

Abstract
We analyzed the FANTOM2 clone set of 60,770 RIKEN full-length mouse cDNA sequences and 44,122 public mRNA sequences. We developed a new computational procedure to identify and classify the forms of splice variation evident in this data set and organized the results into a publicly accessible database that can be used for future expression array construction, structural genomics, and analyses of the mechanism and regulation of alternative splicing. Statistical analysis shows that at least 41% and possibly as much as 60% of multiexon genes in mouse have multiple splice forms. Of the transcription units with multiple splice forms, 49% contain transcripts in which the apparent use of an alternative transcription start (stop) is accompanied by alternative splicing of the initial (terminal) exon. This implies that alternative transcription may frequently induce alternative splicing. The fact that 73% of all exons with splice variation fall within the annotated coding region indicates that most splice variation is likely to affect the protein form. Finally, we compared the set of constitutive (present in all transcripts) exons with the set of cryptic (present only in some transcripts) exons and found statistically significant differences in their length distributions, the nucleotide distributions around their splice junctions, and the frequencies of occurrence of several short sequence motifs.
Numerous databases of alternative splice forms have been generated in recent years (Stamm et al. 1994; Burke et al. 1998; Gelfand et al. 1999; Mangan and Frazer 1999; Mironov et al. 1999; Brett et al. 2000; Croft et al. 2000; Ji et al. 2001; Kan et al. 2001; Kent and Zahler 2001; Modrek et al. 2001), and analysis of their contents indicates a number of general facts about splicing and its mechanisms. First, the frequency of multiexon genes with multiple gene structures is high in mammals (Mironov et al. 1999; Lander et al. 2001; Modrek et al. 2001; Brett et al. 2002; Zavolan et al. 2002). The highest estimates are 59% (Lander et al. 2001) for human genes and 33% (Brett et al. 2002) for mouse genes, raising the question of whether the complexity of gene structure is lower in mouse than in human. Second, most of the splice variation affects the coding region. This has been inferred using (1) a relatively small subset of EST clusters carefully curated (Modrek et al. 2001) and (2) automated analysis of cDNA data (Zavolan et al. 2002).
Exon inclusion into mature mRNA is a complex choice whose outcome is influenced by a variety of factors. Many exons are “cryptic” in the sense that they are included in some but not all transcripts, through mechanisms that are presently not completely understood. Cryptic exons have been reported to be shorter, on average, than constitutive exons (Berget 1995) and to be flanked by weaker splice sites (Stamm et al. 2000). Although exon splice enhancers can compensate for weaker splice sites (Berget 1995; Fairbrother et al. 2002), it is unclear at present whether constitutive and cryptic exons differ in the content of splice enhancers. Finally, studies using full-length cDNA sequences indicate that alternative transcription initiation (Suzuki et al. 2001; FANTOM2 Consortium and the RIKEN GSC Genome Exploration Group 2002; Zavolan et al. 2002) and alternative choice of polyadenylation sites (Zavolan et al. 2002) further enhance proteome diversity.
To make effective use of the large repertoire of transcripts, a cell must be able to tightly regulate their expression. With the completion of the mouse genome sequence (ftp://wolfram.wi.mit.edu/pub/mousecontigs/MGSCV3) and a very large collection of mouse full-length cDNAs (FANTOM2 Consortium and the RIKEN GSC Genome Exploration Group 2002), the body of data available for testing hypotheses about the regulation of alternative splicing has improved, both quantitatively and qualitatively. Full-length cDNA sequences are obtained using mRNA trapping techniques designed to capture polyadenylated transcripts carrying the cap structure (Carninci et al. 1996). Multiple full-length cDNA sequences derived from the same “transcription unit” (a segment of the genome from which transcripts are generated; FANTOM2 Consortium and the RIKEN GSC Genome Exploration Group 2002) reveal the full spectrum of exon combinations that are, indeed, generated by the spliceosome, and they enable analyses of the cis-acting elements that contribute to differential exon usage. Sequences of introns, exons, and upstream and downstream regions are available for a large set of transcripts unambiguously mapped to the genome.
Here we report the results of our analysis of splice variation in the FANTOM2 clone set of 60,770 RIKEN full-length mouse cDNA sequences and 44,122 public mRNA sequences.
RESULTS
Frequency of Alternative Splicing in the Mouse Transcriptome
Of 60,770 RIKEN full-length mouse cDNA sequences and 44,122 public mRNA sequences, 101,356 aligned to 36,617 genomic loci. A locus was defined based on the genomic map of the transcripts: Two transcripts were clustered together if their genomic maps overlapped by at least one nucleotide in at least one exon. We defined the genomic locus of a transcript cluster as the shortest genomic interval that contains the genomic maps of all of the transcripts in the cluster. We found that 77,640 of the cDNA sequences mapped to the genome at >95% identity over their entire length, with every exon being mapped at ≥95% identity or with at most five errors. Of these transcripts, 23,150 were either unspliced or came from single-exon genes. Of the remaining 54,490 multiexon transcripts, 7293 did not cluster with any other transcripts (we called these transcripts singletons), whereas 47,197 formed 11,677 multitranscript clusters, which we analyzed for splice variation. We constructed the union of the genomic mappings of all transcripts in a cluster and denoted the contiguous genomic regions in this union of mappings as “genomic exons.” A genomic exon represented in some, but not all, transcripts in a cluster was denoted “cryptic.” A genomic exon with different 5′ and/or 3′ boundaries in different transcripts, was denoted an exon with “alternative 3′- and/or 5′-splice sites.” We chose not to report cases in which the variation could be explained by an intron inclusion, because we cannot readily distinguish computationally between intron inclusion and incomplete splicing. Genomic exons that were represented with the same splice junctions in all transcripts that were long enough to have contained them were denoted “constitutive.” A cluster with at least one variant genomic exon was denoted “variant.” Of the 11,677 multitranscript clusters, 41% (4750) were variant (Table 1).
Table 1.
Mouse Transcriptome Summary. Flow of the Data Through Our Analysis Procedure
|
Riken sequences only
|
Riken and GenBank sequences
|
|||
|---|---|---|---|---|
| Data set description | Sequences | Clusters | Sequences | Clusters |
| Genome-mapped | 60,301 | 33,936 | 101,356 | 36,617 |
| Quasi-complete mappings | 51,741 | 30,447 | 77,640 | 33,042 |
| Spliced | 31,593 | 16,541 | 54,490 | 18,970 |
| Singletons | 9,260 | 9,260 | 7,293 | 7,293 |
| Multitranscript | 22,333 | 7,281 | 47,197 | 11,677 |
| Alternative splicing | 10,601 | 3,121 | 22,150 | 4,750 |
| Without splice variation | 11,732 | 4,160 | 25,047 | 6,927 |
This value is larger than our previous estimate on a smaller data set of full-length mouse cDNAs (30%; Zavolan et al. 2002), and larger than indicated by ESTs (33%; Brett et al. 2002). The real value is expected to be even higher, as many of the variants generated in vivo are rare, specific to tissues and developmental stages. Furthermore, the RIKEN cDNA cloning strategy, involving sequential subtraction and normalization of libraries, is actually designed to avoid redundant sequencing of cDNAs derived from the same transcription unit (see Discussion). The extent to which this strategy failed is in part attributable to the frequency with which the outputs of individual transcription units vary at the 5′ and 3′ ends, and are therefore clustered independently in Phase 1 sequencing (FANTOM2 Consortium and the RIKEN GSC Genome Exploration Group 2002).
From our (Zavolan et al. 2002) as well as other studies (Kan et al. 2002), it is clear that the estimated frequency of genes with alternative splice forms increases with the depth of sampling, that is, with the average number of transcripts sequenced for a gene. Some of the clusters for which we have not yet found any splice variants will eventually acquire splice forms as more transcripts are sequenced. To account for the limited sampling of transcripts, we developed a Bayesian model (see Methods) that allows us to estimate the probability that a gene has multiple splice forms, even though only identically spliced transcripts were observed in our data. The model's estimate depends crucially on the prior distribution that we assume for the number of splice forms per gene, and the prior for the relative frequencies of occurrence of these splice forms in the cDNA pool. As explained in Methods, we chose these priors to bias the estimate toward a high number of genes with multiple splice forms. As a result, one can consider the estimate from this model as an upper bound on the incidence of genes with multiple splice forms. The value that we obtained is 60%. Therefore, we estimate at present that the frequency of multiexon mouse genes that have multiple splice forms is at least 41% and may be as high as 60%.
Characterization of Splice Variants
Of all 55,534 genomic exons in variant clusters, 9427 (17%) show splice variation. Of the 9427 variant genomic exons, 6635 were cryptic exons, of which 2927 occurred as initial or terminal in a transcript and 3708 occurred in internal positions. The remaining 2792 variant genomic exons were constitutive, with alternative 5′- and/or 3′-splice sites. We developed a Bayesian model (see Methods) to investigate if alternative splice site usage correlates with exon skipping and found a posterior probability Pc = 0.71 for this correlation to exist. Thus, there is little evidence in our data for a correlation between exon skipping and alternative choice of splice signals.
We sought to independently validate the exon variants using mouse EST sequences from the dbEST database. EST mappings were filtered with the same stringency as the cDNA mappings, and were searched for the presence of the variant exon forms. An exon variation was considered confirmed when all the exon forms used to infer the variation were present in the EST data. That is, a confirmed cryptic exon is an exon that is present in some EST but skipped in others. A cryptic exon with alternative 5′-splice sites is confirmed when both forms of splice site usage, as well as the skipped form occur in the EST data. Table 2 summarizes the results. It is not surprising that multiple variations are confirmed at lower rates than single variations. Given that most ESTs are the result of 3′- or 5′-end sequencing, it is also to be expected that internal cryptic exons have lower confirmation rates than initial or terminal cryptic exons. Exons with multiple variations have, however, lower confirmation rates than we would predict if each variation requires independent confirmation. This indicates that certain of the variant exon forms are less common than others. For instance, such forms may be expressed in only a few cell types, or are generated through infrequent errors of the spliceosome (see below). Kan et al. (2002) made similar observations based on analysis of individual splicing events.
Table 2.
Variant Exon Statistics
|
Constitutive exons
|
Cryptic internal exons
|
Cryptic initial/terminal exons
|
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Alternative SSa | Total | EST hit | Confirmed | Total | EST hit | Confirmed | Total | EST hit | Confirmed |
| None | 46,107 | 24,265 (52.6%) | 22,180 (91.4%) | 3523 | 1417 (40.2%) | 118 (8.3%) | 2779 | 586 (21.1%) | 151 (25.8%) |
| 5′ | 1,237 | 668 (54%) | 151 (22.6%) | 70 | 42 (60%) | 3 (7.1%) | 90 | 51 (56.7%) | 7 (13.7%) |
| 3′ | 1,522 | 928 (61%) | 225 (24.2%) | 106 | 59 (55.7%) | 3 (5.1%) | 56 | 27 (48.2%) | 4 (14.8%) |
| 5′ and 3′ | 33 | 19 (56.7%) | 2 (10.5%) | 9 | 7 (77.8%) | 0 (0%) | 2 | 2 (100%) | 0 (0%) |
dbEST sequences were searched for the presence of the variant exon forms that were identified from cDNA sequences. An exon identified from cDNA sequences was considered to have an EST hit when at least one EST from dbEST contained some form of that exon. If all the variant forms were present in the EST data, the exon was considered “confirmed”
SS = splice site
Functional Splice Variation Versus Noise in the Splicing Process
We observed that alternative 5′/3′-splice sites are frequently only a few nucleotides apart, indicating that some of the variation may be caused by random use of 5′- and 3′-splice sites within a short region around the exon boundary. To document this effect, we isolated exons with alternative 5′- and 3′-splice sites that matched the genome perfectly for at least 10 nt from the splice junction, and were preceded/followed by invariant exons with perfect alignment to the genome for at least 10 nt from the splice junction. This selection ensures that the variation in the placement of the splice junction is not caused by sequencing, assembly, or mapping errors. We then computed the distribution of distances between these alternative splice sites (Fig. 1). In 83 (18%) of the 452 cases of alternative 5′-splice sites and 237 (40%) of the 587 cases of alternative 3′-splice sites, the distance between alternative sites was <10 nt.
Figure 1.

Distribution of distances between alternative splice sites: (A) 5′-splice sites; (B) 3′-splice sites. Note that we only show the distribution up to a distance of 100 nt.
Whereas alternative 3′-splice sites seem to be selected for preserving the reading frame, alternative 5′ sites are not. In 310 (52.8%, p-value < 2.2 × 10-16) of the 587 cases of exons with alternative 3′-splice sites, the distance between these sites was a multiple of 3, compared with 159 (35.2%, p-value = 0.4247) of the 452 alternative 5′-splice sites. The asymmetry is, however, entirely due to the frequent use (161 of 587 cases) of alternative 3′-splice sites that are only 3 nt apart. In summary, our data indicate that the spliceosome may frequently “slip” around the splice junction, and that, at the 3′ site, slips that preserve the reading frame are more common than others.
Impact of Splice Variation on the Proteome
Splice variation significantly enhances the proteome diversity. We estimated that 6225 (74%) of 8456 variant exons (cryptic and/or with alternative 5′/3′-splice sites) overlap with the coding region of the transcript. This fraction is similar to the value reported by Modrek et al. (2001) for human transcripts. Localization of splice variation was determined relative to the coding region of the representative sequences (FANTOM2 Consortium and the RIKEN GSC Genome Exploration Group 2002) of each cluster (see Methods) and is summarized in Table 3. Of all variant clusters, 4242 have been annotated as protein-encoding. Of these, 933 (22%) have cryptic initial and/or terminal exons that lie entirely within the coding region. These transcripts, if translated, would miss either initial and/or terminal parts of the protein. In contrast to transcripts that are merely truncated, the presence of an alternative splice in these initial exons indicates that they are not simply the result of nucleotide loss during the experimental procedure. Such variants may have important functional implications, as has been recently shown for the STAT proteins (Henriksen et al. 2002).
Table 3.
Localization of Splice Variations: Statistics
|
Constitutive exons
|
Cryptic internal exons
|
Cryptic initial/terminal exons
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Splice site usage | 5′-UTR | CDS | 3′-UTR | ?a | 5′-UTR | CDS | 3′-UTR | ?a | 5′-UTR | CDS | 3′-UTR | ?a |
| Invariant | 4046 | 36,540 | 2531 | 2990 | 453 | 2632 | 114 | 324 | 802 | 1487 | 140 | 350 |
| Alternative 5′ | 285 | 768 | 55 | 129 | 9 | 51 | 5 | 5 | 40 | 42 | 2 | 6 |
| Alternative 3′ | 211 | 1,104 | 74 | 133 | 19 | 73 | 5 | 9 | 0 | 44 | 9 | 3 |
| Alternative 5′ and 3′ | 2 | 17 | 4 | 10 | 1 | 7 | 0 | 1 | 1 | 0 | 0 | 1 |
Exons from clusters without an annotated protein-coding region
Representation of Gene Ontology Categories in Variant and Invariant Clusters
To explore the functional repertoire of splice variants, we extracted from the variant and invariant clusters 4236 and 6234 representative transcripts (FANTOM2 Consortium and the RIKEN GSC Genome Exploration Group 2002), respectively. Representative transcripts from 3018 (71.2%) variant and 4758 (76.3%) invariant clusters matched to 2522 gene ontology (GO) term assignments (Table 4). Of those, 407 (16.1%) GO terms were associated only with representatives of variant clusters (Supplementary Table 1; available online at www.genome.org), 829 (32.8%) GO terms only with representatives of invariant clusters (Supplementary Table 2), and 1286 (51%) GO terms occurred among representatives of both types of clusters (Supplementary Table 3). The lists of 20 most represented GO terms in variant and invariant clusters share the terms DNA binding, transcription regulation, signal transduction, signal transducer, and kinase. In contrast to Modrek and Lee (2002), who reported that proteins involved in signal transduction or transcription are highly represented among splice variants, we found that these categories occur at high frequency in both variant and invariant clusters. For example, signal transduction occurs with the same frequency (1:10) in variant and invariant clusters. However, when comparing GO terms that were unique to representatives of variant and invariant clusters, we found that GO terms related to enzymes and enzymatic functions occurred more than twice as frequently in invariant (364, 14.4%) as in variant clusters (158, 6.3%). The results imply that there may be only a limited potential for diversification of enzymatic function through splice variation.
Table 4.
Counts of GO Terms Associated with Variant, Nonvariant, and Both Types of Clusters
| GO category | Variant | Variant only | Nonvariant | Nonvariant only | Both |
|---|---|---|---|---|---|
| Molecular function | 916 (54.1%) | 226 (55.5%) | 1198 (56.6%) | 508 (61.3%) | 690 (53.7%) |
| Cell component | 176 (10.4%) | 34 (8.6%) | 215 (10.2%) | 73 (8.8%) | 142 (11.0%) |
| Biological process | 601 (35.5%) | 147 (36.1%) | 702 (33.2%) | 248 (29.9%) | 454 (35.3%) |
| Total | 1693 (100%) | 407 (100.0%) | 2115 (100%) | 829 (100%) | 1286 (100%) |
We additionally distinguished GO terms that were associated exclusively with variant or with nonvariant clusters
Comparative Analysis of Cryptic and Constitutive Exons
The mechanism of exon selection has been extensively studied (Robberson et al. 1990; Talerico and Berget 1990; Hoffman and Grabowski 1992; Zahler et al. 1993; Staknis and Reed 1994; Berget 1995; Wang et al. 1995; Caceres and Krainer 1997; McCullough and Berget 1997; Liu et al. 1998; Eldridge et al. 1999; Tacke and Manley 1999; Blencowe et al. 2000). However, the details of this process are not understood to the extent that a computational tool can predict alternative splice forms for a given locus. There are some indications that cryptic exons are shorter and/or have weaker splice sites (Dominski and Kole 1992; Berget 1995; Stamm et al. 2000). We sought to test these hypotheses in the context of our data set. For this purpose, we extracted a set of constitutive exons that were found to be included as internal exons with invariant splice sites in transcripts from at least four different libraries. This constitutes our reference set of constitutive exons (n = 15,298). We also extracted the set of cryptic internal exons that were present in only two forms in our data set: included with invariant splice sites, and skipped (n = 3468).
A Kolmogorov-Smirnov test indicates that the length distribution of constitutive exons differs significantly from that of cryptic exons (Fig. 2, p-value = 2.075 × 10-12). Contrary to our expectation that cryptic exons would be shorter than constitutive exons (Berget 1995), we observed an enrichment of cryptic exons at both small and large exon lengths. Cryptic exons have a larger mean and also a larger variance (142.4 ± 192.7 vs. 129.2 ± 92.2). The difference between the length distribution of cryptic exons that we find and that reported based on EST analysis may be explained by the fact that very long internal exons are underrepresented in EST data. This would lead to the disappearance of the right-hand tail in the length distribution for cryptic exons in Figure 2.
Figure 2.

Estimated frequency of lengths of constitutive (black) and cryptic (red) exons. For each value of the length L we determined the frequency in our data set of exons with length between 0.8 L and 1.2 L.
Figure 3 shows the distribution of nucleotides found around the splice sites in constitutive and cryptic exons (see Methods). The slightly larger size of the letters in the upper panels indicates that the splice junctions flanking constitutive exons show more conservation than those flaking cryptic exons. Statistically significant differences (see Methods) are apparent at positions +1, +4, and +5 in the 5′-splice site and positions -5 and -6 in the 3′-splice site (Fig. 4). These results are consistent with previous observations that cryptic exons have “weaker” splice sites (Stamm et al. 2000). Exons flanked by weak splice signals and otherwise skipped will be included in the mRNA if the exons are modified to contain exonic splice enhancers (ESEs; Liu et al. 1998; Schaal and Maniatis 1999; Tacke and Manley 1999; Fairbrother et al. 2002). These are short sequence motifs, occurring naturally in the vicinity of the splice signals (Berget 1995; Fairbrother et al. 2002), known to promote the inclusion of exons in the mature mRNA.
Figure 3.

Nucleotide distribution in the (A,C) 5′- and (B,D) 3′-splice signals flanking constitutive (upper panels) and cryptic (lower panels) exons. The relative sizes of the letters indicate the relative frequencies of the nucleotides at that distance from the splice junctions. The absolute sizes of the letters correspond to the information score of the nucleotide distribution at that position.
Figure 4.

Position-specific probability that the nucleotides at (A) 5′- and (B) 3′-splice signals of cryptic exons were generated from different versus the same underlying distribution as those of the constitutive exons.
We sought to identify sequence motifs that are over- or underrepresented in cryptic exons relative to constitutive exons, and are therefore candidate regulatory elements responsible for the inclusion of the cryptic exon in the mature mRNA. We extracted candidate motifs as described in Methods. In short, we select all motifs that occur significantly more frequently in constitutive than cryptic exons (or vice versa), and whose frequency is significantly different from that expected from the mononucleotide frequencies in at least one of the two sets of exons. We classified these motifs based on (1) the neighboring splice site (5′ or 3′); (2) over- or underrepresentation in constitutive versus cryptic; (3) over- or underrepresentation of the motif when compared with the frequency expected from the mononucleotide frequencies in the exon set.
The most striking patterns that emerged are the following. Some of the previously reported splice enhancers (Fairbrother et al. 2002) are overrepresented in constitutive exons compared with cryptic exons. Of these, TGAAG- and AAGAA-containing motifs are overrepresented in both constitutive and cryptic exons relative to their mononucleotide frequencies. In contrast, TGGA-containing motifs are enriched in constitutive exons but depleted in cryptic exons relative to the mononucleotide frequencies. Virtually all motifs that are depleted in both constitutive and cryptic exons relative to the mononucleotide frequencies, but are overrepresented in constitutive exons, are CG-dinucleotide-containing motifs. This enrichment is not due to an overall higher C + Gcontent of constitutive exons (data not shown).
In contrast, cryptic exons are enriched in pyrimidine-rich motifs, reminiscent of the SRp20-specific motifs described by Schaal and Maniatis (1999), or perhaps of motifs to which the polypyrimidine-tract-binding protein binds (Fairbrother and Chasin 2000). Additionally, a number of our motifs resemble binding sites of hRNP proteins A1 (Burd and Dreyfuss 1994; Chabot et al. 1997), F and H (Min et al. 1995), and of p54nrb (Basu et al. 1997). These binding sites are G-rich, frequently containing an AGGG core, or containing alternating A and Gnucleotides. These results indicate that cryptic exons not only lack splice enhancers, but also contain signals that inhibit splicing. The complete set of significant motifs that we extracted is given in Supplementary Figure 1.
Examples
The entire data set of splice variants can be viewed at http://genomes.rockefeller.edu/MouSDB. In addition to information stored locally in our database, we have provided links to external resources such as the FACTS database of functional associations (http://facts.gsc.riken.go.jp; Nagashima et al. 2003) and the SMART (http://smart.emblheidelberg.de) tool for analysis of protein domains. These will facilitate exploration of the functional impact of the splice variants in our data set.
Here we present one example to illustrate the breadth of strategies by which transcript and protein diversity appear to be achieved in mouse. In separate analyses, we have noted that alternative splicing is relatively common among the members of the very large zinc finger family of transcription factors, commonly leading to inclusion or exclusion of individual zinc fingers or other functional domains (Ravasi et al. 2003). It is arguable that if splice variation plays a major role in the complexity of mammalian development, then a relative overrepresentation of splice variation among regulatory proteins, especially those at the apex of regulatory hierarchies, is to be expected. Indeed, we observe that a relatively large number of examples in our splice variant database are transcriptional regulators.
As an example, Figure 5 shows different splice forms of the polypyrimidine-tract-binding protein (PTB), also known as hnRNP1, that are observed in our data set. This is an RNA-binding protein that controls 3′-end processing, internal initiation of translation, and RNA localization, in addition to splice acceptor recognition. As the figure indicates, this gene, involved in the control of alternative splicing, is itself extensively alternatively spliced. Human PTB is known to be expressed in at least three isoforms that differ by the insertion of 19 and 26 amino acids, respectively, between the second and the third RNA-recognition-motif domains (Romanelli et al. 2000). The latter variant is present in our data set. The different isoforms contribute differently to alternative splicing (Wollerton et al. 2001). Figure 5 shows a large number of transcripts encoding the mouse polypyrimidine-tract-binding protein, and a number of new splice forms present in the FANTOM2 data set. As is the case with many other examples that we inspected, here splice variation gives rise to transcripts encoding truncated proteins. This is caused by exon skipping in transcript AK088126 (riE430004K05) and an alternative choice of splice sites in transcript AK036745 (ri9830169E20). The potential intron inclusion in transcript AK037607 (riA130029H05) would also produce a truncated protein by introducing an in-frame stop codon. Alternative choices of 3′-splice site in transcript AK036745 (ri9830169E20) do not change the reading frame; it only truncates the first RNA-recognition motif. It will be particularly interesting to see whether any of these isoforms are expressed in a tissue-specific manner and contributes to tissue-specific alternative splicing of other genes.
Figure 5.

Splice variants of polypyrimidine-tract-binding protein.
MouSDB: The Database of Splice Variants Identified in the Mouse Transcriptome
All multitranscript clusters that we identified in this data set have been deposited in a Postgres database that can be queried via a Web interface at http://genomes.rockefeller.edu/MouSDB. We expanded our data set to include not only cDNA sequences, but also EST sequences that we mapped to the mouse genome with the same stringency as the full-length cDNAs. These include 1,440,717 ESTs from the dbEST section of GenBank, 547,149 RIKEN 5′-end, and 1,442,236 RIKEN 3′-end sequences. In addition to browsing precomputed multitranscript clusters, a user may perform splice analysis and annotation of a set of genes of interest.
DISCUSSION
Our study provides the first database and analysis of alternative splice forms identified in a large set of full-length cDNAs. The data set used in this analysis is unique in the following ways. With ESTs one generally has to infer the full transcript by combining sequence from several ESTs, and there is no guarantee that the inferred transcript occurs in vivo. In contrast, full-length cDNAs directly reveal the spectrum of splice forms that are realized in vivo. Additionally, the full-length cDNAs and 5′- and 3′-end EST sequences generated by the FANTOM project (FANTOM2 Consortium and the RIKEN GSC Genome Exploration Group 2002) allow us to identify transcription start and termination in the genome, which is important for the identification of alternative promoters and polyadenylation signals. Alternative transcription is of interest not only for the purpose of understanding gene expression, but also because it can induce variation in the use of splice sites.
One complication for the analysis of splice variation in this data set is that the sequences have been generated during the course of a project aimed at maximizing the nonredundant coverage of the mouse transcriptome. For this reason, the data set does not cover transcripts in proportion to their representation in the body (Konno et al. 2001). As we mentioned above, sequencing of transcripts with 3′ and 5′ ends identical to already sequenced transcripts is suppressed. Note, however, that this bias cannot be very large because the majority of multiple-transcript clusters shows no splice variation, that is, there are still more copies of identical transcripts in the data than alternative transcripts of the same gene. However, we must take into account the possibility that internal splice variation is somewhat underrepresented in our data set, and we designed our statistical tests such that such a bias would not affect the general conclusions. The only numbers that are potentially affected are the relative frequencies of initial/terminal exons among cryptic exons (which we potentially overestimate) and the frequency with which splice variation affects the coding region (which we potentially underestimate).
We developed a novel computational tool to automate the analysis of splice variation in cDNA and EST sequences. It maps the sequences to the genome, clusters sequences mapping to overlapping regions in the genome, produces multiple alignments of the sequences within a cluster, and annotates them in terms of splice variation. Our methods are highly conservative: To reduce the frequency of false positives, we only use sequences that map to the genome at very high percentage identity and coverage. This methodology can be applied to any set of EST or cDNA sequences for which the corresponding genome has been sequenced.
We organized the data into a publicly accessible resource that can be used to study specific genes of interest, for expression array construction, and for further analyses of the mechanism and regulation of alternative splicing. To increase our coverage of splice forms in the mouse transcriptome, we also incorporated 5′- and 3′-end RIKEN EST sequences and EST sequences from dbEST. Both data sets (cDNA sequences only and cDNA + EST sequences) are accessible at http://genomes.rockefeller.edu/MouSDB. The database provides information about transcripts derived from individual transcription units and can be searched in various ways, including by gene name. The information includes multiple sequence alignments, functional annotation, the tissues from which the transcripts have been derived, and annotation of the splice variation observed in the transcripts. One may, for instance, use the database to discover novel splice forms for genes of interest, and to explore the tissue-specificity of expression patterns of various splice forms. For each cDNA sequence, our interface provides annotation of the coding region and links to external databases such as the FACTS functional annotation database and the SMART tool for protein domain identification, allowing one to evaluate the effects of the splice variation at the protein level. The database can also be used to identify alternative promoters and polyadenylation signals, which, as our analysis indicates, may be an important source of proteome diversity. Using the genomic mapping coordinates that we provide, one can extract the sequence of putative alternative promoters from the publicly available draft of the mouse genome to search for upstream regulatory elements. Recognizing that sequence data continue to accumulate, we allow users to perform these analyses on new sets of sequences by submitting them to the Web interface of our programs.
We found that 41% of the loci for which multiple spliced transcripts were present in the data set had multiple splice forms. We took advantage of the functional annotation of these sequences to confirm that most of the splice variation occurs inside the protein-coding region. Interestingly, in almost half of all transcription units that show splice variation, we found cases in which an apparent alternative transcription start (stop) site is associated with an alternative splice in the initial (terminal) exon. The use of alternative transcription in these cases indicates that the alternative splice forms are differentially expressed and are, therefore, functional. We also found numerous examples of exons whose alternative splice sites are only a few nucleotides apart, indicating that the spliceosome can slide around the splice junction.
Finally, given the high frequency of alternative splicing in mammalian genomes, the regulation of alternative splicing has now become a prominent question. How does the cell ensure that the appropriate splice forms are expressed in the right place and at the right time? Our analysis confirms that the splice junctions around cryptic exons deviate from those flanking constitutive exons, and we identified the precise positions at which significant deviations occur. We also found compositional differences between cryptic and constitutive exons, one notable difference being the enrichment of pyrimidine-rich motifs in cryptic exons. Other motifs, previously reported to act as splice enhancers (Fairbrother et al. 2002), are found at significantly higher frequencies in constitutive exons. In contrast to previous reports that cryptic exons tend to be shorter than constitutive exons, we find that both short and large exon lengths are overrepresented in the cryptic set. This could indicate that different mechanisms are involved in the recognition of short versus long exons. Further analyses of the relative lengths of the cryptic exons and their flanking introns may reveal that the inclusion of cryptic exons in the mature mRNA is sometimes due to exon definition and other times to intron definition (Berget 1995). One of the most important applications that we envision for our database of alternative splice forms is to provide the data on which such hypotheses can be tested.
METHODS
Data Sets and Mapping
The cDNA collection used in this study comprised the RIKEN set of 60,770 full-length mouse cDNA sequences and 44,122 public domain mouse mRNA sequences from LocusLink and from the non-EST divisions of the Mouse Gene Index and GenBank (FANTOM2 Consortium and the RIKEN GSC Genome Exploration Group 2002). The procedure for constructing high-content, full-length libraries has been described in detail in Carninci et al. (1996). This procedure takes advantage of biochemical properties of the cap and poly(A) tail, and produces libraries in which >94% of the clones are full-length. We aligned the sequences to the mouse genome assembly (ftp://wolfram.wi.mit.edu/pub/mousecontigs/MGSCV3) using Paracel BLAST (pb BLASTALL -p BLASTN -e-5). We then extracted, for each cDNA sequence, the genomic region to which the largest number of cDNA nucleotides could be mapped (Kondo et al. 2001). Finally, we aligned each cDNA to its genomic region using Sim4 (Florea et al. 1999) with standard parameter settings.
EST sequences from dbEST (May 2002 release) and from the RIKEN data set of 5′- and 3′-end sequences were repeat-masked using the Paracel filtering package (Paracel Inc.) and aligned to the mouse genome using Paracel BLAST and BLAT (Kent 2002). We again selected the best locus for each sequence (Kondo et al. 2001) and aligned the ESTs to their regions using Sim4 (Florea et al. 1999).
Transcript Clustering Based on the Genome Mapping
Ideally, if the genome assembly were complete and correct, each transcript would map to exactly one locus (unless, of course, the genome contains identical copies of the same gene). In addition, if the genome can be divided into a number of genes, these genes should be clearly separated by intergenic regions, and transcripts whose genomic maps overlap should represent copies of the same transcription unit. Our clustering method directly reflects this view. Two transcripts were placed in a cluster if they mapped in the same orientation, and if their genomic maps overlapped by at least one nucleotide in one exon. Using the annotated representative transcript set (FANTOM2 Consortium and the RIKEN GSC Genome Exploration Group 2002), we estimated that only 84 of the 11,677 (<1%) of our clusters contained more than one representative transcript. Thus, cases of overlapping transcription units are sufficiently rare that our simple clustering method correctly retrieves the transcription units.
Characterization of Splice Variation
For the analysis of splice variation we selected transcripts that mapped quasicompletely to the genome assembly: Only initial and terminal nucleotides were allowed to remain unmapped, >95% of the cDNA sequence had to be involved in perfect matches with the genome, and every exon had to be ≥95% identical (or contain at most five errors) to the genome. These transcripts were clustered as described above. Single-exon and unspliced transcripts were discarded as they do not reveal any information about alternative splicing.
Clusters with multiple spliced transcripts were analyzed for the presence of cryptic exons and exons with alternative 5′/3′-splice sites. We compared all exons of all transcripts in a cluster in a pairwise fashion to identify those that used alternative 5′- and 3′-splice sites (Fig. 6). For each exon of a transcript, we tabulated all splice sites from the other transcript falling inside the exon. If the first of these splice sites was a 3′-splice site, both exons were marked as having alternative 3′-splice sites. If the last of these splice sites was a 5′-splice site, both exons were marked as having alternative 5′-splice sites. Note that this scheme does not classify intron inclusions as alternative splicing, because the above condition will not be satisfied. Our classification scheme did not consider intron inclusions as alternative splicing, because we could not distinguish them (computationally) from incomplete mRNA processing. Additionally, we identified cryptic exons, defined as exons that are present in some and skipped in other transcripts, in a cluster. We distinguished between cryptic exons internal to a transcript and initial or terminal cryptic exons. We believe that initial and terminal cryptic exons often occur in transcripts with alternative transcription, and we wanted to recognize these as a separate category for further investigation.
Figure 6.

Annotation of variant exons. The splice sites responsible for the annotation of the associated exons as variant are indicated by vertical bars. Exons with invariant splice sites are shown in green, exons with alternative 5′ sites are shown in yellow, and those with alternative 3′ sites in blue. Cryptic exons are indicated by a black box surrounding the exon. Introns are shown in red.
To avoid biases arising from cDNA library construction and cDNA amplification, we reported variation at the level of genomic exons. A genomic exon is defined as the union of all overlapping exons identified in the cDNAs of a cluster. We propagated the alternative splice information from transcript exons to the level of genomic exons as follows. A genomic exon is considered cryptic or constitutive depending on whether or not it is skipped in some of the transcripts in the cluster. In cases in which the cryptic exon occurred in both internal and initial/terminal positions in the transcripts, we called the genomic exon cryptic internal. If the genomic exon contained transcript exons with alternative 3′- and/or 5′-splice sites, the genomic exon was considered to have alternative 5′- and/or 3′-splice sites.
Estimation of the Frequency of Alternative Splicing in the Mouse Transcriptome
We set out to estimate the number of mouse genes that have alternative splice forms even though no alternative splice forms were found in our data. To this end, we need to calculate the probability P(n k) that a gene has n splice forms given that k identically spliced transcripts have been observed. In a Bayesian framework, this probability depends on the prior probability P(n) that a gene has n splice forms and on the prior probability P(p1, p2, ···, pn n) for the relative frequencies with which these splice forms (1 through n) occur in the cDNA pool from which our data have been generated.
We assume that the prior P(n) is a so-called scale-prior, P(n) ∝ (1/n). This prior is uniform in log(n), and it is also the unique prior that is invariant under scale transformations n λn and transformations of the form n nλ. Note that this prior puts almost all a priori probability on there being a large number of splice forms.
For the prior P(p1, p2, ···, pn n), we take a uniform distribution. This again puts a relatively high a priori weight on very skewed frequencies pi, and thus increases the a priori probability of observing only one splice form even if alternative splice forms exist. Thus, our calculation can be interpreted as providing an upper bound on the number of mouse genes that have alternative splice forms even though only one splice form occurred in the data. On the other hand, assuming that none of these genes has alternative splice forms provides a lower bound.
Given n and pi, the probability of observing k times the same splice form is given by 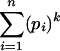 . Thus, the probability P(k n) of observing k times the same splice form given that the gene has, in fact, n splice forms is obtained by integrating out the nuisance parameters pi:
. Thus, the probability P(k n) of observing k times the same splice form given that the gene has, in fact, n splice forms is obtained by integrating out the nuisance parameters pi:
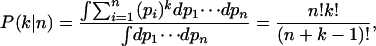 |
where the integrals are over the simplex 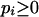 ,
, 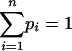 .
.
The posterior probability that the gene has n distinct splice forms given that k identically spliced transcripts have been observed is
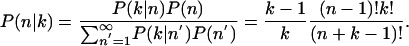 |
The probability that there is exactly one splice form given that k identical transcripts have been sampled is P(n = 1 k) = 1 - (1/k). Thus, the probability that the gene has multiple splice forms is P(n > 1 k) = (1/k). We then sum this probability for all multiple-transcript clusters that have only identical transcripts. In this way, we obtain an estimate for the number of genes that have multiple splice forms, even though only identical transcripts were observed. For the 6927 invariant multiple-transcripts clusters, this leads to an expected value of 1870 clusters (27%) with multiple splice forms. Together with the clusters for which we already detected splice variation, this yields an expected frequency of 57% of spliced genes with multiple splice forms.
This calculation assumes that each transcript in our data is a sample of an independent mature mRNA. However, because we cannot exclude that the same transcript was amplified multiple times through PCR, only transcripts from different libraries are guaranteed to be independent. Assuming that only transcripts from different libraries are independent, we have 5996 multiple-transcript invariant clusters of which an expected 37% have splice variation according to our model. Combining this again with the transcripts for which splice variation was observed, we obtain an overall upper bound of 60% for the frequency of splice variation.
Note that transcripts that differ by an intron inclusion are also guaranteed to be independent and are counted as such, and that single-transcript clusters are excluded from all these calculations because they do not contain any information about splice variation. Finally, note that our “upper bound” is not a formal upper bound; for example, it is in principle conceivable (although extremely unlikely) that every gene has an alternative splice form that occurs at such low frequencies that it would never be observed in our data set.
Relationship Between Exon Skipping and Alternative Splice Site Usage
To assess if alternative splice site usage correlates with exons being cryptic, we used the following Bayesian analysis. We calculate the posterior probability for a model in which exon “crypticness” is independent of alternative splice site usage (model 1), and for a model in which these forms of exon variation are correlated (model 2).
Let n00, n10, n01, and n11 be the number of constitutive exons with invariant splice sites, cryptic exons with invariant splice sites, constitutive exons with alternative splice site usage, and cryptic exons with alternative splice site usage. Under model 1, let p be the probability that an exon is cryptic and q the probability that it has alternative splice sites. The likelihood of the observed data D given p and q is
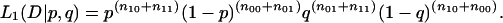 |
Integrating over p and q with a uniform prior yields
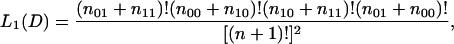 |
where n = n00 + n01 + n10 + n11 is the total number of exons. Under model 2, let p00, p01, p10, p11 be the probabilities of exons in the corresponding categories to be generated. The likelihood of the observed data D is
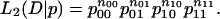 |
Integrating over the unknown values p with a uniform prior, we obtain
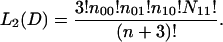 |
The posterior probability that the exon variations are correlated is then given by
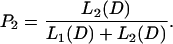 |
Note that this Bayesian test is almost identical to a Fisher exact test.
Impact of Alternative Splicing on the Proteome
We associated each splice-variant cluster with a representative transcript as follows. If a cluster contained sequences from the representative set of transcripts (FANTOM2 Consortium and the RIKEN GSC Genome Exploration Group 2002), the longest of these sequences was chosen as the representative for the cluster. If a cluster contained no sequence from the representative set (1207 clusters), the longest cDNA in the cluster was chosen as the cluster representative. Using the genome mapping of the representative transcript associated with the cluster and the FANTOM-annotated coding region (CDS; Furuno et al. 2003), we determined the genomic coordinates corresponding to the start and end of the coding region. If these coordinates were not covered by the sequence alignment, we chose the shortest genomic interval that bounded the CDS. If initial and terminal nucleotides were not mapped and such an interval could not be constructed, we chose the first and last mapped nucleotide of the transcript as the boundary of the CDS.
Distinct proteins forms will be generated when cryptic exons are present within the genomic map of the CDS, and when alternative 5′- and/or 3′-splice sites fall within the genomic map of the CDS. All genomic exons falling into one of these categories were counted as “CDS exons.” Genomic exons mapped upstream of the translation start of the representative transcript were counted as “5′-UTR exons,” and exons mapped downstream of the translation stop were counted as “3′-UTR exons.” Some clusters did not contain any transcripts with a CDS annotation, and for these, CDS status was deemed “unknown.”
EST Confirmation of Splice Variants
To independently confirm the splice forms in our variant clusters, we compared the variant exons against mouse mRNA sequences in the dbEST division of GenBank. We mapped dbEST sequences to the mouse genome using a strategy similar to that for mapping full-length cDNAs: We identified the locus using Paracel BLAST or BLAT (Kent 2002), and then we identified intron/exon boundaries using Sim4 (Florea et al. 1999). ESTs that mapped with ≥95% identity to the genome, with every exon mapped at 95% identity or ≤5 gaps or mismatches, were searched for the presence of the exon forms that we observed in the cDNA data. Variant exons were considered confirmed if each of the variant forms was found in at least one independent EST. For instance, a cryptic exon was considered confirmed if we found both an EST in which the exon was included and an EST in which the exon was skipped. Table 2 shows the results.
Comparison of Splice Signals Flanking Cryptic and Constitutive Exons
We extracted a data set of 15,298 internal exons that were found in at least four different libraries and that had no evidence of splice variation. The number of libraries is somewhat arbitrary, chosen only to enhance our confidence that these exons are, indeed, constitutive. These constitute our set of constitutive exons. The set of 3468 cryptic exons consists of internal exons that were present in at least one transcript and were skipped in at least one other transcript, but did not have any evidence of alternative splice site usage. We extracted 15 intronic nucleotides from the 5′- and 15 intronic nucleotides from the 3′-splice junction of these exons, and computed the frequencies f iα with which the nucleotides α ε 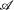 = A, C, G, T appear at distance i from the splice junction. For each position i we computed the information score Ii of the distribution f iα of nucleotides at that position, which is defined as
= A, C, G, T appear at distance i from the splice junction. For each position i we computed the information score Ii of the distribution f iα of nucleotides at that position, which is defined as
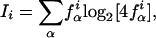 |
and is a measure of the deviation of the distribution of nucleotides at position i from a random distribution of nucleotides. The results are shown in Figure 3. The absolute size of the letters at each position corresponds to the information score Ii, and the relative sizes correspond to the relative frequencies f iα.
We additionally want to identify positions in the splice signal for which there is a significant difference between the nucleotide distributions of constitutive and cryptic exons. To this end, we computed for each position i the likelihoods of the observed nucleotide frequencies assuming that (1) the two data sets had different underlying nucleotide frequencies at that position, and (2) the two data sets were generated using the same underlying nucleotide frequencies at that position. Under the first model, there are independent frequencies p iα and q iα with which the nucleotides α ε = A, C, G, T occurat distance i from the splice junction in constitutive and cryptic exons. With n iα and m iα the number of observed nucleotides α at position i in the two data sets, the likelihood for the observed nucleotide counts given the frequencies p iα and q iα is
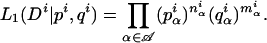 |
Using a uniform prior for the unknown frequencies pαi and qαi, we obtain
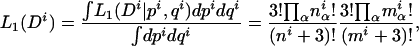 |
where 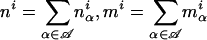 , and the integrals are over the simplex
, and the integrals are over the simplex 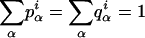 .
.
Similarly, if we assume that the nucleotide counts at position i in the constitutive and cryptic exons derive from the same underlying distribution piα, we obtain
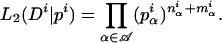 |
Using again a uniform prior over pαi, we obtain
 |
The likelihood ratio L1(Di)/L2(Di) for each position relative to both 3′- and 5′-splice junctions is plotted in Figure 4.
Comparative Analysis of Motif Composition in Cryptic and Constitutive Exons
For each motif of eight or fewer nucleotides, we computed the frequency fco of occurrence in the set of 15,298 constitutive exons, and their frequency fcr in the set of 3468 cryptic exons. We focused our analysis on 40 nucleotides around the 5′- and 3′-splice junctions of these exons. Under the assumption that the probabilities of occurrence of a motif in different exons and different positions around a given splice junction within an exon are independent, the number of occurrences of the motif is binomially distributed. Approximating the binomial distribution by a Gaussian, we calculated, for each motif, the z-statistics to determine if the motif is significantly over- or underrepresented in one category of exons (cryptic or constitutive). For each length l we select all motifs for which the p-value corresponding to this z-statistic is <4-l.
We further purged this list of motifs by removing those that are contained within longer motifs that also occur in the list. Finally, we compare the frequencies of the remaining motifs with the frequencies that we would expect based on the nucleotide frequencies in constitutive and cryptic exons. We again calculate the z-statistics and only retain the motifs that are significantly over- or underrepresented in at least one set of exons with respect to a mononucleotide frequency model.
In short, we collect all motifs that have significantly different frequencies of occurrence in constitutive versus cryptic exons, and whose frequencies of occurrence are significantly different from the frequencies expected based on the mononucleotide frequencies in at least one set of exons. The resulting 90 motifs are shown in Supplementary Figure 1.
Acknowledgments
M.Z. thanks Henry Prince and Ben Snyder for assistance in developing the database, Magda Konaska for valuable suggestions, and Erik van Nimwegen for critically reading the manuscript. This research has been supported in part by National Cancer Institute Grant R33-CA84699 and National Science Foundation Grant DBI9984882 to T.G., and by the Rockefeller University Lita Annenberg Hazen Presidential Fellowship (M.Z.). The RIKEN data sets of full-length cDNA sequences and 5′- and 3′-end EST sequences have been generated by the Genomic Sciences Center RIKEN Yokohama Institute in Japan and by the Functional annotation of the Mouse Genome (FANTOM) Consortium.
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.1017303.
Footnotes
[Supplemental material is available online at www.genome.org.]
References
- Basu, A., Dong, B., Krainer, C., and Howe, A.R. 1997. The intracisternal A-particle proximal enhancer-binding protein activates transcription and is identical to the RNA- and DNA-binding protein p54nrb/NonO. Mol. Cell. Biol. 17: 677-686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berget, S. 1995. Exon recognition in vertebrate splicing. J. Biol. Chem. 270: 2411-2414. [DOI] [PubMed] [Google Scholar]
- Blencowe, B., Bauren, G., Eldridge, A., Issner, R., Nickerson, J., Rosonina, E., and Sharp, P. 2000. The SRm160/300 splicing coactivator subunits. RNA 6: 111-120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brett, D., Hanke, J., Lehmann, G., Haase, S., Delbruck, S., Krueger, S., Reich, J., and Bork, P. 2000. EST comparison indicates 38% of human mRNAs contain possible alternative splice forms. FEBS Lett. 474: 83-86. [DOI] [PubMed] [Google Scholar]
- Brett, D., Pospisil, H., Valcarcel, J., Reich, J., and Bork, P. 2002. Alternative splicing and genome complexity. Nat. Genet. 30: 29-30. [DOI] [PubMed] [Google Scholar]
- Burd, C. and Dreyfuss, G. 1994. RNA binding of hnRNP A1: Significance of hnRNP A1 high-affinity binding sites in pre-mRNA splicing. EMBO J. 13: 1197-1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke, J., Wang, H., Hide, W., and Davison, D. 1998. Alternative gene form discovery and candidate gene selection from gene indexing projects. Genome Res. 8: 276-290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caceres, J. and Krainer, A. 1997. Mammalian pre-mRNA splicing factors. In Eukaryotic mRNA processing (ed. A. Krainer), pp. 174-182.
- Oxford IRL Press, New York. Carninci, P., Kvam, C., Kitamura, A., Ohsumi, T., Okazaki, Y., Itoh, M., Kamiya, M., Shibata, K., Sasaki, N., Izawa, M., et al. 1996. High-efficiency full-length cDNA cloning by biotinylated CAP trapper. Genomics 37: 327-336. [DOI] [PubMed] [Google Scholar]
- Chabot, B., Blanchette, M., Lapierre, I., and La Branche, H. 1997. An intron element modulating 5′ splice site selection in the hnRNP A1 pre-mRNA interacts with hnRNP A1. Mol. Cell. Biol. 17: 1776-1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croft, L., Schandorff, S., Clark, F., Burrage, K., Arctander, P., and Mattick, J. 2000. ISIS, the intron information system, reveals the high frequency of alternative splicing in the human genome. Nat. Genet. 24: 340-341. [DOI] [PubMed] [Google Scholar]
- Dominski, Z. and Kole, R. 1992. Cooperation of pre-mRNA sequence elements in splice site selection. Mol. Cell. Biol. 12: 2108-2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eldridge, A., Li, Y., Sharp, P., and Blencowe, B. 1999. The SRm160/300 splicing coactivator is required for exon-enhancer function. Proc. Natl. Acad. Sci. 96: 6125-6130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairbrother, W. and Chasin, L. 2000. Human genomic sequences that inhibit splicing. Mol. Cell. Biol. 20: 6816-6825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairbrother, W., Yeh, R., Sharp, P., and Burge, C. 2002. Predictive identification of exonic splicing enhancers in human genes. Science 297: 1007-1013. [DOI] [PubMed] [Google Scholar]
- FANTOM2 Consortium and the RIKEN GSC Genome Exploration Group. 2002. Analysis of the mouse transcriptome based upon functional annotation of 60,770 full length cDNAs. Nature 420: 563-573. [DOI] [PubMed] [Google Scholar]
- Florea, L., Hartzell, G., Zhang, Z., Rubin, G., and Miller, W. 1999. A computer program for aligning a cDNA sequence with a genomic DNA sequence. Genome Res. 8: 967-974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuno, M., Kasukawa, T., Saito, R., Adachi, J., Suzuki, H., Baldarelli, R., Hayashizaki, Y., and Okazaki, Y. 2003. CDS annotation in full-length cDNA sequence. Genome Res. (this issue). [DOI] [PMC free article] [PubMed]
- Gelfand, M., Dubchak, I., Dralyuk, I., and Zorn, M. 1999. ASDB: Database of alternatively spliced genes. Nucleic Acids Res. 27: 301-302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henriksen, M.A., Betz, A., Fuccillo, M., and Darnell, J. 2002. Negative regulation of STAT92E by an N-terminally truncated STAT protein derived from an alternative promoter site. Genes & Dev. 16: 2379-2389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman, B. and Grabowski, P. 1992. U1 snRNP targets an essential splicing factor, U2AF65, to the splice site by a network of interactions spanning the exon. Genes & Dev. 6: 2554-2568. [DOI] [PubMed] [Google Scholar]
- Ji, H., Zhou, Q., Wen, F., Xia, H., Lu, X., and Li, Y. 2001. AsMamDB: An alternative splice database of mammals. Nucleic Acids Res. 29: 260-263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kan, Z., Rouchka, E., Gish, W., and States, D. 2001. Gene structure prediction and alternative splicing analysis using genomically aligned ESTs. Genome Res. 11: 889-900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kan, Z., States, D., and Gish, W. 2002. Selecting for functional alternative splices. Genome Res. 12: 1837-1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent, W. 2002. BLAT—The BLAST-like alignment tool. Genome Res. 12: 656-664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent, W. and Zahler, A. 2001. The intronerator: Exploring introns and alternative splicing in Caenorhabditis elegans. Nucleic Acids Res. 28: 91-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo, S., Shinagawa, A., Saito, T., Kiyosawa, H., Yamanaka, I., Aizawa, K., Fukuda, S., Hara, A., Itoh, M., Kawai, J., et al. 2001. Computational analysis of full-length mouse cDNAs compared with human genome sequences. Mamm. Genome 12: 673-677. [DOI] [PubMed] [Google Scholar]
- Konno, H., Fukunishi, Y., Shibata, K., Itoh, M., Carninci, P., Sugahara, Y., and Hayashizaki, Y. 2001. Computer-based methods for the mouse full-length cDNA encyclopedia: Real-time sequence clustering for construction of a nonredundant cDNA library. Genome Res. 11: 281-289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander, E.S., Linton, L.M., Birren, B., Nusbaum, C., Zody, M.C., Baldwin, J., Devon, K., Dewar, K., Doyle, M., FitzHugh, W., et al. 2001. Initial sequencing and analysis of the human genome. Nature 409: 860-921. [DOI] [PubMed] [Google Scholar]
- Liu, H., Zhang, M., and Krainer, A. 1998. Identification of functional exonic splicing enhancer motifs recognized by individual SR proteins. Genes & Dev. 12: 1998-2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangan, M. and Frazer, K. 1999. An extensive list of genes that produce alternative transcripts in the mouse. Bioinformatics 15: 170-171. [DOI] [PubMed] [Google Scholar]
- McCullough, A. and Berget, S. 1997. Gtriplets located throughout a class of small vertebrate introns enforce intron borders and regulate splice site selection. Mol. Cell. Biol. 17: 4562-4571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min, H., Chan, R., and Black, D. 1995. The generally expressed hnRNP F is involved in a neural-specific pre-mRNA splicing event. Genes & Dev. 9: 2659-2671. [DOI] [PubMed] [Google Scholar]
- Mironov, A., Fickett, J., and Gelfand, M. 1999. Frequent alternative splicing of human genes. Genome Res. 9: 1288-1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modrek, B. and Lee, C. 2002. A genomic view of alternative splicing. Nat. Genet. 30: 13-19. [DOI] [PubMed] [Google Scholar]
- Modrek, B., Resch, A., Grasso, C., and Lee, C. 2001. Genome-wide detection of alternative splicing in expressed sequences of human genes. Nucleic Acids Res. 29: 2850-2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagashima, T., Silva, D.G., Petrovsky, N., Socha, L.A., Suzuki, H., Saito, R., Kasukawa, T., Kurochkin, I.V., Konagaya, A., and Schönbach, C. 2003. Inferring higher functional information for RIKEN mouse full-length cDNA clones with FACTS. Genome Res. (this issue). [DOI] [PMC free article] [PubMed]
- Ravasi, T., Huber, T., Zavolan, M., Forrest, A., Gaasterland, T., Grimmond, S., RIKEN GER Group and GSL Members, and Hume, D.A. 2003. Systematic characterization of the zinc-finger-containing proteins in the mouse transcriptome. Genome Res. (this issue). [DOI] [PMC free article] [PubMed]
- Robberson, B., Cote, G., and Berget, S. 1990. Exon definition may facilitate splice site selection in RNAs with multiple exons. Mol. Cell. Biol. 10: 84-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romanelli, M., Lorenzi, P., and Morandi, C. 2000. Organization of the human gene encoding heterogeneous nuclear ribonucleoprotein type I (hnRNP I) and characterization of hnRNP I related pseudogene. Gene 255: 267-272. [DOI] [PubMed] [Google Scholar]
- Schaal, T. and Maniatis, T. 1999. Selection and characterization of pre-mRNA splicing enhancers: Identification of novel SR protein-specific enhancer sequences. Mol. Cell. Biol. 19: 1705-1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staknis, D. and Reed, R. 1994. SR proteins promote the first specific recognition of pre-mRNA and are present together with the U1 small nuclear ribonucleoprotein particle in a general splicing enhancer complex. Mol. Cell. Biol. 14: 7670-7682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamm, S., Zhang, M., Marr, T., and Helfman, D. 1994. A sequence compilation and comparison of exons that are alternatively spliced in neurons. Nucleic Acids Res. 22: 1515-1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamm, S., Zhu, J., Nakai, K., Stoilov, P., Stoss, O., and Zhang, M. 2000. An alternative-exon database and its statistical analysis. DNA Cell Biol. 19: 739-756. [DOI] [PubMed] [Google Scholar]
- Suzuki, Y., Taira, H., Tsunoda, T., Mizushima-Sugano, J., Sese, J., Hata, H., Ota, T., Isogai, T., Tanaka, T., Morishita, S., et al. 2001. Diverse transcriptional initiation revealed by fine, large-scale mapping of mRNA start sites. EMBO Rep. 2: 388-393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tacke, R. and Manley, J. 1999. Determinants of SR protein specificity. Curr. Opin. Cell Biol. 11: 358-362. [DOI] [PubMed] [Google Scholar]
- Talerico, M. and Berget, S. 1990. Effect of 5′ splice site mutations on splicing of the preceding intron. Mol. Cell. Biol. 10: 6299-6305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Z., Hoffman, H., and Grabowski, P. 1995. Intrinsic U2AF binding is modulated by exon enhancer signals in parallel with changes in splicing activity. RNA 1: 335-346. [PMC free article] [PubMed] [Google Scholar]
- Wollerton, M., Gooding, C., Robinson, F., Brown, E., Jackson, R., and Smith, C. 2001. Differential alternative splicing activity of isoforms of polypyrimidine tract binding protein (PTB). RNA 7: 819-832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahler, A., Neugebauer, K., Lane, W.S., and Roth, M. 1993. Distinct functions of SR proteins in alternative pre-mRNA splicing. Science 260: 219-222. [DOI] [PubMed] [Google Scholar]
- Zavolan, M., van Nimwegen, E., and Gaasterland, T. 2002. Splice variation in mouse full-length cDNAs identified by mapping to the mouse genome. Genome Res. 12: 1377-1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
WEB SITE REFERENCES
- ftp://wolfram.wi.mit.edu/pub/mousecontigs/MGSCV3; draft of the mouse genome sequence.
- http://facts.gsc.riken.go.jp; Functional Association/annotation of cDNA clones from Text/sequence Sources (FACTS).
- http://genomes.rockefeller.edu/MouSDB; database of alternative splice forms in the mouse transcriptome.
- http://smart.embl-heidelberg.de; Simple Modular Architecture Research Tool (SMART). [DOI] [PMC free article] [PubMed]