

Abstract
This study discusses three software tools, the first two aid in integrating sequence with an FPC physical map and the third automatically selects a minimal tiling path given genomic draft sequence and BAC end sequences. The first tool, FSD (FPC Simulated Digest), takes a sequenced clone and adds it back to the map based on a fingerprint generated by an in silico digest of the clone. This allows verification of sequenced clone positions and the integration of sequenced clones that were not originally part of the FPC map. The second tool, BSS (Blast Some Sequence), takes a query sequence and positions it on the map based on sequence associated with the clones in the map. BSS has multiple uses as follows: (1) When the query is a file of marker sequences, they can be added as electronic markers. (2) When the query is draft sequence, the results of BSS can be used to close gaps in a sequenced clone or the physical map. (3) When the query is a sequenced clone and the target is BAC end sequences, one may select the next clone for sequencing using both sequence comparison results and map location. (4) When the query is whole-genome draft sequence and the target is BAC end sequences, the results can be used to select many clones for a minimal tiling path at once. The third tool, pickMTP, automates the majority of this last usage of BSS. Results are presented using the rice FPC map, BAC end sequences, and whole-genome shotgun from Syngenta.
FPC (FingerPrinted Contigs; Soderlund et al. 1997) maps are built from fingerprinted clones and annotated with markers scored against the clones in the map. The contigs are ordered by genetic markers that have also been scored against the clones in the map. FPC maps are often used interactively to select an MTP (minimal tiling path) of clones to sequence. As sequence becomes available, it can be used to further annotate the map and aid in the sequencing project. Toward this end, we have developed the FSD (FPC Simulated Digest) tool and the BSS (Blast Some Sequence) tool, where the sequence for BSS can be from two different classes. The first class is sequence associated with clones in the map: draft-sequenced clones, finished-sequenced clones, and BAC end sequences (BES). The second class is sequence that is not associated with clones in the map: whole-genome shotgun (WGS), gene-rich contigs (GRC), and the sequence of markers.
Adding Sequenced Clones to the Map
A clone fingerprint produced by agarose gel (Marra et al. 1997) is analyzed by Image (Sulston et al. 1989; www.sanger.ac.uk/software/Image), which generates the migration rate for each band, and each migration rate is converted to a size. FPC can assemble clones into contigs by use of either rates or sizes, but migration rates are typically used. The FSD tool runs a simulated digest on a sequenced clone and converts the size to a migration rate. The SD (simulated digest) clones can be added to the FPC map in the exact same manner as any other fingerprinted clone. The benefits of this procedure are as follows: confirmation of the location and assembly of a sequenced clone, annotation of the map with finished sequence, anchor information for contigs, and an integrated map of sequence from many sources.
Electronically Adding Markers to the Map
Scoring markers to find what clones they hit is time consuming when done by standard approaches such as hybridization or PCR. Once marker sequences become available, they can be added electronically. Using the BESs and any sequenced (draft or finished) clones, it has been standard to add PCR-based markers to the map using ePCR (Schuler 1998), as was done by the International Human Genome Mapping Consortium (2001). For other types of markers in which the complete sequence is known, they can be added to the map by use of sequence-similarity programs such as BLAST (Altschul et al. 1997) or MegaBLAST (Zhang et al. 2000). For sequences that have introns spliced out, programs like BLAT (Kent 2002) are used to align the spliced sequence to genomic sequence. We have developed a tool called BSS (Blast Some Sequence) that makes it easy to add electronic markers. It runs within FPC, creates a report of hits, shows alignments if requested, and adds markers to the map in either interactive or batch mode. It has the option of running BLAST, MegaBLAST, or BLAT.
A Hybrid Approach of WGS and BAC-Based Sequencing
The BAC-based sequencing approach versus the WGS approach was first debated in 1997 by Green (1997) and Weber and Myers (1997). In 2001, papers on the human genome sequence were published using the BAC-based approach (The International Human Sequencing Consortium 2001) and the WGS approach (Venter et al. 2001). The discussion continues in papers by Waterston et al. (2002), Green (2002), and Myers et al. (2002). A hybrid-sequencing strategy is emerging that combines the strengths of the BAC based with the strengths of the WGS. The WGS is faster and cheaper, but results in thousands of sequenced contigs, the majority of which are not anchored. The BAC based inherently has most sequences anchored, as the clones are selected from the physical map, plus the clones are finished to a 1 in 10,000 error rate. The finishing stage is the most time consuming, but it produces sequence that is much more valuable as a resource (Mardis et al. 2002). This hybrid approach is being used for the sequencing of the mouse genome, in which a BAC map was built, the clones end sequenced (Gregory et al. 2002), and WGS data is being generated (http://www.ensembl.org/Mus_musculus/).
Although not intentional, the sequencing of rice has taken on a hybrid approach. The rice genome (Oryza sativa L. ssp japonica) has an FPC map (Chen et al. 2001), and the BACs have been end sequenced (Mao et al. 2000). The International Rice Genome Sequencing Project (IRGSP; Sasaki and Burr 2000) has embarked on providing a complete, finished, anchored sequence using the BAC-based approach (see http://www.genome.arizona.edu/shotgun/rice/). In the spring of 2001, Monsanto announced the draft sequence of the japonica rice genome using the BAC-based approach (Barry 2001). In the spring of 2002, Syngenta published a 6× draft sequence of the japonica rice genome using the WGS approach (Goff et al. 2002). Also, in the spring of 2002, the Beijing Genomics Institute (BGI), in collaboration with several other laboratories, published a 4.2× draft sequence of the indica rice genome using the WGS approach (Yu et al. 2002). All three drafts are in many sequenced contigs, and although they give partial information of genetic content, they are no substitute for the complete sequence. Given the international importance of rice and the fact that its genome is compact (430 Mb), it is a good model genome with which to compare other cereal genomes that are only sparsely sequenced. Therefore, the IRGSP is continuing to finish the genome, and chromosomes 1, 4, and 10 are now complete (Feng et al. 2002; Sasaki et al. 2002; The Rice Chromosome 10 Sequencing Consortium 2003). To support this effort, both Syngenta and Monsanto have made their data available to the IRGSP for targeted regions. This data is used in conjunction with reads produced in the laboratory to increase the coverage for clones. Consequently, the rice genome is being sequenced mainly by the BAC-based approach, but augmented by the draft sequence for a hybrid approach. To aid in this effort, BSS has been used to locate draft sequence near a clone in order to use its reads for finishing, close sequence gaps, and close gaps between contigs (Y. Yu, pers. comm).
For many plants, in which the genomes are often large and very repetitive, there is not a large enough benefit to justify sequencing the whole genome. The hybrid-sequencing approach of using a BAC map, BESs, and draft sequence is ideal for providing an initial landscape of the genome, which can later be followed by BAC-based sequencing of the interesting gene-rich regions. A variation on this theme is being sponsored by the NSF Plant Genome Program for sequencing the maize genome (Chandler and Brendel 2002), but in this case, the draft sequence is targeted toward gene-rich regions by using methyl-filtered (Rabinowicz et al. 1999) and high-complexity Cot clones (Peterson et al. 2002). These gene-rich sequences will be assembled with other sequences from maize to form gene-rich contigs. We anticipate that BSS will be used effectively to add the GRCs to the map as markers, which will elucidate the gene-rich regions for subsequent complete BAC sequencing.
Selecting a Minimal Tiling Path
Three paradigms are used for selecting minimally overlapping clones for sequencing. The first is a map-based approach as used by the Caenorhabditis elegans project (Coulson et al. 1986) and human chromosomes 1, 6, 20, 22, and X (Bentley et al. 2001). Fingerprints of clone pairs that appear to have a minimum overlap are analyzed in the FPC Gel Image display. Viewing the gel images of neighboring clones helps identify false-positive and false-negative bands. With this method, a complete MTP can be picked before any sequencing is started, so that all clones can be sequenced in parallel. However, the amount of overlap may be large, because a band is on the average 4096 bases, and clones need to share multiple bands to have enough evidence of overlap. Manual selection of one minimally overlapping pair takes ∼15 min per pair for an experienced person. The International Human Genome Mapping Consortium (2001) used a map-based approach with an automatic MTP program to select the clones to sequence, but in order to use it, the clones in the map had to be ordered manually. The resulting MTP had average overlaps of 47.5 kb.
The BES-based approach (Venter et al. 1996) bypasses building a physical map, but a BES library for the clones is required and fingerprints are advantageous. A seed clone is picked, sequenced, and finished. The BES library is queried for hits to the finished sequence. Fingerprints of candidate MTP clones are compared with overlapping clones to ensure internal consistency. The new set of MTP clones is sequenced, and new clones are picked off the ends of this set. This process is repeated until the region is sequenced. By use of sequence comparison, the amount of overlap required for MTP pairs is reduced drastically compared with the map-based approach. However, as no map is available, the overlap cannot be verified by clone positions on the map, therefore, the risk of false positives is high, especially when considering repeats and errors in the low-quality BES. It is also time consuming and error prone to look through pages of BLAST output, where the majority of hits are false positives. For example, when running the sequence of rice BAC clone accession number AC107619 against the rice BES database using a BLAST expectation value of 1e-100, only 17 of the 500 hits are true hits. It is typical to select many seed clones to initially sequence and then select minimally overlapping clones from the seeds; this introduces a degree of parallelism for sequencing, but not to the same extent as the map-based selection.
The third approach is a hybrid of the first two and has been used by various sequencing projects such as Arabidopsis (Marra et al. 1999) and Drosophila (Hoskins et al. 2000). A map is built and the ends of the clones are sequenced. The seed clone picking and extending process is similar to that used in the BES-based approach. However, the map is used to verify overlap, so the risk of false-positive overlaps is reduced drastically. To make this approach much easier for the user, the FPC tool BSS may be used to perform the seed sequence→BES comparisons and show hits as they relate to the FPC map.
We have recently developed a fourth method for picking an MTP. It uses the output of the BSS WGS→BES comparison, runs a shortest path algorithm (Aho et al. 1983) to find all MTPs, and then filters for the best ones. By use of this approach, most MTP clones can be picked automatically in one execution of the algorithm, thereby making parallelized sequencing possible and reducing manual selection tremendously. No manual reordering of the clones is needed to use this approach. The development of the pickMTP algorithm was precipitated by the large amount of draft data that became available to the rice project. If the hybrid sequencing trend continues, in which large projects have FPC maps, BESs, and draft data available, and if it is desirable to sequence to completion regions of the genome, then this tool will both save user time and avoid large overlaps, as will be described in the Results section.
The amount of data used in FPC has grown enormously, yet the original graphics for the contig display are still used. Often markers and remarks run off the display, and not all frameworks are shown if there are too many. We have recently developed a new contig display that allows the user to define tracks of data based on filters of name substring, remarks, and attributes. This new display will greatly simplify viewing the various types of data provided by these tools.
Table 1 shows a summary of the different functions just described. The functions that are marked with an asterisk are further described in the Results and Methods sections.
Table 1.
Summary of Functions
| Program | Actiona | Modeb | Results |
|---|---|---|---|
| Add Sequence | |||
| *FSD + FPC functions | Simulated digest | Confirm location of clone | |
| Add clones from other maps | |||
| Anchor contigs to chromosomes | |||
| *BSS | Marker→BES | Batch or interactive | Electronic markers |
| Marker→Sequence | Merge FPC contigs | ||
| BSS | GRC→BES | Batch | Place GRCs on the map to eludicate gene rich regions. |
| GRC→Sequence | Merge FPC contigs | ||
| Selecting MTPc | |||
| *BSS | Sequence→BES | Interactive | Manual selection |
| *BSS + pickMTP | WGS→BES | Batch | Automatic selection |
| Finishingc | |||
| BSS | Draft→Sequence | Interactive | Locate draft (WGS or BAC-based) sequence that overlaps clone in order to use the reads and close sequencing gaps |
| BSS | WGS→BES | Interactive or batch | Merge FPC contigs |
(FSD) FPC Simulated Digest, (BSS) Blast Some Sequence, (BES) BAC End Sequence, (GRC) Gene Rich Contig, (WGS) Whole Genome Shotgun.
The GRC, WGS, and Draft would all be treated as markers in BSS, i.e., use the BSS Marker→Sequence and Marker→BES. Sequence refers to BAC-based sequence.
Interactive mode allows the user to add one marker at a time after confirming the marker by the BSS report and sequence alignment. Batch mode adds all markers at once, based on a user specified filter.
A marker added for a BES or for draft sequence may not be of interest for the release version of the database, in which case, a copy of the FPC database can be made for the intermediate results.
Features discussed in Results and Methods.
RESULTS
Adding Simulated Digest
FSD is a supplemental program to FPC that performs a complete digest in silico on a sequence and produces the sizes of the fragments. The sizes are converted into migration rates so that they can be assembled into an FPC map built with migration rates. Nightly, we download from GenBank any modified or new rice sequences, and automatically add them to the rice FPC map. The Web-based FPC display is updated nightly as well, and can be viewed at http://www.genome.arizona.edu/fpc/rice. Each SD clone is compared with all other clones, and is assigned a position based on the clone it matches best (given that there is a match satisfying a cutoff of 1e-10).
One benefit of adding the SD clone is to confirm the sequenced clone. The SD clones should automatically position very close to the agarose fingerprint. If this is not the case, the correct clone was not sequenced or the sequence is incorrect or unfinished. On the rice FPC map, there are 1567 sequenced clones, 180 of which do not match their corresponding fingerprinted clones at a 1e-10 cutoff. Of these 180, 155 are not yet finished. Two of the remaining 25 were sequenced by the Arizona Genomics Institute and had match values of 9e-10 and 3e-07. Inspection of the fingerprints showed that most of the bands between the simulated and real fingerprint would match if a slightly larger tolerance were used, indicating that the original fingerprints were skewed. A second benefit of FSD is that there is a large amount of rice data available publicly from GenBank, and many of the sequenced clones are not from our FPC map. By use of the SD clones generated by FSD, 1997 clones were integrated into our map from other sources. Figure 1 shows clones from the Japanese tiling path incorporated into an FPC contig.
Figure 1.

SD clones and electronic markers. The clones in blue are simulated digest clones from the Japanese minimal tiling path; most of the original clones are not in FPC. The markers in blue are electronic markers supplied by Gramene. As described in the text, these markers could have been added by BSS. The contig display is from FPC V7. Tracks can be added, resized, and moved around.
The combination of genetic markers and sequenced clones verifies the location of a contig on a chromosome. A function in FPC V6.4 assigns contigs to chromosomes using this information. For rice, 308 contigs have been assigned to chromosomes, of which 284 contigs have genetic markers that give them a position on the chromosome. Eleven contigs have strong evidence suggesting a position on more than one chromosome, which indicates incorrect joins. The rules for contig to chromosome assignment are given in the Methods section.
Overview of BSS
The Methods section describes the details of using BSS for adding electronic markers and selecting the next clone for sequencing. As BSS has many uses, the different modes and execution types are somewhat complicated. This section provides a brief overview. BSS has three search modes, Marker→BES, Marker→Sequence, Sequence→BES, and it can be run per contig or for the whole map. The query and target database may be one or more files in a directory. A report is generated for each of the query files and can be viewed in the BSS report window. An alignment can be viewed by selecting a hit from the report. The hits can be added as markers either (1) interactively for each hit, (2) one file at a time, or (3) for all BSS files in a directory. A variety of ways are provided for the user to manipulate the results for maximum flexibility. For example, the user can edit the BSS report interactively to delete some unwanted hits, and then add the rest of the hits as electronic markers. BSS also provides a filter so that only the markers with given attributes will be added. Figure 2 shows the various windows associated with BSS. The content of these four will vary depending on the mode, search program, and whether it is run per contig or on the whole FPC file.
Figure 2.

The windows that would be shown for a Marker→Sequence search using MegaBLAST in the Batch mode, which runs the search on all contigs. Values are entered in the Setup and Batch BSS windows and the search started. The results can be viewed in the BSS Report window. If the results are to be automatically added as markers using the Add as FPC Markers function, the Marker Add Conditions may first be altered.
Adding Electronic Markers
For the Gramene project (Ware et al. 2002), electronic markers were identified from the Japanese Rice Genomic Research Program (JRGP, Harushima et al. 1998, http://rgp.dna.affrc.go.jp/Publicdata.html) and the rice draft clone sequence, and have been added to the rice FPC. They used the search tool BLAT, with parameter minScore = 120, along with three additional screens on the resulting hits as follows: (1) 80% or more of the total marker length must be matched, (2) the largest target gap size must be less than 3000, and (3) the clone that was hit must be located on the chromosome to which the marker was mapped (cf. http://www.gramene.org/documentation/Alignment_docs, and the Methods section below). We have studied this strategy and found it to be a useful alternative to the traditional BLAST E-value search; therefore, functionality to carry out the first two of these screens has been incorporated into BSS. The results of our study are summarized below; details and further discussion can be found at www.genome.org and http://www.genome.arizona.edu/software/fpc/gr2003_supplemental. Note that we had previously compared MegaBLAST to BLAST for this data set, and found MegaBLAST to be faster without any performance loss, therefore, this study compared BLAT to MegaBLAST.
Screening for the percent match makes sense because one is searching for exact embeddings of the marker in the target. A 50% match to a long marker may have a very low E-value, but still should not be considered a good hit; conversely, a 100% match to a short marker may not produce a particularly low E-value, but it is still the best possible match that this marker can have.
If the markers are EST markers, then the percent-match strategy necessitates the use of BLAT, which automatically joins together the hits for consecutive exons; in constrast, MegaBLAST output will have a separate high-scoring pair for each exon, making it difficult to tell the true percentage of the marker that was matched. If BLAT is used, then it is also desirable to place a limit on the maximum allowed target gap; this limit should be related to the typical intron size of the genome in question.
Our tests made use of the full rice genome draft sequence, and markers developed by the JRGP. We compared BLAT with the percent match and MegaBLAST with E-value only. On the 263 sequenced-tagged sites (STSs) in this set, the percent-match strategy was greatly superior, outperforming E-value by a factor of >2. On the 2191 EST markers, percent-match was also superior, but by only 20%. The difference between the STS and EST results is possibly due to differing quality in the marker sequences; poor quality sequence, which tends to occur on the ends, will have a very detrimental effect on the percent-match strategy. The extent of these errors can be estimated by observing how many markers fail to score any hits at a high match percentage; if the number scoring hits at 90% match is considerably lower than would be expected on the basis of coverage of the target sequence, then an E-value screen may be needed as well.
We also tested these strategies using the same marker set, but with rice BESs as the target. In this case, one no longer expects the full marker sequences to be found, therefore, the percent-match strategy loses much of its rationale. Our tests did not show a significant difference between the percentage-match and E-value strategies in this case.
Manually Selecting the Next Clone to Sequence by Use of BESs
Blasting a clone sequence against the BESs associated with all FPC contigs often produces hundreds of hits. Furthermore, without map information, the orientation of a clone is not known, therefore, a clone may be selected that totally overlaps the sequenced clone. Using FPC resolves both of these problems and using BSS automates much of the process. Unless the user specifies otherwise, BSS uses only the BESs from the contig that contain the sequenced clone, as this restriction greatly reduces the number of hits. The BES hits are added to the contig, which makes it easy to view what clones to consider and to determine the orientation of a clone (see Fig. 3). From the BSS report, a hit can be selected to see the alignment. The Arizona Genomics Institute (AGI) has selected 209 clones for sequencing. The clones that were selected with both BES and FPC data all have correct overlap. AGI started using BSS about half-way through the selection of clones, found it of considerable help, and continued to get consistently accurate overlaps (Y. Yu, pers. comm.). Intuitively, this would be expected, as the dual constraints of a BES hitting a clone and being near the clone in the FPC map are strong evidence of a correct minimal overlap. The one problem still encountered was the bottleneck in waiting for the sequence of clones to be finished to the extent of having ordered contigs, so that BESs hitting the ends of the clone can be determined. Using draft sequence as described in the next section can reduce this bottleneck.
Figure 3.

Selecting the next clone for sequencing. Clone AP005522 has been sequenced. It was blasted against all of the BESs in the contig. The clone was added as a marker attached to all of the clones that had a BES hit. Selecting the AP005522 marker highlights these clones. Some clones are obviously contained in the sequenced clone, so can be ignored. The BSS report file can be viewed to see details of the hits.
Automatically Selecting a Minimal Tiling Path by Use of Draft Data and BESs
BSS can be used to map WGS-sequenced contigs to the FPC map by blasting all sequenced contigs against the BESs. Because draft-sequenced contigs are generally not associated with a clone, they need to be blasted against all BESs. The draft-sequenced contigs are much shorter than a BAC clone (avg. ∼5000 bases for rice indica; Yu et al. 2002), therefore, the number of hits is significantly less, and a region in the map can often be unambiguously identified as that of the sequenced contig. In such cases, a sequenced contig can be anchored to the FPC map by adding it as an electronic marker via BSS. With this information, one could determine when two neighboring FPC clones hit the same sequenced contig, and a minimal tiling path could be constructed on the basis of the amount of overlap of the clone pairs given by the map and the sequence alignment. However, the amount of data is overwhelming; for example, for a 22-Mb sequence, there are over 8000 initial pairs. Hence, pickMTP was developed to automate this method.
The pickMTP algorithm can be divided into several sections as follows: (1) use BSS to blast the draft sequence against the BES library, (2) for all possible pairs of hits in the BSS report, filter out pairs on the basis of various rules, and output a list of overlapping clone pairs, (3) from the overlapping pairs, identify all possible expressways, where each expressway is a path of minimally overlapping clones, (4) greedily pick the set of expressways that span each FPC contig, and (5) remove excess clones to minimize the junctions between expressways, where a junction is the overlap between expressways. The clones used in each expressway may have positive or negative overlaps, in which a negative overlap is bridged by a sequenced contig. The overlaps for clones within expressways have multiple constraints; first, the BESs of two clones must hit the same sequenced contig; second, the BLAST results must confirm that the clones extend in opposite directions; and third, the two clones must be near each other in the FPC map. Although results have only been verified on simulated data, these constraints are strong enough that we do not feel it is necessary for the user to inspect the overlap of clones within expressways. It is necessary to inspect the overlaps between expressways, but this can be done after all clones in the expressways are sequenced, as these will provide additional information for selecting the remaining MTP clones.
Simulated Data
To test the pickMTP algorithm, a simulated data set was generated from the complete 22.4-Mb japonica sequence of chromosome 10 (The Rice Chromosome 10 Sequencing Consortium 2003). Table 2 shows the sizes of the different types of data generated. There was no error introduced into the data, although a tolerance of seven was used for assembly, making the positions inexact (Soderlund et al. 2000). A total of 41 FPC contigs were generated; there was no manual editing done on these contigs. The BES and draft data were not screened for repeats. The BSS BLAST search was run at an expectation value of 1e-100, resulting in 34,557 hits. The filtering step removed hits with lowconfidence alignments, and discarded sequenced contigs with ambiguous locations, after which there remained 5227 hits. These hits produced 8883 pairs, in which each pair is two BESs that hit the same sequenced contig. A total of 5397 of these were rejected on the basis of the orientations of their alignments, as matching orientations indicate that the two clones cover the same region (see Methods). Map position information was used to eliminate 262 of the remaining pairs, leaving a total of 3224 acceptable pairs. PickMTP selected 145 clones for the MTP, which covered 82% of the FPC map. Both overlapping and bridging pairs were allowed. A summary of the results is given in Table 3 and the distribution of the expressway lengths is shown in Figure 4. To verify the correctness of the identified pairs, the positions of the sequenced contigs and BESs along the complete sequence were compared. Of the 3224 pairs, 11 were incorrectly identified due to repetitive sequence occurring in 5 regions of the chromosome, leaving 99.7% of the pairs correctly identified. Three of these incorrect pairs were included in the MTP, causing unexpected gaps or overlaps in the finished sequence.
Table 2.
Summary of Test Data
| Data | Coverage | Average size | Comments |
|---|---|---|---|
| FPC map | Redundancy | Clone | No. contigs |
| simulated | 20× | 135,000 | 41 |
| rice | 20× | 135,000 | 20 |
| BES | Genome | BES | % of clones |
| simulated | 20% | 675 | 100 |
| rice | 18% | 690 | 89 |
| Draft | Genome | seqCtg | No. seqctg |
| Simulated | 85%b | 4.1 kb | 4745 |
| Monsantoa | 53%b | 4.2 kb | 2882 |
| Syngentaa | 91%b (93%)c | 12 kb | 1729 |
| BGI | 84%b (92%)c | 2.9 kb | 127,561 |
Only seqCtgs assigned to chromosome 10 by Monsanto and Syngenta, respectively.
Computed assuming unique seqCtgs and a chromosome 10 size of 22.4 Mb.
Published coverage.
Table 3.
Coverage and Sizes for the Test Data Sets
|
|
Number of expressways
|
Number of clones in expressways
|
Average positive overlap of clones in expressways
|
Average negative overlap of clones in expressways
|
Average expressway length
|
||
| Simulated (unmasked) | 31 | 145 | 1.7 kb | 3.4 kb | 687 kb | ||
| Simulated (masked) | 35 | 142 | 1.8 kb | 4.1 kb | 605 kb | ||
| Syngenta | 37 | 132 | 4.9 kb | 8.4 kb | 452 kb | ||
| BGI | 40 | 104 | 3.0 kb | 1.5 kb | 321 kb | ||
| Monsanto
|
17
|
35
|
5.2 kb
|
6.2 kb
|
273 kb
|
||
| Data set | Number of junctions | Number of gaps | Average length of junctions | Average length of gaps | Number of bad pairs | Coverage | |
| Simulated (unmasked) | 2 | 21 | 53 kb | 50 kb | 11 | 82% | |
| Simulated (masked) | 5 | 23 | 56 kb | 62 kb | 4 | 81% | |
| Syngenta | 10 | 27 | 41 kb | 103 kb | ? | 80% | |
| BGI | 7 | 32 | 23 kb | 168 kb | ? | 62% | |
| Monsanto | 1 | 16 | 12 kb | 400 kb | ? | 24% | |
Figure 4.

The distribution of expressway lengths.
To reduce the number of bad hits, RepeatMasker (http://ftp.genome.washington.edu/RM/RepeatMasker.html) was run on the BESs, using rice repeats provided by The Institute for Genomic Research (TIGR; http://www.tigr.org/tdb/e2k1/osa1/blastsearch.shtml). This reduced the number of incorrectly identified pairs to 4, of which only 1 was incorporated into the MTP, resulting in an unexpected gap of around 1000 bases. This incorrect MTP pair, as well as the three others, result from a region of repetitive sequence with high conservation (97%) not identified by RepeatMasker, with the two copies almost 1000 bases from each other. The performance difference between the masked and unmasked data can be compared in Table 3. It can be concluded that the algorithm picks overlapping pairs quite accurately for a large portion of the FPC map, and that the overlaps between clones in expressways are quite small. Any errors are the result of highly conserved neighboring repeats, artifacts that most likely could not be identified by manual inspection. Furthermore, masking the BESs reduces the number of bad hits, but causes a small degradation in expressway length and map coverage.
Real Data
We only have draft sequence from Monsanto and Syngenta for chromosome 10, therefore, we created an FPC database and the file of BESs from only chromosome 10 data. The rice FPC database has been edited manually only to merge or split contigs; that is, there is no manual rearranging of the clones. Chromosome 10 has an estimated size of 22.4 MB (The Rice Chromosome 10 Sequencing Consortium 2003). A total of 17 of the 20 FPC contigs contain >50 clones. The Syngenta WGS draft-sequenced contigs assigned to chromosome 10 were compared against the rice BESs to produce the BSS hits file. PickMTP produced 8747 pairs, of which 2106 remained after filtering (5182 rejected on the basis of orientation; 1451 rejected on the basis of map position; 8 rejected on the basis of other criteria, see Methods). PickMTP automatically selected 132 clones for the MTP, covering 80% of the FPC map. Results are summarized in Table 3. After repeat-masking the BESs, the number of clones picked decreased to 129, and the coverage dropped to 78%.
The algorithm was also run on the Beijing Genomics Institute (BGI) WGS data from rice subspecies indica, and on the Monsanto clone by clone draft data for clones assigned to chromosome 10. This data produced comparable, but inferior results. For the BGI data, the discrepancy is presumably due to subspecies differences; for the Monsanto data, it is presumably caused by sparse coverage (53%) on chromosome 10 of the provided Monsanto draft. PickMTP produced map coverage of chromosome 10 for the BGI and Monsanto data sets of 62% and 24%, respectively. Additional figures showing results are at www.genome.org and http://www.genome.arizona.edu/software/fpc/gr2003_supplemental.
By use of the BGI whole-genome draft sequence with the entire rice FPC file that contains 72 k clones, the average runtime on a Sun 280R with 2GB RAM is 30 sec. When using the Syngenta data and only the chromosome 10 FPC contigs (4.2 k clones), the runtime is 7 sec.
DISCUSSION
This study covers a number of problems in which the solutions and results interleave. To be specific:
Task 1: Annotating the physical map with sequence data.
Task 2: Confirming and finishing the sequence of a clone.
Task 3: Detecting incorrectly merged contigs and contigs that can be merged.
Task 4: Selecting minimal tiling path clones for sequencing in order to reduce human time and to minimize overlaps.
We have developed a set of FPC-compatible tools to aid in these tasks.
The FSD tool creates a simulated fingerprint from a sequenced clone. The addition of these SD clones to FPC annotates the map with the sequence, allowing the community to know what regions are sequenced (task 1). If the fingerprint of the SD clone does not match with the original fingerprinted clone, the sequence may be assembled wrong, or the clone may be misnamed (task 2). The GenBank record generally specifies the chromosome for the sequenced clone. This information is used to help anchor contigs to chromosomes. If the contig has existing information anchoring it to a different chromosome, the contig may be chimeric (task 3). An additional benefit is that this information places sequence from other maps onto the FPC map. For example, using FSD, we have added 3573 sequenced clones to the rice FPC map, of which 1997 are from external sources. Many of the external ones were selected from the Japanese physical map (Saji et al. 2001). Not only does this help integrate the two maps, but since the clones are represented in FPC, their sequence can be used for placing electronic markers by BSS.
The BSS tool was written to increase the user efficiency in selecting a clone for sequencing using both the FPC map and sequence similarity from BESs and the genomic sequence of a seed clone (task 4). It can be run from a seed clone's contig so that the search is limited to the BESs from that contig, which considerably limits the output. Furthermore, the BSS report further limits the output the user must scan through, as it provides a summary of each hit. As an alternative to constraining the search to the contig, it can be constrained to use only BESs from the ends of contigs. This constraint can identify potential contigs to join (task 3).
BSS was extended to map marker sequence to the map by blasting it against the genomic sequences or BESs associated with clones in the map (task 1). An option was added to run MegaBLAST, as it runs much faster than BLAST on nucleotide sequences, and provides good results on similar sequence. Another option was added to run BLAT, as it works well for markers that are based on cDNAs that may have spliced introns. For BLAT, three filters were added. The first is the percent match that ensures that most of the sequence is matched, the second is the maximum intron length, and the third is the BLAT score. Using this feature, electronic markers can be automatically added to the map. If there are low-quality bases on the ends of the ESTs, or if they are being screened against the BESs, there will be incomplete hits. In this case, MegaBLAST sometimes finds hits that BLAT does not find. The one type of marker that this does not work for is PCR based; a future enhancement would be to run ePCR (Schuler 1998) within BSS.
For the options of Marker→Sequence and Marker→BES, the marker files can be any sequence, such as gene-rich contigs or draft sequence. For example, the draft sequence can be BSS'd against a clone in the process of being sequenced. The results will identify the sequenced contigs that hit the clone, and the reads from these sequenced contigs can be used in finishing the sequence of the clone (task 2). Regardless of whether the sequences are markers, gene-rich contigs, or draft, if the results are added as markers, this will elucidate potential joins of contigs and regions of possible repetitive sequence (task 3). Note that the draft sequence can be added as markers in order to aid in ordering the draft contigs, but a program such as GigAssembler (Kent and Haussler 2001) is written specifically for this application, and therefore, should be the program of choice for this task.
Because there are many sequencing projects that have an FPC map, BESs, and draft sequence available, we have developed an algorithm called pickMTP that automatically selects clones for sequencing by use of this set of data. This involves running BSS, finding overlapping pairs of clones, and executing a shortest-path algorithm. This does not result in one long path through each contig, as the large gaps between BESs cause gaps in the path. Therefore, all of the possible paths, called expressways, are generated, and the best set of expressways is selected. To test this technique on ideal data, we performed a simulation using data generated from the rice chromosome 10 sequence (The Rice Chromosome 10 Sequencing Consortium 2003). Negative overlaps were allowed, as the gap between the two BESs can be closed with the bridging sequenced contig. Results show an average positive clone overlap of 1.7 kb and an average negative overlap of 3.4 kb, with 86% total coverage of the chromosome. There was one false-positive overlap due to a highly conserved sequence repeated within 1000 bases. We also ran this algorithm using the real BAC end sequences, rice FPC map, and Syngenta draft sequence, which resulted in average overlaps of +4.9 kb and –8.4 kb, covering 80% of the chromosome. A total of 132 clones were automatically selected from the 20 contigs covering 22 MB. The multiple constraints of sequence similarity, orientation, and relative location of the clones in FPC provide very strong evidence for the correctness of these overlaps. There were 10 junctions between expressways that need to be inspected by a human.
The main FPC program is typically used by the biologists who are manipulating the map or selecting clones for sequencing; this will also be the case for the programs just described. For the general user who is only viewing the map, we have developed a Web-based FPC called WebFPC that is written in Java (Soderlund et al. 2003). We have also developed a Web-based form of BSS, called WebBSS, so that the user can have a sequence blasted against all of the BESs or genomic sequence associated with an FPC map, with the results shown in a BSS style report. The report includes the contig number of each clone, which can be selected to view the contig in WebFPC. These two Web tools are available for the rice FPC map (http://www.genome.arizona.edu/fpc/rice).
Current Work
We are working on automatically finding minimal overlapping clones by using the fingerprints only. These resulting pairs can go into the shortest path algorithm used in pickMTP. We are adding a graphical interface in order to find minimal tiling path clones with either or both methods. The user will then be able to interactively view the selected clones for any given contig along with the scores of the overlaps.
Availability
The software is freely available from www.genome.arizona.edu/software/fpc/ along with manuals for the BSS tool and the pick-MTP tool. FPC V6.5 contains the version of BSS with BLAT. FPC V7 has the new contig display.
METHODS
Adding Sequenced Clones
FSD takes as input one or more sequences, producing bands and sizes files based on a specified restriction enzyme. To convert the sizes to migration rates, the standard file is used. The standard file is created at the beginning of the fingerprinting project. When a gel is run, the set of standard markers (i.e., fragments) are also run. These markers have known rates and sizes, so that the rates of the new clones can be normalized by Image (Sulston et al. 1989). FSD fits a cubic spline curve to the standard values. It then converts the sizes to migration rates using this spline curve.
For our rice project, a cronjob downloads an incremental update file from GenBank every evening, which contains all of the previous day's updates to GenBank. This file is scanned for GenBank entries pertaining to the organism Oryza sativa. Each entry is parsed out and put in a separate file named by the GenBank accession number. These files are then run through FSD to generate SD clones for that sequence. A remark file is generated that contains the name of the clone, the associated chromosome, and name of the first author of the GenBank submission. The clone name is the GenBank accession number followed by sd1; if the sequence is over 180 kb, it is split up into overlapping sequences labeled sd2, etc.
The SD clones and remarks are added to FPC, and the SD clones are compared with all other clones and given the same position as its highest hitting clones, and the Process SD Clones function is run. This last function sets the sequencing state of all SD clones, makes sure they are not buried, and reports any sequenced clones that do not match their original clone in FPC. The FPC commands to perform these steps are:
fpc rice -batch updcor
fpc rice -batch mergerem <remarks.ace file>
fpc rice -batch -web
These commands are run in the nightly cronjob after the download and FSD.
Assigning Contigs to Chromosome
In FPC V6.4, on the main window is the Ctg→Chr button that initiates a window that has the Assign Ctg→Chr function on it. When it evaluates each contig, all SD clones and FW (Frame-Work) markers are considered, in which a FW marker is an ordered marker with a chromosome assignment and position, and each SD clone generally has a chromosome assignment. Each SD and FW counts as one point for a chromosome. The chromosome with the most points is the winning chromosome, and the contig is assigned to that chromosome. For example, a contig remark:
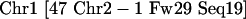 |
indicates a total of 48 points, 47 of them assigned to Chr1 and one assigned to Chr2. Also, for each chromosome, the number of clone hits is counted. That is, the sum of clone hits for all frameworks for a given chromosome plus the SD clones. Contigs are not assigned to chromosomes in the following two cases: (1) There is only one clone hit; that is, one framework hits one clone, or there is one SD clone. For example, the contig remark
 |
indicates that there is one framework on Chr4 with one clone. The minus (–) sign indicates no assignment. (2) The number of clone hits to the winning chromosome is less than four times the number of hits to the clones hitting other chromosomes. For example,
 |
indicates that chromosome 1 has four pieces of evidence, but it is not strong enough for an assignment. Note that a contig can be assigned a chromosome even if there is evidence for other chromosomes. Any contig that has evidence from multiple chromosomes needs to be inspected manually, as there could be a false join, false-positive markers, or incorrect sequence assignments.
Modifications to BSS for BLAT
Before running the BLAT search, the command-line parameter minScore may be specified (this takes the place of the E-value setting for BLAST searches). The pslx output of BLAT, which is used by BSS, consists of one line for each match, giving the number of bases matched, block sizes and locations, matching sequences, and other data for the alignment. When the user clicks on a particular hit, BSS uses this data to display the alignment, including all exons, in a BLAST-like format. The markers can be further filtered when they are added to FPC using the Marker Add Conditions window. Three filters are available, including %match and max_intron, which were discussed in the Results. The third filter is score, which is the same as minScore from the BLAT command line, except that, as the score is not included in the BLAT output, it is recomputed for the filter using the following formula:
 |
The PickMTP Algorithm
The following describes the 5 steps used by pickMTP for selecting an MTP:
BLAST the Draft Sequence Against the BES Library
BLAST searches are performed using the previously described BSS feature found in FPC V5 and higher. The draft sequenced contigs are BLASTed against the BES library of clones in FPC. The summary report that is generated associates BES hits with their clone and FPC contig number. After the report is filtered for hits of low confidence, it becomes the input for the next step.
Compile a List of Overlapping Clone Pairs
Any pair of BESs from different clones hitting a common sequenced contig is a potential MTP pair. Most false-positive pairs can be removed by examining the FPC map; if a pair contains clones from two different contigs, they are immediately rejected. (NB A pair of clones that are at the ends of two different contigs can be used to identify contigs to merge, a procedure not covered in this study). The remaining pairs fit one of the five cases depicted in Fig. 5. If a sequenced contig hits the BES of two clones close to one another in the map, either the clones overlap (case 1) or are bridged clones (case 2), in which the definition of bridged clones is that they do not overlap, but are bridged by a sequenced contig that can be used to fill the gap. If there is sufficient coverage by BES and draft sequence, it may not be necessary to use bridging clones, in which case such pairs may be rejected. The overlaps depicted in cases 3–5 are rejected as follows (additional details are provided in the Supplemental Material).
Figure 5.

The five different types of pairs. Case 1 pairs consist of two overlapping clones, with the exact amount of overlap identified by the seqCtg. Case 2 clones have a gap between them; they are bridged by a seqCtg. In Case 3, the two clones cover a similar area. Case 4 pairs result from false positive hits. Case 5 pairs result from two small clones hitting on opposite ends of a long seqCtg; note this is similar to Case 1, but a large seqCtg may result in a large overlap.
Case 3 occurs when two left ends or two right ends of the clones hit the sequenced contig. To eliminate this case, we consider the orientation of the hits with respect to the sequenced contig (see Fig. 6). The true orientation of the clone in relation to the chromosome is not known. However, the BES is always read and written starting from the end of a clone and progressing toward the middle, and the orientation of the BES pair is known with respect to the sequenced contig. Therefore, a case 1 or case 2 hit requires that the BES of one clone be reverse complemented to match the sequenced contig, whereas the BES of the other clone must not be. If the BLAST output has two Plus/Plus or two Plus/Minus for the pair, it is rejected.
Figure 6.

(A) S1 and S2 are sequenced contigs, and C1, C2, and C3 are clones. The underlined sequence represents the BESs and where they hit on the seqCtg. Note, all lengths of sequences are tremendously reduced to fit on the page. (B) There are four possible pairs, shown in the four rows in the table. A + indicates the BES was not reverse complemented to match the seqCtg. An rc indicates that it was reverse complemented. The seqCtg-BES hits for the first two pairs both have the same orientations, whereas the orientations for the next two are different. The first two are rejected as candidate MTP pairs, wheras the second two are retained.
Case 4 occurs when a sequenced contig hits two clones in the same contig that are not near each other, which may occur from a repetitive sequence. To eliminate resulting bad pairs, the distance between the two clones in FPC is checked. If a sequence contig is repetitive, it may hit many contigs; these sequenced contigs are rejected unless requested otherwise.
Case 5 occurs when the draft sequence contains very long sequenced contigs, such that a pair may overlap more than suitable for a minimal tiling path. All pairs are rejected that have such an overlap (note, in the shortest paths algorithm described below, large overlaps are avoided when possible).
The pairs passing all tests are candidate MTP pairs and are written to a file that is the input for a C program used to pick expressways based on the clone pairs.
Identify all Possible Expressways
The single source shortest paths problem is defined as follows: given a directed graph G = (V, E) in which each edge has a positive weight and one vertex is specified as the source, determine the cost of the shortest path from the source vertex to every other vertex in V, in which the length of a path is the sum of the weights on the edges of the path (Aho et al. 1983). This algorithm is used to compute all possible expressways for each contig. There are four steps to building the expressways: (1) construct a set of disconnected DAGs (Directed Acyclic Graphs), (2) find the set S of source vertices and the set T of target vertices for each subgraph, (3) run the shortest paths algorithm on each source si in S, and (4) save each shortest path Pij from a source si to a target tj as an expressway. We use Dijkstra's algorithm (Dijkstra 1959) for the shortest paths problem, which has a known complexity of O(E log V) in a standard implementation with a sparse matrix and priority queue, in which E represents the number of edges in the graph and V is the number of vertices.
The set of DAGs are constructed as follows: every clone that is in a candidate MTP pair is a vertex in V, and there exists an edge in E between each candidate MTP pair. The edge is directed from the left to the right clone, as defined by the FPC map. The edge weight is determined by the absolute difference between the positions of the BESs on the sequenced contig, and the weight for bridging clones is multiplied by a factor of 10 in order to make selecting overlapping clones take precedence over bridging clones. Figure 7 shows how graphs are constructed from the pair information. Note that this step creates many subgraphs, as will be explained in step 4.
Figure 7.

Construction of DAGs from overlapping clone pairs. (A) The amount of overlap or distance is recorded for all good clone pairs, shown as w, x, y, and z. (B) Clones define vertices, and pairs define edges, with edge weight determined by overlap or distance. If there is a gap between the clones, then the value is multiplied by 10. An MTP, shown in gray, is picked from the expressways. Note the junction from C to F, in which there is no sequence evidence that the clones overlap.
Once the graphs are constructed, all vertices with no incoming edges are marked as source vertices. From each source si, the vertex representing a clone furthest to the right of the contig, and reachable from si, is marked as the target. The shortest paths algorithm is run on each subgraph for all S source vertices (for details, see Aho et al. 1983). Every shortest path Pij from source si to target tj is saved as an expressway.
Greedily Pick the Set of Expressways That Span Each Contig
Once all possible expressways have been identified for each contig, a subset is picked to cover that contig. Optimally, there will be one expressway spanning the entire contig. Unfortunately, this is usually not the case, as gaps in the draft sequence, poor quality BESs, missing BESs, incorrectly discarded pairs, and sparse clone coverage can all contribute to non-contiguous expressways through contigs. With a 20× map coverage, 150-kb clones, BESs of length 650, and a random distribution, the BESs will be distributed at an average distance of 3100 bases. Consequently, it is very possible for a reasonably sized sequenced contig to hit zero or one BES, thus producing no pairs for that region. Also, a pair p may occur in the middle of a clone that is part of one expressway, but pair p starts a new expressway, as no sequence evidence exists to link the two clones.
The clones within an expressway have minimal overlap, so the primary objective is to pick long expressways to span the contig. A secondary objective is to reduce the number of junctions and gaps. A greedy approach is used that gives priority to the longest expressways, and each subsequent expressway selected must have at least 80% of their span not yet covered by any previously included expressways. Overlapping expressways may overlap by more than one clone, therefore, excess clones are removed while retaining a minimum of 5 CB units between overlapping expressways.
Display Results in FPC
A file of sequence status is created that will set the status of all MTP clones to TILE. These files can be loaded into FPC. The reader is referred to the FPC manual for more information regarding remarks and sequence status (Soderlund 1999) and the tutorial by Engler and Soderlund (2003).
Acknowledgments
F.E. is partially supported by an NSF Plant Genome no. 0213764 and partially supported by a USDA-IFAFS no. 11180. J.H. is partially supported by USDA-IFAFS no. 11180. W.N. is supported by NSF Plant Genome no. 0211851. J.S., who is a graduate student in computer science at the University of Arizona, developed the Web-based BSS; he is supported by a USDA-IFAFS no. 11180. We thank Rod Wing and Yeisoo Yu for fruitful discussions that contributed to this approach, and for their comments on the manuscript. We also thank David Kudrna and Brad Barbuzuk for feedback on the manuscript. We thank Lenny Teytelman for discussions on BLAT and screens that he performed. And, we also thank Jim Kent for answering all of our questions on BLAT.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
[Supplemental material is available online at www.genome.org and http://www.genome.arizona.edu/software/fpc/gr2003_supplemental.]
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.1068603. Article published online before print in August 2003.
References
- Aho, A., Hopcroft, J., and Ullman, J. 1983. Data structures and algorithms. pp. 203–208. Addison-Wesley, Reading, MA.
- Altschul, S., Madden, T., Schaffer, A., Zhang, J., Zhang, Z., Miller, W., and Lipman, D. 1997. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 25: 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Arabidopsis Genome Initiative. 2000. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 408: 769–815. [DOI] [PubMed] [Google Scholar]
- Barry, G. 2001. The use of the Monsanto draft rice genome sequence in research. Plant Phys. 125: 1164–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentley, D.R., Deloukas, P., Dunham, A., French, L., Gregory, S.G., Humphray, S.J., Mungall, A.J., Ross, M.T., Carter, N.P., Dunham, I., et al. 2001. The physical maps for sequencing human chromosomes 1, 6, 9, 10, 13, 20 and X. Nature 409: 942–943. [DOI] [PubMed] [Google Scholar]
- Chandler, V.L. and Brendel, V. 2002. Maize genome sequencing project. Plant Phys. 130: 1594–1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, M., Presting, G., Barbazuk, W., Goicoechea, J., Blackmon, B., Fang, G., Kim, H., Frisch, D., Yu, Y., Higingbottom, S., et al. 2001. An integrated physical and genetic map of the rice genome. Plant Cell 14: 537–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulson, A., Sulston, J., Brenner, S., and Karn, J. 1986. Towards a physical map of the genome of the nematode C. elegans. Proc. Natl. Acad. Sci. 83: 7821–7825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dijkstra, E.W. 1959. A note on two problems in connexion with graphs. Numerische Mathematik 1: 269–271. [Google Scholar]
- Engler, F. and Soderlund, C. 2003. Software for physical maps. In Genome Mapping and Sequencing (ed. I. Dunham), pp. 20–236. Horizon Scientific Press, Genome Technology Series, Norfolk, UK.
- Feng, Q., Zhang, Y., Hao, P., Wang, S., Fu, G., Huang, Y., Li, Y., Zhu, J., Liu, Y., Hu, X., et al. 2002. Sequence and analysis of rice chromosome 4. Nature 420: 316–320. [DOI] [PubMed] [Google Scholar]
- Goff, S.A., Ricke, D., Lan, T.H., Presting, G., Wang, R., Dunn, M., Glazebrook, J., Sessions, A., Oeller, P., Varma, H., et al. 2002. A draft sequence of the rice genome (Oryza sativa L. ssp. Japonica). Science 296: 92–100. [DOI] [PubMed] [Google Scholar]
- Green, P. 1997. Against a whole-genome shotgun. Genome Res. 7: 410–417. [DOI] [PubMed] [Google Scholar]
- ____. 2002. Whole-genome disassembly. Proc. Natl. Acad. Sci. 99: 4143–4144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory, S.G., Sekhon, M., Marra, M., Zhao, S., Osoegawa, K., Scott, C.E., Evans, R.S., Burridge, P.W., Cox, T.V., Fox, C.A., et al. 2002. A physical map of the mouse genome. Nature 418: 743–750. [DOI] [PubMed] [Google Scholar]
- Harushima, Y., Yano, M., Shomura, A., Sato, M., Shimano, T., Kuboki, Y., Yamamoto, T., Lin, S.Y., Antonio, B.A., Parco, A., et al. 1998. A High-density rice genetic linkage map with 2275 markers using a single F2 population. Genetics 148: 479–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoskins, R., Nelson, C., Berman, B., Laverty, T., George, R., Ciesiolka, L., Naeemuddin, M., Arenson, A., Durbin, J., David, R., et al. 2000. A BAC-based physical map of the major autosomes of Drosophila melanogaster. Science 287: 2271–2274. [DOI] [PubMed] [Google Scholar]
- The International Human Genome Mapping Consortium. 2001. A physical map of the human genome. Nature 409: 934–941. [DOI] [PubMed] [Google Scholar]
- The International Human Sequencing Consortium. 2001. Initial sequencing and analysis of the human genome. Nature 409: 860–920.11237011 [Google Scholar]
- Kent, J. 2002. BLAT—The BLAST-like alignment tool. Genome Research 12: 656–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent, J. and Haussler, D. 2001. Assembly of the working draft of the human genome with GigAssembler Genome Res. 11: 1541–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao, L., Wood, T., Yu, Y., Budiman, M., Woo, S., Sasinowski, M., Goff, S., Dean, R., and Wing, R. 2000. Rice transposable elements: A survey of 73,000 sequence-tagged-connectors (BESs). Genome Res. 10: 982–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mardis, E., McPherson, J., Martienssen, R., Wilson, R., and McCombie, W. 2002. What is finished, and why does it matter. Genome Res. 12: 669–671. [DOI] [PubMed] [Google Scholar]
- Marra, M., Kucaba, T., Dietrich, N., Green, E., Brownstein, B., Wilson, R., McDonald, K., Hillier, L., McPherson, J., and Waterston, R. 1997. High-throughput fingerprint analysis of large-insert clones. Genome Res. 7: 1072–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marra, M., Kucaba, T., Sakhon, M., Hillier, L., Martienssen, R., Chinwalla, A., Crockett, J., Fedele, J., Grover, H., Gund, C., et al. 1999. zA map for sequence analysis of the Arabidopsis thaliana genome. Nat. Genet. 22: 265–275. [DOI] [PubMed] [Google Scholar]
- Myers, E.W., Sutton, G.G., Smith, H.O., Adams, M.D., and Venter, J.C. 2002. On the sequencing and assembly of the human genome. Proc. Natl. Acad. Sci. 99: 4145–4146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson, D., Schulze, S., Sciara, E., Lee, S., Bowers, J., Nagel, A., Jiang, N., Tibbitts, D., Wessler, S., and Paterson, A. 2002. Integration of Cot analysis, DNA cloning, and high-throughput sequencing facilitates genome characterization and gene discovery. Genome Res. 12: 795–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinowicz, P., Schutz, K., Dedhia, N., Yordan, C., Parnell, L., Stein, L., McCombi, W., and Martienssen, R. 1999. Differential methoylation of genes and retrotransposons facilitates shotgun sequencing of the maize genome. Nat. Genet. 23: 305–308. [DOI] [PubMed] [Google Scholar]
- The Rice Chromosome 10 Sequencing Consortium. 2003. In-depth view of structure, activity, and evolution of rice chromosome 10. Science 300: 1566–1569. [DOI] [PubMed] [Google Scholar]
- Saji, S., Umehara, Y., Antonio, B., Yamane, H., Tanoue, H., Baba, T., Aoki, H., Ishige, N., Wu, J., Koike, K., et al. 2001. A physical map with yeast artificial chromosome (YAC) clones covering 63% of the 12 rice chromosomes. Genome 44: 32–37. [DOI] [PubMed] [Google Scholar]
- Sasaki, T. and Burr, B. 2000. International rice genome sequencing project: The effort to completely sequence the rice genome. Curr. Opin. Plant Biol. 3: 138–141. [DOI] [PubMed] [Google Scholar]
- Sasaki, T., Matsumoto, T., Yamamoto, K., Sakata, K., Baba, T., Katayose, Y., Wu, J., Niimura, Y., Cheng, Z., Nagamura, Y., et al. 2002. The genome sequence and structure of rice chromosome 1. Nature 420: 312–316. [DOI] [PubMed] [Google Scholar]
- Schuler, G. 1998. Electronic PCR: Bridging the gap between genome mapping and genome sequencing. Trend Biotechnol. 16: 456–459. [DOI] [PubMed] [Google Scholar]
- Soderlund, C. 1999. FPC V4 User's manual. The Sanger Centre, Technical Report SC-01–SC-99.
- Soderlund, C., Longden, I., and Mott, R. 1997. FPC: A system for building contigs from restriction fingerprinted clones. Computat. Appl. Biosci. 13: 523–535. [DOI] [PubMed] [Google Scholar]
- Soderlund, C., Humphrey, S., Dunhum, A., and French, L. 2000. Contigs built with fingerprints, markers and FPC V4.7. Genome Res. 10: 1772–1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soderlund, C., Engler, F., Hatfield, J., Blundy, S., Chen, M., Yu, Y., and Wing, R. 2003. Mapping sequence to Rice FPC. In Computational biology and genome informatics (eds. P. Wang, J. Wang, and C. Wu), pp. 59–80. World Scientific Publishing, Singapore.
- Sulston, J., Mallett, R., Durbin, F., and Horsnell, T. 1989. Image analysis of restriction enzyme fingerprints autoradiograms. Computat. Appl. Biosci. 5: 101–132. [DOI] [PubMed] [Google Scholar]
- Venter, J.C., Smith, H.O., and Hood, L. 1996. A new strategy for genome sequencing. Nature 381: 364–366. [DOI] [PubMed] [Google Scholar]
- Venter, J.C., Adams, M.D., Myers, E.W., Li, P.W., Mural, R.J., Sutton, G.G., Smith, H.O., Yandell, M., Evans, C.A., Holt, R.A., et al. 2001. The sequence of the Human Genome. Science 291: 1304–1351. [DOI] [PubMed] [Google Scholar]
- Ware, D., Jaiswal, P., Ni, J., Yap, I., Pan, X., Clark, K., Teytelman, L., Schmidt, S., Zhao, W., Chang, K., et al. 2002. Gramene, a tool for grass genomics. Plant Physiol. 130: 1606–1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterston, R.H., Lander, E.S., and Sulston, J.E. 2002. On the sequencing of the human genome. Proc. Natl. Acad. Sci.. 99: 3712–3716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber, L. and Myers, E. 1997. Human whole-genome shotgun sequencing. Genome Res. 7: 410–409. [DOI] [PubMed] [Google Scholar]
- Yu, J., Hu, S., Wang, J., Wong, G.K., Li, S., Liu, B., Deng, Y., Dai, L., Zhou, Y., Zhang, X., et al. 2002. A draft sequence of the rice genome (Oryza sativa L. ssp. indica). Science 296: 79–91. [DOI] [PubMed] [Google Scholar]
- Zhang, Z., Schwartz, S., Wagner, L., and Miller, W. 2000. A greedy algorithm for aligning DNA sequences. J. Comp. Biol. 7: 203–214. [DOI] [PubMed] [Google Scholar]
WEB SITE REFERENCES
- http://ftp.genome.washington.edu/RM/RepeatMasker.html; Smit, A.F.A. and Green, P., RepeatMasker.
- http://rgp.dna.affrc.go.jp/Publicdata.html; Japanese Rice Genome Research Program site for genetic markers and sequence.
- http://www.ensembl.org/Mus_musculus/; Ensembl Mouse Genome Server.
- http://www.genome.arizona.edu/fpc/rice; Rice Physical Mapping Home Page.
- http://www.genome.arizona.edu/fpc/rice/bss.html; Web-based BSS for rice.
- http://www.genome.arizona.edu/shotgun/rice/; ACWW Rice Genome Sequencing Consortium Home Page.
- http://www.genome.arizona.edu/software/fpc/; FPC and WebFPC Download Site.
- http://www.genome.arizona.edu/software/fpc/gr2003_supplemental; F. Engler, W. Nelson, and C. Soderlund, provides supplemental information for this manuscript.
- http://www.genome.arizona.edu/software/fpc/userGuide/bss-tutorial/tutorial1.htm; BSS Tutorial.
- http://www.gramene.org/documentation/Alignment_docs/rice_rflp.html; Listing of the rules by which the JGRP and Cornell rice markers were added to the rice FPC.
- www.sanger.ac.uk/software/Image; Image—the fingerprint image analysis system.
- http://www.tigr.org/tdb/e2k1/osa1/blastsearch.shtml; TIGR rice repeat database.