimize health care costs by an order of magnitude.89 In order to realize this new paradigm, there is a critical need to deepen our understanding of the early life origins and natural history of disease, explore new paradigms using a life course perspective, and put humankind on a long-range strategic and economically sustainable path to better health.
The prenatal period is particularly vulnerable to environmental exposures because of rapid cellular differentiation and development. Developmental programming and the establishment of the epigenome (regulation of gene expression) is most sensitive during the prenatal period.69 Advances in a wide range of biological, behavioral, and social sciences are expanding our understanding of how early environmental influences and genetic predispositions affect adult diseases. The new science suggests that epigenetic mechanisms could be an important interface that links genetic factors, environmental factors, and phenotypic expression.90,91 Yet, further studies are needed to determine whether the measurements of epigenetic marks in early life can be used as biomarkers to identify individuals who have experienced environmental perturbations during development, and thus who are more likely to develop obesity and metabolic disease in later life.
Longitudinal studies, in particular, birth cohort studies, are lacking, yet critically needed to identify the links among early life factors, genetics, and the development of metabolic disorders in children, adolescents, and adults. The value of birth cohorts has received great attention from the scientific community.92 As compared to cross-sectional or case-control studies, birth cohorts can be used for testing a wide range of hypotheses on risk factors and outcomes, and exploring temporal, dose-response and causal relationships to gain important insights into the epigenetic mechanisms by which environmental exposures and genetic predisposition interact to affect growth, development and disease over the life course and across generations.
Conclusion
There is limited and inconsistent evidence regarding the relationship between early life exposure to specific environmental toxicants and MetS in later life. Available studies on prenatal exposure to EDCs and their association with later obesity and MetS had small sample sizes and were performed mostly in non-U.S. populations. The study findings were often inconsistent, which may be due to variations in length of exposure, dose, sex and other confounders (e.g. exposure to other un-measured toxicants). The data is particularly lacking on prenatal exposure to metals and the development of MetS later in life. Even less data is available on early life exposure to toxicants and epigenetics alterations in the offspring that may regulate gene expression throughout the life course. Given that an individual is often exposed to a mixture of environmental toxicants, there is a particular need for prospective birth cohort studies on the metabolic consequences of multiple exposures, including EDCs and metals (e.g. arsenic, mercury, selenium and lead), in utero and in early childhood. Prospective birth cohort studies could help to delineate temporal and dose-response relationships, evaluate differences in the effects of various environmental toxicants and their joint effect on MetS, and identify epigenetic mechanisms underlying the trans-generational effects of specific toxicants such as EDCs and metals.
Acknowledgments
Dr. Xiaobin Wang is supported in part by the National Institutes of Health (NIH) grants (R01 HD041702 and R21HD066471).
Footnotes
Compliance with Ethics Guidelines
Conflict of Interest
Guoying Wang, Zhu Chen, Tami Bartell, and Xiaobin Wang declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
| Name | Mailing address | Phone | Fax | |
|---|---|---|---|---|
| Guoying Wang | 615 N. Wolfe St, Rm#E4001, Baltimore, MD 20215 | guwang@jhsph.edu | 4105028973 | 4105025831 |
| Zhu Chen | 615 N. Wolfe St, Rm#E4006, Baltimore, MD 20215 | zchen@jhsph.edu | 4105028946 | 4105025831 |
| Tami Bartell | 225 E. Chicago Avenue, Box 157, Chicago, Illinois 60611 | tbartell@luriechildrens.org | 3122277087 | 3122279523 |
| Xiaobin Wang | 615 N. Wolfe St, Rm#E4032, Baltimore, MD 20215 | xiwang@jhsph.edu | 4109555824 | 4105025831 |
References
Papers of particular interest, published recently, have been highlighted as:
•Of importance
••Of major importance
- 1.Alberti KG, Zimmet P, Shaw J. The metabolic syndrome--a new worldwide definition. Lancet. 2005;366(9491):1059–1062. doi: 10.1016/S0140-6736(05)67402-8. [DOI] [PubMed] [Google Scholar]
- 2.Ford ES, Giles WH, Dietz WH. Prevalence of the metabolic syndrome among US adults: findings from the third National Health and Nutrition Examination Survey. Jama. 2002;287(3):356–359. doi: 10.1001/jama.287.3.356. [DOI] [PubMed] [Google Scholar]
- 3.Wassink AM, van der Graaf Y, Olijhoek JK, Visseren FL. Metabolic syndrome and the risk of new vascular events and all-cause mortality in patients with coronary artery disease, cerebrovascular disease, peripheral arterial disease or abdominal aortic aneurysm. Eur Heart J. 2008;29(2):213–223. doi: 10.1093/eurheartj/ehm582. [DOI] [PubMed] [Google Scholar]
- 4.Eckel RH, Grundy SM, Zimmet PZ. The metabolic syndrome. Lancet. 2005;365(9468):1415–1428. doi: 10.1016/S0140-6736(05)66378-7. [DOI] [PubMed] [Google Scholar]
- 5.Teslovich TM, Musunuru K, Smith AV, et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature. 2010;466(7307):707–713. doi: 10.1038/nature09270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6•.Boney CM, Verma A, Tucker R, Vohr BR. Metabolic syndrome in childhood: association with birth weight, maternal obesity, and gestational diabetes mellitus. Pediatrics. 2005;115(3):e290–296. doi: 10.1542/peds.2004-1808. This study examined the associaton of maternal obesity and gestational diabetes and metabolic syndrome in children. [DOI] [PubMed] [Google Scholar]
- 7.Barker DJ, Winter PD, Osmond C, Margetts B, Simmonds SJ. Weight in infancy and death from ischaemic heart disease. Lancet. 1989;2(8663):577–580. doi: 10.1016/s0140-6736(89)90710-1. [DOI] [PubMed] [Google Scholar]
- 8.Barker DJ, Gluckman PD, Godfrey KM, Harding JE, Owens JA, Robinson JS. Fetal nutrition and cardiovascular disease in adult life. Lancet. 1993;341(8850):938–941. doi: 10.1016/0140-6736(93)91224-a. [DOI] [PubMed] [Google Scholar]
- 9.Karmaus W, Osuch JR, Eneli I, et al. Maternal levels of dichlorodiphenyl-dichloroethylene (DDE) may increase weight and body mass index in adult female offspring. Occup Environ Med. 2009;66(3):143–149. doi: 10.1136/oem.2008.041921. [DOI] [PubMed] [Google Scholar]
- 10.Lee BE, Park H, Hong YC, et al. Prenatal bisphenol A and birth outcomes: MOCEH (Mothers and Children’s Environmental Health) study. Int J Hyg Environ Health. 2013 doi: 10.1016/j.ijheh.2013.07.005. [DOI] [PubMed] [Google Scholar]
- 11.DiVall SA. The influence of endocrine disruptors on growth and development of children. Curr Opin Endocrinol Diabetes Obes. 2013;20(1):50–55. doi: 10.1097/MED.0b013e32835b7ee6. [DOI] [PubMed] [Google Scholar]
- 12.Grun F, Blumberg B. Endocrine disrupters as obesogens. Mol Cell Endocrinol. 2009;304(1–2):19–29. doi: 10.1016/j.mce.2009.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.WHO. DDT and its derivatives: environmental aspects. Environmental Health Criteria. 1989 [Google Scholar]
- 14.Woolley DETG. Distribution of DDT, DDD, and DDE in tissues of neonatal rats and in milk and other tissues of mother rats chronically exposed to DDT. Toxicol Appl Pharmacol. 1971;18:907–916. doi: 10.1016/0041-008x(71)90238-9. [DOI] [PubMed] [Google Scholar]
- 15.Sala M, Ribas-Fitó N, Cardo E, et al. Levels of hexachlorobenzene and other organochlorine compounds in cord blood: exposure across placenta. Chemosphere. 2001;43(4–7):895–901. doi: 10.1016/s0045-6535(00)00450-1. [DOI] [PubMed] [Google Scholar]
- 16.Apostoli P, Magoni M, Bergonzi R, et al. Assessment of reference values for polychlorinated biphenyl concentration in human blood. Chemosphere. 2005;61(3):413–421. doi: 10.1016/j.chemosphere.2005.02.034. [DOI] [PubMed] [Google Scholar]
- 17.Reed L, Buchner V, Tchounwou PB. Environmental toxicology and health effects associated with hexachlorobenzene exposure. Rev Environ Health. 2007;22(3):213–243. doi: 10.1515/reveh.2007.22.3.213. [DOI] [PubMed] [Google Scholar]
- 18.Tsukimori K, Morokuma S, Hori T, et al. Characterization of placental transfer of polychlorinated dibenzo-p-dioxins, dibenzofurans and polychlorinated biphenyls in normal pregnancy. J Obstet Gynaecol Res. 2013;39(1):83–90. doi: 10.1111/j.1447-0756.2012.01906.x. [DOI] [PubMed] [Google Scholar]
- 19.Butler Walker J, Seddon L, McMullen E, et al. Organochlorine levels in maternal and umbilical cord blood plasma in Arctic Canada. Sci Total Environ. 2003;302(1–3):27–52. doi: 10.1016/s0048-9697(02)00319-4. [DOI] [PubMed] [Google Scholar]
- 20.Mendez MA, Garcia-Esteban R, Guxens M, et al. Prenatal organochlorine compound exposure, rapid weight gain, and overweight in infancy. Environmental health perspectives. 2011;119(2):272. doi: 10.1289/ehp.1002169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Valvi D, Mendez MA, Martinez D, et al. Prenatal concentrations of polychlorinated biphenyls, DDE, and DDT and overweight in children: a prospective birth cohort study. Environ Health Perspect. 2012;120(3):451–457. doi: 10.1289/ehp.1103862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cupul-Uicab LA, Hernández-Ávila M, Terrazas-Medina EA, Pennell ML, Longnecker MP. Prenatal exposure to the major DDT metabolite 1, 1-dichloro-2, 2-bis (p-chlorophenyl) ethylene (DDE) and growth in boys from Mexico. Environmental research. 2010;110(6):595–603. doi: 10.1016/j.envres.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gladen BC, Ragan NB, Rogan WJ. Pubertal growth and development and prenatal and lactational exposure to polychlorinated biphenyls and dichlorodiphenyl dichloroethene. J Pediatr. 2000;136(4):490–496. doi: 10.1016/s0022-3476(00)90012-x. [DOI] [PubMed] [Google Scholar]
- 24.Smink A, Ribas-Fito N, Garcia R, et al. Exposure to hexachlorobenzene during pregnancy increases the risk of overweight in children aged 6 years. Acta Paediatr. 2008;97(10):1465–1469. doi: 10.1111/j.1651-2227.2008.00937.x. [DOI] [PubMed] [Google Scholar]
- 25.Tang-Peronard JL, Heitmann BL, Andersen HR, et al. Association between prenatal polychlorinated biphenyl exposure and obesity development at ages 5 and 7 y: a prospective cohort study of 656 children from the Faroe Islands. The American journal of clinical nutrition. 2013 doi: 10.3945/ajcn.113.066720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Behl M, Rao D, Aagaard K, et al. Evaluation of the association between maternal smoking, childhood obesity, and metabolic disorders: a national toxicology program workshop review. Environ Health Perspect. 2013;121(2):170–180. doi: 10.1289/ehp.1205404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Verhulst SL, Nelen V, Hond ED, et al. Intrauterine exposure to environmental pollutants and body mass index during the first 3 years of life. Environ Health Perspect. 2009;117(1):122–126. doi: 10.1289/ehp.0800003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Calafat AM, Kuklenyik Z, Reidy JA, Caudill SP, Ekong J, Needham LL. Urinary concentrations of bisphenol A and 4-nonylphenol in a human reference population. Environ Health Perspect. 2005;113(4):391–395. doi: 10.1289/ehp.7534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wolff MS, Teitelbaum SL, Windham G, et al. Pilot study of urinary biomarkers of phytoestrogens, phthalates, and phenols in girls. Environ Health Perspect. 2007;115(1):116–121. doi: 10.1289/ehp.9488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang M, Kim SY, Chang SS, Lee IS, Kawamoto T. Urinary concentrations of bisphenol A in relation to biomarkers of sensitivity and effect and endocrine-related health effects. Environ Mol Mutagen. 2006;47(8):571–578. doi: 10.1002/em.20230. [DOI] [PubMed] [Google Scholar]
- 31.Miyamoto K, Kotake M. Estimation of daily bisphenol a intake of Japanese individuals with emphasis on uncertainty and variability. Environ Sci. 2006;13(1):15–29. [PubMed] [Google Scholar]
- 32.Calafat AM, Ye X, Wong LY, Reidy JA, Needham LL. Exposure of the U.S. population to bisphenol A and 4-tertiary-octylphenol: 2003–2004. Environ Health Perspect. 2008;116(1):39–44. doi: 10.1289/ehp.10753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vandenberg LN, Chahoud I, Heindel JJ, Padmanabhan V, Paumgartten FJ, Schoenfelder G. Urinary, circulating, and tissue biomonitoring studies indicate widespread exposure to bisphenol A. Environ Health Perspect. 2010;118(8):1055–1070. doi: 10.1289/ehp.0901716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lakind JSND. Daily intake of bisphenol A and potential sources of exposure: 2005–2006 National Health and Nutrition Examination Survey. J Expo Sci Enviro Epidemiol. 2011;21(3):272–279. doi: 10.1038/jes.2010.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yamada H, Furuta I, Kato EH, et al. Maternal serum and amniotic fluid bisphenol A concentrations in the early second trimester. Reprod Toxicol. 2002;16(6):735–739. doi: 10.1016/s0890-6238(02)00051-5. [DOI] [PubMed] [Google Scholar]
- 36.Takahashi O, Oishi S. Disposition of orally administered 2,2-Bis (4-hydroxyphenyl)propane (Bisphenol A) in pregnant rats and the placental transfer to fetuses. Environ Health Perspect. 2000;108(10):931–935. doi: 10.1289/ehp.00108931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schonfelder G, Wittfoht W, Hopp H, Talsness CE, Paul M, Chahoud I. Parent bisphenol A accumulation in the human maternal-fetal-placental unit. Environ Health Perspect. 2002;110(11):A703–707. doi: 10.1289/ehp.110-1241091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zalko D, Soto AM, Dolo L, et al. Biotransformations of bisphenol A in a mammalian model: answers and new questions raised by low-dose metabolic fate studies in pregnant CD1 mice. Environ Health Perspect. 2003;111(3):309–319. doi: 10.1289/ehp.5603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ikezuki Y, Tsutsumi O, Takai Y, Kamei Y, Taketani Y. Determination of bisphenol A concentrations in human biological fluids reveals significant early prenatal exposure. Hum Reprod. 2002;17(11):2839–2841. doi: 10.1093/humrep/17.11.2839. [DOI] [PubMed] [Google Scholar]
- 40.Lee YJ, Ryu HY, Kim HK, et al. Maternal and fetal exposure to bisphenol A in Korea. Reprod Toxicol. 2008;25(4):413–419. doi: 10.1016/j.reprotox.2008.05.058. [DOI] [PubMed] [Google Scholar]
- 41.Foster W, Chan S, Platt L, Hughes C. Detection of endocrine disrupting chemicals in samples of second trimester human amniotic fluid. J Clin Endocrinol Metab. 2000;85(8):2954–2957. doi: 10.1210/jcem.85.8.6850. [DOI] [PubMed] [Google Scholar]
- 42.Gerona RR, Woodruff TJ, Dickenson CA, et al. Bisphenol-A (BPA), BPA Glucuronide, and BPA Sulfate in Midgestation Umbilical Cord Serum in a Northern and Central California Population. Environ Sci Technol. 2013;47(21):12477–12485. doi: 10.1021/es402764d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nachman R, Lees P, Groopman J. Early life metabolism of bisphenol: a systematic review of the literature. Current Environmental Health Reports. doi: 10.1007/s40572-013-0003-7. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Alonso-Magdalena P, Vieira E, Soriano S, et al. Bisphenol A exposure during pregnancy disrupts glucose homeostasis in mothers and adult male offspring. Environ Health Perspect. 2010;118(9):1243–1250. doi: 10.1289/ehp.1001993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wei J, Lin Y, Li Y, et al. Perinatal exposure to bisphenol A at reference dose predisposes offspring to metabolic syndrome in adult rats on a high-fat diet. Endocrinology. 2011;152(8):3049–3061. doi: 10.1210/en.2011-0045. [DOI] [PubMed] [Google Scholar]
- 46.Sikorski R, Paszkowski T, Szprengier-Juszkiewicz T. Mercury in neonatal scalp hair. Sci Total Environ. 1986;57:105–110. doi: 10.1016/0048-9697(86)90015-x. [DOI] [PubMed] [Google Scholar]
- 47.Foldspang A, Hansen JC. Dietary intake of methylmercury as a correlate of gestational length and birth weight among newborns in Greenland. Am J Epidemiol. 1990;132(2):310–317. doi: 10.1093/oxfordjournals.aje.a115660. [DOI] [PubMed] [Google Scholar]
- 48.Chen Z, Myers R, Wei T, et al. Placental Transfer and Concentrations of Cadmium, Mercury, Lead, and Selenium in Mothers, Newborns, and Young Children. J Expo Sci Environ Epidemiol. doi: 10.1038/jes.2014.26. under review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kuo CC, Moon K, Thayer KA, Navas-Acien A. Environmental chemicals and type 2 diabetes: an updated systematic review of the epidemiologic evidence. Curr Diab Rep. 2013;13(6):831–849. doi: 10.1007/s11892-013-0432-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Adomako EE, Williams PN, Deacon C, Meharg AA. Inorganic arsenic and trace elements in Ghanaian grain staples. Environ Pollut. 2011;159(10):2435–2442. doi: 10.1016/j.envpol.2011.06.031. [DOI] [PubMed] [Google Scholar]
- 51.Navas-Acien A, Silbergeld EK, Pastor-Barriuso R, Guallar E. Arsenic exposure and prevalence of type 2 diabetes in US adults. Jama. 2008;300(7):814–822. doi: 10.1001/jama.300.7.814. [DOI] [PubMed] [Google Scholar]
- 52.Zierold KM, Knobeloch L, Anderson H. Prevalence of chronic diseases in adults exposed to arsenic-contaminated drinking water. Am J Public Health. 2004;94(11):1936–1937. doi: 10.2105/ajph.94.11.1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jones MR, Tellez-Plaza M, Sharrett AR, Guallar E, Navas-Acien A. Urine arsenic and hypertension in US adults: the 2003–2008 National Health and Nutrition Examination Survey. Epidemiology. 2011;22(2):153–161. doi: 10.1097/EDE.0b013e318207fdf2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang SL, Chang FH, Liou SH, Wang HJ, Li WF, Hsieh DP. Inorganic arsenic exposure and its relation to metabolic syndrome in an industrial area of Taiwan. Environ Int. 2007;33(6):805–811. doi: 10.1016/j.envint.2007.03.004. [DOI] [PubMed] [Google Scholar]
- 55.Waalkes MP, Liu J, Diwan BA. Transplacental arsenic carcinogenesis in mice. Toxicol Appl Pharmacol. 2007;222(3):271–280. doi: 10.1016/j.taap.2006.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Smith AH, Marshall G, Liaw J, Yuan Y, Ferreccio C, Steinmaus C. Mortality in young adults following in utero and childhood exposure to arsenic in drinking water. Environ Health Perspect. 2012;120(11):1527–1531. doi: 10.1289/ehp.1104867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Current Environmental Health Reports. Arsenic-associated changes to the epigenome: what are the functional consquences? In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.He K, Xun P, Liu K, Morris S, Reis J, Guallar E. Mercury exposure in young adulthood and incidence of diabetes later in life: the CARDIA Trace Element Study. Diabetes Care. 2013;36(6):1584–1589. doi: 10.2337/dc12-1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Choi B, Yeum KJ, Park SJ, Kim KN, Joo NS. Elevated serum ferritin and mercury concentrations are associated with hypertension; analysis of the fourth and fifth Korea national health and nutrition examination survey (KNHANES IV-2, 3, 2008–2009 and V-1, 2010) Environmental toxicology. 2013 doi: 10.1002/tox.21899. [DOI] [PubMed] [Google Scholar]
- 60.Mozaffarian D, Shi P, Morris JS, et al. Methylmercury exposure and incident diabetes in u.s. Men and women in two prospective cohorts. Diabetes Care. 2013;36(11):3578–3584. doi: 10.2337/dc13-0894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lippman SM, Klein EA, Goodman PJ, et al. Effect of selenium and vitamin E on risk of prostate cancer and other cancers: the Selenium and Vitamin E Cancer Prevention Trial (SELECT) JAMA: the journal of the American Medical Association. 2009;301(1):39–51. doi: 10.1001/jama.2008.864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mueller AS, Mueller K, Wolf NM, Pallauf J. Selenium and diabetes: an enigma? Free radical research. 2009;43(11):1029–1059. doi: 10.1080/10715760903196925. [DOI] [PubMed] [Google Scholar]
- 63.Stranges S, Sieri S, Vinceti M, et al. A prospective study of dietary selenium intake and risk of type 2 diabetes. BMC Public Health. 2010;10:564. doi: 10.1186/1471-2458-10-564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Navas-Acien A, Guallar E, Silbergeld EK, Rothenberg SJ. Lead exposure and cardiovascular disease--a systematic review. Environmental Health Perspectives. 2007;115(3):472–482. doi: 10.1289/ehp.9785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Moon SS. Additive effect of heavy metals on metabolic syndrome in the Korean population: the Korea National Health and Nutrition Examination Survey (KNHANES) 2009–2010. Endocrine. 2013 doi: 10.1007/s12020-013-0061-5. [DOI] [PubMed] [Google Scholar]
- 66.Bird A. Perceptions of epigenetics. Nature. 2007;447(7143):396–398. doi: 10.1038/nature05913. [DOI] [PubMed] [Google Scholar]
- 67.Chmurzynska A. Fetal programming: link between early nutrition, DNA methylation, and complex diseases. Nutr Rev. 2010;68(2):87–98. doi: 10.1111/j.1753-4887.2009.00265.x. [DOI] [PubMed] [Google Scholar]
- 68.Lillycrop KA, Phillips ES, Jackson AA, Hanson MA, Burdge GC. Dietary protein restriction of pregnant rats induces and folic acid supplementation prevents epigenetic modification of hepatic gene expression in the offspring. J Nutr. 2005;135(6):1382–1386. doi: 10.1093/jn/135.6.1382. [DOI] [PubMed] [Google Scholar]
- 69••.Wang D, Liu X, Zhou Y, et al. Individual variation and longitudinal pattern of genome-wide DNA methylation from birth to the first two years of life. Epigenetics. 2012;7(6):594–605. doi: 10.4161/epi.20117. This study offered an epigenetic insight for this trans-generational effect. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Liu XCQ, Tsai HJ, Wang G, Hong X, Zhou Y, Zhang CL, Liu CY, Liu R, Wang H, Zhang S, Yu Y, Mestan K, Pearson C, Otlans P, Zuckerman B, Wang X. Maternal Preconception Body Mass Index and Offspring Cord Blood DNA Methylation: Exploration of Early Life Origins of Disease. Environmental and Molecular Mutagenesis. doi: 10.1002/em.21827. Accepted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Toperoff G, Aran D, Kark JD, et al. Genome-wide survey reveals predisposing diabetes type 2-related DNA methylation variations in human peripheral blood. Hum Mol Genet. 2012;21(2):371–383. doi: 10.1093/hmg/ddr472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yang BT, Dayeh TA, Volkov PA, et al. Increased DNA methylation and decreased expression of PDX-1 in pancreatic islets from patients with type 2 diabetes. Mol Endocrinol. 2012;26(7):1203–1212. doi: 10.1210/me.2012-1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ling C, Del Guerra S, Lupi R, et al. Epigenetic regulation of PPARGC1A in human type 2 diabetic islets and effect on insulin secretion. Diabetologia. 2008;51(4):615–622. doi: 10.1007/s00125-007-0916-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ruchat SM, Houde AA, Voisin G, et al. Gestational diabetes mellitus epigenetically affects genes predominantly involved in metabolic diseases. Epigenetics. 2013;8(9) doi: 10.4161/epi.25578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.West NA, Kechris K, Dabelea D. Exposure to Maternal Diabetes in Utero and DNA Methylation Patterns in the Offspring. Immunometabolism. 2013;1:1–9. doi: 10.2478/immun-2013-0001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Casati L, Sendra R, Poletti A, Negri-Cesi P, Celotti F. Androgen receptor activation by polychlorinated biphenyls: Epigenetic effects mediated by the histone demethylase Jarid1b. Epigenetics. 2013;8(10) doi: 10.4161/epi.25811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bastos Sales L, Kamstra JH, Cenijn PH, van Rijt LS, Hamers T, Legler J. Effects of endocrine disrupting chemicals on in vitro global DNA methylation and adipocyte differentiation. Toxicol In Vitro. 2013;27(6):1634–1643. doi: 10.1016/j.tiv.2013.04.005. [DOI] [PubMed] [Google Scholar]
- 78.Casati L, Sendra R, Colciago A, et al. Polychlorinated biphenyls affect histone modification pattern in early development of rats: a role for androgen receptor-dependent modulation? Epigenomics. 2012;4(1):101–112. doi: 10.2217/epi.11.110. [DOI] [PubMed] [Google Scholar]
- 79.Bromer JG, Zhou Y, Taylor MB, Doherty L, Taylor HS. Bisphenol-A exposure in utero leads to epigenetic alterations in the developmental programming of uterine estrogen response. FASEB J. 2010;24(7):2273–2280. doi: 10.1096/fj.09-140533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Rusiecki JA, Baccarelli A, Bollati V, Tarantini L, Moore LE, Bonefeld-Jorgensen EC. Global DNA hypomethylation is associated with high serum-persistent organic pollutants in Greenlandic Inuit. Environ Health Perspect. 2008;116(11):1547–1552. doi: 10.1289/ehp.11338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kim KY, Kim DS, Lee SK, et al. Association of low-dose exposure to persistent organic pollutants with global DNA hypomethylation in healthy Koreans. Environ Health Perspect. 2010;118(3):370–374. doi: 10.1289/ehp.0901131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Arai Y, Ohgane J, Yagi S, et al. Epigenetic assessment of environmental chemicals detected in maternal peripheral and cord blood samples. J Reprod Dev. 2011;57(4):507–517. doi: 10.1262/jrd.11-034a. [DOI] [PubMed] [Google Scholar]
- 83.Hanna CW, Bloom MS, Robinson WP, et al. DNA methylation changes in whole blood is associated with exposure to the environmental contaminants, mercury, lead, cadmium and bisphenol A, in women undergoing ovarian stimulation for IVF. Hum Reprod. 2012;27(5):1401–1410. doi: 10.1093/humrep/des038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Pilsner JR, Hall MN, Liu X, et al. Associations of plasma selenium with arsenic and genomic methylation of leukocyte DNA in Bangladesh. Environ Health Perspect. 2011;119(1):113–118. doi: 10.1289/ehp.1001937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Pilsner JR, Hu H, Ettinger A, et al. Influence of prenatal lead exposure on genomic methylation of cord blood DNA. Environ Health Perspect. 2009;117(9):1466–1471. doi: 10.1289/ehp.0800497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Dosunmu R, Alashwal H, Zawia NH. Genome-wide expression and methylation profiling in the aged rodent brain due to early-life Pb exposure and its relevance to aging. Mech Ageing Dev. 2012;133(6):435–443. doi: 10.1016/j.mad.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wang G, Walker SO, Hong X, Bartel TR, Wang X. Epigenetics and Early Life Origins of Chronic Non-Communicable Diseases. J Adolesc Health. doi: 10.1016/j.jadohealth.2012.04.019. in press. [DOI] [PubMed] [Google Scholar]
- 88.Drong AW, Lindgren CM, McCarthy MI. The genetic and epigenetic basis of type 2 diabetes and obesity. Clinical pharmacology and therapeutics. 2012;92(6):707–715. doi: 10.1038/clpt.2012.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zerhouni E. Annual Meeting of NIH Roadmap Multidisciplinary Clinical Research Career Development Program. 2007. [Google Scholar]
- 90.Hong X, Caruso D, Kumar R, et al. IgE, but not IgG4, antibodies to Ara h 2 distinguish peanut allergy from asymptomatic peanut sensitization. Allergy. 2012;67(12):1538–1546. doi: 10.1111/all.12047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wang G, Walker SO, Hong X, Bartell TR, Wang X. Epigenetics and early life origins of chronic noncommunicable diseases. Journal of Adolescent Health. 2013;52(2):S14–S21. doi: 10.1016/j.jadohealth.2012.04.019. [DOI] [PubMed] [Google Scholar]
- 92.Lawlor DA, Andersen A-MN, Batty GD. Birth cohort studies: past, present and future. International journal of epidemiology. 2009;38(4):897–902. doi: 10.1093/ije/dyp240. [DOI] [PubMed] [Google Scholar]
- 93.Hertz-Picciotto I, Charles MJ, James RA, Keller JA, Willman E, Teplin S. In utero polychlorinated biphenyl exposures in relation to fetal and early childhood growth. Epidemiology. 2005;16(5):648–656. doi: 10.1097/01.ede.0000173043.85834.f3. [DOI] [PubMed] [Google Scholar]
- 94.Jovanovic D, Rasic-Milutinovic Z, Paunovic K, Jakovljevic B, Plavsic S, Milosevic J. Low levels of arsenic in drinking water and type 2 diabetes in Middle Banat region, Serbia. Int J Hyg Environ Health. 2013;216(1):50–55. doi: 10.1016/j.ijheh.2012.01.001. [DOI] [PubMed] [Google Scholar]
- 95.He K, Xun P, Liu K, Morris S, Reis J, Guallar E. Mercury Exposure in Young Adulthood and Incidence of Diabetes Later in Life The CARDIA Trace Element Study. Diabetes care. 2013;36(6):1584–1589. doi: 10.2337/dc12-1842. [DOI] [PMC free article] [PubMed] [Google Scholar]