

Abstract
An enantioselective method for the synthesis of 1,2-anti diols has been developed. A cyclometallated chiral-at-Ru complex catalyzes the asymmetric ring opening/cross metathesis of di–oxygenated cyclobutenes, resulting in functionally rich synthetic building blocks. Syntheses of the insect pheromone (+)-endo brevicomin and monosaccharide ribose demonstrate the synthetic utility of the 1,2-anti diol fragments generated in the title reaction.
Keywords: olefin metathesis, vicinal diol, monosaccharides, pheromones, asymmetric ring opening/cross metathesis
The formation of multiple stereocenters in a single catalytic transformation is a powerful approach to the synthesis of stereochemically complex targets. While the development of such a transformation must overcome the challenge of simultaneously controlling diastereo- and enantioselectivity, the end result can reduce the step count of a synthesis and improve its atom economy. One commonly encountered motif is the vicinal diol, which is pervasive throughout natural products and ligands for asymmetric transformations. While the problem of introducing vicinal diols in high enantiopurity has largely been solved by the Sharpless asymmetric dihydroxylation (AD), the formation of 1,2-anti diols remains challenging due to the low enantioselectivity observed in the AD of cis-1,2 disubstituted alkenes.[1] Accordingly, a number of methods have been developed for the enantioselective formation of 1,2-anti diols, including asymmetric epoxidation/hydrolysis,[2] glycolate aldol,[3] iterative cross metathesis/allylic substitution,[4] nucleophilic addition to aldehydes,[5] desymmetrizing monofunctionalization,[6] and allene hydroboration/aldehyde allylation.[7] In contrast to many of these methods, an asymmetric ring opening/cross metathesis (AROCM) approach (Scheme 1) would consolidate the transformation into a single step and generate a differentiated 1,5-diene fragment in a convergent manner.
Scheme 1.
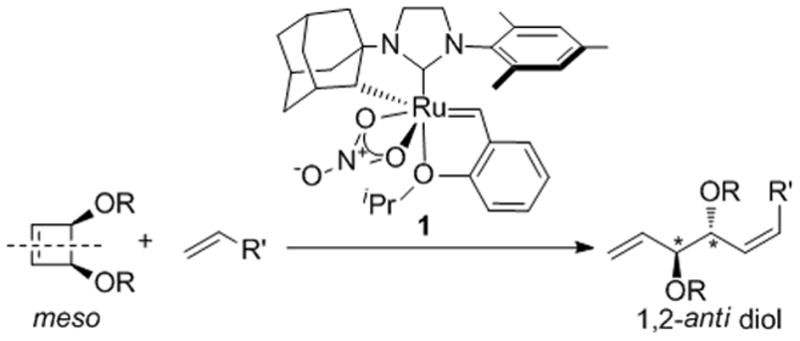
AROCM Reaction to Afford Highly Enantioenriched 1,2-anti diols
Asymmetric olefin metathesis is a powerful C–C bond forming reaction and has enabled the synthesis of stereochemically complex bioactive compounds.[8] Advances in stereoselective olefin metathesis have resulted in the development of catalysts capable of forming products with high diastereo-[9] and enantioselectivity.[10] Although the ROCM of cyclobutenes to form racemic products has been demonstrated,[11] previous studies of their AROCM reactions have afforded products with low enantioenrichment.[10i]
It was envisioned that the desymmetrization of suitably substituted meso cyclobutenes in AROCM would afford the 1,2-anti diol motif in perfect anti diastereoselectivity and potentially high enantioselectivity upon application of a newly developed cyclometalated metathesis catalyst (1, Scheme 1).[12] The resultant 1,5-diene would be a versatile synthetic intermediate due to the differential reactivity of the two alkenes, paving the way for further chemoselective transformations. Herein, we report the successful application of 1 to afford highly enantioenriched 1,2-anti diols and demonstrate the versatility of these products in the synthesis of the insect pheromone (+)-endo brevicomin and a derivative of the monosaccharide L-ribose. Pest control strategies utilizing insect pheromones have become a promising alternative to the application of broad-spectrum insecticides, underscoring the importance of rapid synthetic routes to (+)-endo brevicomin and related bioactive compounds.[13][14]
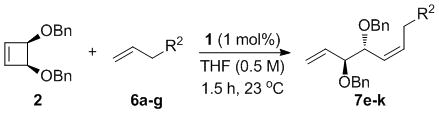
Initial attempts to form 1,2-anti diols were carried out with complex 1, allyl acetate (3), and cis-3,4-dibenzyloxycyclobutene (2, Table 1), which was synthesized by substitution of commercially available cis-3,4-dichlorocyclobutene with sodium phenylmethanolate.[15] Solvent had no effect on selectivity of the AROCM reaction except for slightly diminished enantioselectivity in CH2Cl2 (entry 1, Table 1); yield was highest in THF (entry 4). The effect of stoichiometry in AROCM has been explored for a number of catalysts.[10b; 10i; 16] In the current study, an excess of terminal olefin was optimal (7 equiv, entry 4); as the equivalents of terminal olefin were reduced, the yield of the reaction dropped, yet a modest yield of 29% could be obtained with 1.2 equivalents of 3. No di-cross products were observed. Reducing the concentration also resulted in lower yield, leading to the optimal conditions of 7 equiv. of terminal olefin 3 in THF at a concentration of 0.5 M in 2 with 1 mol% 1 for 1.5 h. It is worth noting that although alternative solvents or stoichiometry negatively impacted reaction efficiency, the diastereo- and enantioselectivity remained consistently high, demonstrating the robustness of the reaction.
Table 1.
Optimization of the AROCM of Cyclobutene 2 with 3.
| ||||||
|---|---|---|---|---|---|---|
| entry | Equiv 3 | Conc (M) | Solvent | Yield[a] | % Z[a] | ee (Z)[b] |
| 1 | 7 | 0.5 | CH2Cl2 | 35 | 83 | 93 |
| 2 | 7 | 0.5 | Benzene | 49 | 84 | 95 |
| 3 | 7 | 0.5 | Toluene | 52 | 84 | 95 |
| 4 | 7 | 0.5 | THF | 79 | 85 | 95 |
| 5 | 5 | 0.5 | THF | 71 | 84 | 96 |
| 6 | 3 | 0.5 | THF | 63 | 85 | 96 |
| 7 | 1.2 | 0.5 | THF | 29 | 87 | 97 |
| 8 | 7 | 0.3 | THF | 72 | 85 | 95 |
| 9 | 7 | 0.1 | THF | 43 | 85 | 95 |
Determined by GC.
Determined by chiral SFC.
While the synthesis of a 1,2-anti alkoxy motif had been demonstrated, inclusion of alternative protecting groups on the diol motif strengthens the synthetic protocol. These modifications would allow a synthetic sequence to be designed taking into account the feasibility of removing the protecting groups in the presence of other functionality. Moreover, modulation of the size and electronics of the groups on the cyclobutene and terminal olefin reactants would provide a better understanding of the factors contributing to selectivity.
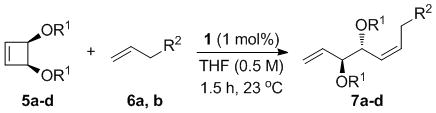
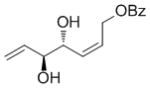
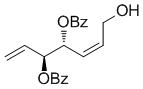
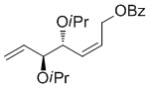
A complement of commonly used hydroxyl protecting groups were tolerated on the cyclobutene and terminal olefin reactants,[17] but enantio- and diastereoselectivity were affected by the choice of substituents (Tables 2 and 3). The increased bulkiness of the tert-butyldimethylsilyl ether resulted in improved Z selectivity and remarkable enantioselectivity (88% Z, 99% ee, 7a, Table 2), while hydroxyls and benzoates on the cyclobutene reactant led to Z products with 91% and 96% ee, respectively. The same enantioinduction was observed in products 7a and 7b. Isopropoxy substituents on the cyclobutene resulted in abrogation of catalyst activity presumably due to the formation of a stable chelating complex.[18]
Table 2.
Scope of the AROCM Reaction with respect to Cyclobutene Substitution.[a]
| |||||
|---|---|---|---|---|---|
| R1 | R2 | Product | Yield[b] | %Z[c] | ee Z[d] (ee E)[d] |
| TBS | OH |
7a
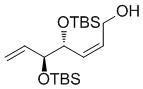
|
66 | 88 | 99 (nd) |
| H | OBz |
7b
|
67 | 75 | 91 (67) |
| Bz | OH |
7c
|
69 | 75 | 96 (82) |
| iPr | OBz |
7d
|
<5 | nd | nd |
0.1 mmol cyclobutene, 0.7 mmol terminal olefin.
Combined isolated yield of E and Z products.
Determined by 500 MHz 1H NMR analysis of crude reaction mixture.
Determined by chiral SFC.
Table 3.
Scope of the AROCM Reaction with respect to Terminal Olefin[a]
| ||||
|---|---|---|---|---|
| R2 | Product | Yield [b] | % Z[c] | ee Z[d] (ee E)[d] |
| OH |
7e
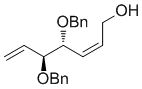
|
62 | 89 | 93 (86) |
| OBz |
7f
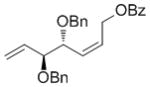
|
61 | 88 | 97 (88) |
| OTBS |
7g
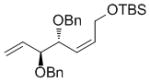
|
68[e] | 87 | 89 (77) |
| OBn |
7h
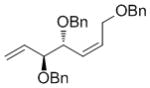
|
64 | 86 | 91 (nd) |
| 4-MeOPh |
7i
|
76 | 90 | 93 (79) |
| CH2C(O)CH3 |
7j
|
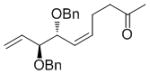
65 | 90 | 92 (84) |
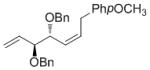
| BPin |
7k
|
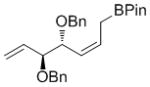
50 | nd[f] | 91 (nd) |
0.1 mmol cyclobutene, 0.7 mmol terminal olefin.
Combined isolated yield of E and Z products.
Determined by 500 MHz 1H NMR analysis of crude reaction mixture.
Determined by chiral SFC.
Yield determined after derivatization to 7e.
Not determined due to instability of E product.
High enantioselectivities were obtained with a wide range of terminal olefins. Among the O-protecting groups surveyed (Table 3, 7e–h), the tert-butyldimethylsilyl group resulted in high enantioselectivity (89% ee, 7g), but the more electron-withdrawing benzoate ester was optimal, resulting in the highest enantioselectivity (97% ee, 7f). Terminal olefins bearing alkyl substitution resulted in higher diastereoselectivity and yield with similar levels of enantioselectivity (7i, j). The chiral allylation reagent 7k was synthesized in 91% ee, affording a functionally useful building block. Z and E isomers were isolable from each other by flash or thin layer chromatography in all cases except 7i.
We next explored the synthetic utility of the 1,2-anti diol fragments produced in the AROCM reaction. Cyclic ketals derived from the 1,2-anti diol motif feature prominently in the structures of several natural products.[19] Accordingly, we targeted this structure in the context of a synthesis of the insect pheromone (+)-endo brevicomin (11, Scheme 2).[20]
Scheme 2.
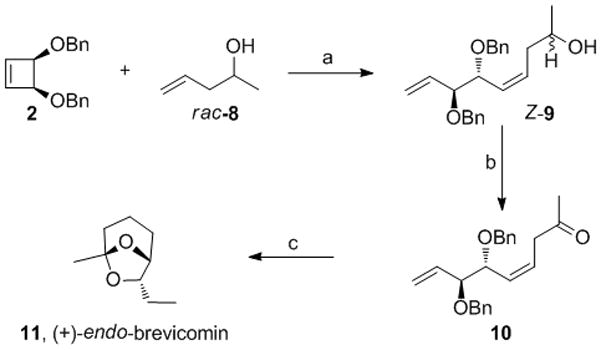
Enantioselective Synthesis of (+)-endo Brevicomin. a) 1 (1 mol%), rac-8 (7 equiv), THF, 23°C, 85% yield, 91% Z, 1:1 dr. b) Dess-Martin periodinane (2 equiv), 0–23°C, 88% yield, 95% ee. c) H2 (1 atm), Pd/C (10%), MeOH/aq. 1N HCl, 67% yield.
(+)-Endo-brevicomin is a male produced component of the attractive pheromone system of Dendroctonus frontalis (southern pine beetle),[19a] a tree-killing insect found in southern North America and Central America. It was envisioned that AROCM of 2 with 4-penten-2-ol would set the relative and absolute stereochemistry in the synthesis of (+)-endo brevicomin.
An expedient three-step synthesis of (+)-endo brevicomin was accomplished featuring the AROCM of 2 with racemic 8 to afford 9 (91% Z) in 85% yield as an inconsequential mixture of diastereomers (Scheme 2). [21] The mixture of epimeric alcohols was cleanly oxidized to the desired ketone by Dess-Martin periodinane in 88% yield. Z-10 was obtained in 95% ee, indicating high enantioselectivity in the AROCM reaction. Hydrogenation of Z-10 in acidic methanol resulted in concomitant reduction of the alkenes, hydrogenolysis of the benzyl groups and cyclization to form (+)-endo brevicomin in 67% yield in a one-pot transformation.[22]
It was envisioned that the synthetic utility of the 1,5-dienes produced in the AROCM of cyclobutenes could be further underscored by chemoselective functionalization of the two alkenes. For example, the introduction of additional hydroxyl groups would enable the rapid synthesis of monosaccharides. In this fashion, a succinct and highly enantioselective synthesis of biologically relevant monosaccharides could function as a robust route to starting materials for complex polysaccharides.
The synthesis of ribose derivative 13 was carried out to demonstrate the conversion of AROCM products such as 7 into useful monosaccharides (Scheme 3). Dihydroxylation of Z-7f catalyzed by OsO4 afforded a 66% yield of differentially protected pentanol 12 in 9:1 dr.[23] Ozonolysis of the remaining double bond afforded the differentially protected L-ribose lactol, which was isolated as methyl glycoside 13 in 47% yield over two steps.[24] It is hypothesized that a broader collection of monosaccharides will be accessible from the AROCM products by the modification of this synthetic sequence.
Scheme 3.
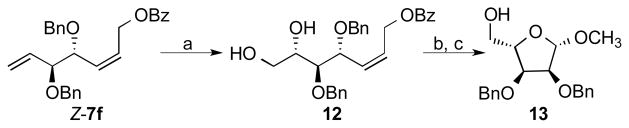
Enantioselective Synthesis of an L-Ribose Derivative. a) OsO4 (5 mol%), K3Fe(CN)6, K2CO3, tBuOH/water, 66% yield, 9:1 dr. b) O3 then Me2S. c) HCl in MeOH (anh.), 47% yield (over two steps).
In conclusion, the highly enantioselective synthesis of 1,2-anti diols was accomplished by the application of catalyst 1 to the AROCM of cis-dioxygenated cyclobutenes. The reaction is robust, tolerating modifications in reaction conditions and substitution on the reactants. Enantioenrichment of the major Z isomers was exceptionally high, ranging from 89–99% ee. The rapid synthesis of insect pheromone (+)-endo brevicomin was accomplished, affording the natural product in 95% ee. A 1,5-diene generated by the AROCM reaction was chemoselectively functionalized to afford ribose derivative 13, demonstrating the utility of the building blocks afforded by the title reaction.
Experimental Section
In a glovebox, cyclobutene 2 (26.6 mg, 0.1 mmol) and allyl benzoate (113 mg, 0.7 mmol, 7 equiv) were dissolved in 0.15 mL THF. To this solution was added 50 μL of a stock solution (0.02 M in THF) of catalyst 1. The reaction vial was capped and stirred for 1.5 h and then quenched with an excess of ethyl vinyl ether outside of the glove box. The reaction mixture was concentrated and subjected to flash chromatography to afford the desired AROCM product (7f, 25.9 mg, 61% isolated yield, 88:12 Z/E, 97% ee (Z product), 88% ee (E product)).
Supplementary Material
Acknowledgments
This work was financially supported by the NIH (5R01GM031332-27 to R.H.G.). The authors thank Materia, Inc. for donation of metathesis catalysts and Drs. Jeffrey Cannon, Bill Morandi and Mr. Zach K. Wickens for helpful discussion.
Footnotes
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
References
- 1.Kolb HC, Vannieuwenhze MS, Sharpless KB. Chem Rev. 1994;94:2483–2547. [Google Scholar]
- 2.a) Lim SM, Hill N, Myers AG. J Am Chem Soc. 2009;131:5763–5765. doi: 10.1021/ja901283q. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Albrecht L, Jiang H, Dickmeiss G, Gschwend B, Hansen SG, Jorgensen KA. J Am Chem Soc. 2010;132:9188–9196. doi: 10.1021/ja103538s. [DOI] [PubMed] [Google Scholar]
- 3.a) Mukaiyama T, Iwasawa N. Chem Lett. 1984:753–756. [Google Scholar]; b) Evans DA, Gage JR, Leighton JL, Kim AS. J Org Chem. 1992;57:1961–1963. [Google Scholar]; c) Notz W, List B. J Am Chem Soc. 2000;122:7386–7387. [Google Scholar]; d) Crimmins MT, McDougall PJ. Org Lett. 2003;5:591–594. doi: 10.1021/ol034001i. [DOI] [PubMed] [Google Scholar]; e) Northrup AB, MacMillan DWC. Science. 2004;305:1752–1755. doi: 10.1126/science.1101710. [DOI] [PubMed] [Google Scholar]; f) Northrup AB, Mangion IK, Hettche F, MacMillan DWC. Angew Chem. 2004;116:2204–2206. doi: 10.1002/anie.200453716. [DOI] [PubMed] [Google Scholar]; Angew Chem, Int Ed. 2004;43:2152–2154. doi: 10.1002/anie.200453716. [DOI] [PubMed] [Google Scholar]; g) Denmark SE, Chung WJ. Angew Chem. 2008;120:1916–1918. doi: 10.1002/anie.200705499. [DOI] [PubMed] [Google Scholar]; Angew Chem, Int Ed. 2008;47:1890–1892. doi: 10.1002/anie.200705499. [DOI] [PubMed] [Google Scholar]
- 4.a) Park JK, McQuade DT. Angew Chem. 2012;124:2771–2775. [Google Scholar]; Angew Chem, Int Ed. 2012;51:2717–2721. doi: 10.1002/anie.201107874. [DOI] [PubMed] [Google Scholar]; b) Kim D, Lee JS, Kong SB, Han H. Angew Chem. 2013;125:4297–4300. [Google Scholar]; Angew Chem, Int Ed. 2013;52:4203–4206. doi: 10.1002/anie.201209112. [DOI] [PubMed] [Google Scholar]
- 5.a) El-Sayed E, Anand NK, Carreira EM. Org Lett. 2001;3:3017–3020. doi: 10.1021/ol016431j. [DOI] [PubMed] [Google Scholar]; b) Luanphaisarnnont T, Ndubaku CO, Jamison TF. Org Lett. 2005;7:2937–2940. doi: 10.1021/ol050881k. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Han SB, Han H, Krische MJ. J Am Chem Soc. 2010;132:1760–1761. doi: 10.1021/ja9097675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhao Y, Rodrigo J, Hoveyda AH, Snapper ML. Nature. 2006;443:67–70. doi: 10.1038/nature05102. [DOI] [PubMed] [Google Scholar]
- 7.Brown HC, Narla G. J Org Chem. 1995;60:4686–4687. [Google Scholar]
- 8.Hoveyda AH, Malcolmson SJ, Meek SJ, Zhugralin AR. Angew Chem. 2010;122:38–49. doi: 10.1002/anie.200904491. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem, Int Ed. 2010;49:34–44. [Google Scholar]
- 9.For a recent review, see Fürstner A. Science. 2013;341:1229713. doi: 10.1126/science.1229713.For leading references, see Endo K, Grubbs RH. J Am Chem Soc. 2011;133:8525–8527. doi: 10.1021/ja202818v.Keitz BK, Endo K, Patel PR, Herbert MB, Grubbs RH. J Am Chem Soc. 2012;134:693–699. doi: 10.1021/ja210225e.Rosebrugh LE, Herbert MB, Marx VM, Keitz BK, Grubbs RH. J Am Chem Soc. 2013;135:1276–1279. doi: 10.1021/ja311916m.Flook MM, Jiang AJ, Schrock RR, Mueller P, Hoveyda AH. J Am Chem Soc. 2009;131:7962–7963. doi: 10.1021/ja902738u.Meek SJ, O’Brien RV, Llaveria J, Schrock RR, Hoveyda AH. Nature. 2011;471:461–466. doi: 10.1038/nature09957.Khan RKM, Torker S, Hoveyda AH. J Am Chem Soc. 2013;135:10258. doi: 10.1021/ja404208a.
- 10.For a recent review, see Kress S, Blechert S. Chem Soc Rev. 2012;41:4389–4408. doi: 10.1039/c2cs15348c.for leading references, see Berlin JM, Goldberg SD, Grubbs RH. Angew Chem. 2006;118:7753–7757. doi: 10.1002/anie.200602469.Angew Chem, Int Ed. 2006;45:7591–7595. doi: 10.1002/anie.200602469.Funk TW, Berlin JM, Grubbs RH. J Am Chem Soc. 2006;128:1840–1846. doi: 10.1021/ja055994d.Savoie J, Stenne B, Collins SK. Adv Synth Catal. 2009;351:1826–1832.Stenne B, Timperio J, Savoie J, Dudding T, Collins SK. Org Lett. 2010;12:2032–2035. doi: 10.1021/ol100511d.Tiede S, Berger A, Schlesiger D, Rost D, Luhl A, Blechert S. Angew Chem. 2010;122:4064–4067. doi: 10.1002/anie.201000940.Angew Chem, Int Ed. 2010;49:3972–3975. doi: 10.1002/anie.201000940.Kannenberg A, Rost D, Eibauer S, Tiede S, Blechert S. Angew Chem. 2011;123:3357–3360. doi: 10.1002/anie.201007673.Angew Chem, Int Ed. 2011;50:3299–3302. doi: 10.1002/anie.201007673.Khan RKM, O’Brien RV, Torker S, Li B, Hoveyda AH. J Am Chem Soc. 2012;134:12774–12779. doi: 10.1021/ja304827a.Yu M, Ibrahem I, Hasegawa M, Schrock RR, Hoveyda AH. J Am Chem Soc. 2012;134:2788–2799. doi: 10.1021/ja210946z.
- 11.a) Randall ML, Tallarico JA, Snapper ML. J Am Chem Soc. 1995;117:9610–9611. [Google Scholar]; b) Snapper ML, Tallarico JA, Randall ML. J Am Chem Soc. 1997;119:1478–1479. [Google Scholar]; c) Tallarico JA, Randall ML, Snapper ML. Tetrahedron. 1997;53:16511–16520. [Google Scholar]; d) Schrader TO, Snapper ML. J Am Chem Soc. 2002;124:10998–11000. doi: 10.1021/ja027154u. [DOI] [PubMed] [Google Scholar]; e) White BH, Snapper ML. J Am Chem Soc. 2003;125:14901–14904. doi: 10.1021/ja037656n. [DOI] [PubMed] [Google Scholar]
- 12.Hartung J, Grubbs RH. J Am Chem Soc. 2013;135:10183–10185. doi: 10.1021/ja4046422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Howse PE, Stevens IDR. Insect Pheromones and their Use in Pest Management. Chapman & Hall; New York: 1998. [Google Scholar]
- 14.Recent work from this group has demonstrated the application of racemic 1 to the synthesis of Lepidoptera female sex pheromones, see Herbert MB, Marx VM, Pederson RL, Grubbs RH. Angew Chem. 2013;125:328–332. doi: 10.1002/anie.201206079.Angew Chem, Int Ed. 2013;52:310–314.
- 15.Kirmse W, Scheidt F, Vater HJ. J Am Chem Soc. 1978;100:3945–3946. [Google Scholar]
- 16.La DS, Ford JG, Sattely ES, Bonitatebus PJ, Schrock RR, Hoveyda AH. J Am Chem Soc. 1999;121:11603–11604. [Google Scholar]
- 17.Attempts to use cyclic protecting groups (ex: benzylidene acetal) resulted in low conversion.
- 18.In preliminary stoichiometric experiments with 1 and 5c, we observe the formation of a kinetically stable intermediate analogous to one described in a recent report on an enantiopure ruthenium alkylidene complex, see Khan RKM, Zhugralin AR, Torker S, O’Brien RV, Lombardi PJ, Hoveyda AH. J Am Chem Soc. 2012;134:12438–12441. doi: 10.1021/ja3056722.
- 19.a) Silverstein RM, Brownlee RG, Bellas TE, Wood DL, Browne LE. Science. 1968;159:889–891. doi: 10.1126/science.159.3817.889. [DOI] [PubMed] [Google Scholar]; b) Yasumoto T, Murata M, Oshima Y, Sano M, Matsumoto GK, Clardy J. Tetrahedron. 1985;41:1019–1025. [Google Scholar]; c) Uemura D, Chou T, Haino T, Nagatsu A, Fukuzawa S, Zheng SZ, Chen HS. J Am Chem Soc. 1995;117:1155–1156. [Google Scholar]; d) Chou T, Kamo O, Uemura D. Tetrahedron Lett. 1996;37:4023–4026. [Google Scholar]; e) Chou T, Haino T, Kuramoto M, Uemura D. Tetrahedron Lett. 1996;37:4027–4030. [Google Scholar]
- 20.For catalytic asymmetric syntheses, see Oehlschlager AC, Johnston BD. J Org Chem. 1987;52:940–943.Burke SD, Muller N, Beaudry CM. Org Lett. 1999;1:1827–1829. doi: 10.1021/ol9910971.Kim SG, Park TH, Kim BJ. Tetrahedron Lett. 2006;47:6369–6372.Singh S, Guiry PJ. J Org Chem. 2009;74:5758–5761. doi: 10.1021/jo901019u.for syntheses relying on stoichiometric chiral reagents, see Bernardi R, Fuganti C, Grasselli P. Tetrahedron Lett. 1981;22:4021–4024.Mori K, Seu YB. Tetrahedron. 1985;41:3429–3431.Sato F, Takahashi O, Kato T, Kobayashi Y. J Chem Soc, Chem Commun. 1985:1638–1641.Hatakeyama S, Sakurai K, Takano S. J Chem Soc, Chem Commun. 1985:1759–1761.Yusufoglu A, Antons S, Scharf HD. J Org Chem. 1986;51:3485–3487.Mulzer J, Angermann A, Munch W. Liebigs Ann Chem. 1986:825–838.Redlich H, Bruns W, Francke W, Schurig V, Payne TL, Vite JP. Tetrahedron. 1987;43:2029–2034.Chong JM, Mar EK. Tetrahedron. 1989;45:7709–7716.Noda Y, Kikuchi M. Chem Lett. 1989:1755–1756.Ramaswamy S, Oehlschlager AC. J Org Chem. 1989;54:255–257.Matsumoto K, Suzuki N, Ohta H. Tetrahedron Lett. 1990;31:7163–7166.Pedrocchifantoni G, Servi S. J Chem Soc, Perkin 1. 1991:1764–1765.Cere V, Mazzini C, Paolucci C, Pollicino S, Fava A. J Org Chem. 1993;58:4567–4571.Soderquist JA, Rane AM. Tetrahedron Lett. 1993;34:5031–5034.Gypser A, Flasche M, Scharf HD. Liebigs Ann Chem. 1994:775–780.Kim MJ, Choi GB, Kim JY, Kim HJ. Tetrahedron Lett. 1995;36:6253–6256.Vettel S, Lutz C, Knochel P. Synlett. 1996:731–733.Gallos JK, Kyradjoglou LC, Koftis TV. Heterocycles. 2001;55:781–784.Lee HY, Jung Y, Moon H. Bull Korean Chem Soc. 2009;30:771–772.
- 21.Attempts to utilize 4-penten-2-one resulted in recovered starting material, likely due to deactivation of the catalyst by formation of a stable chelating species.
- 22.The absolute configurations of the AROCM products in this study were assigned by analogy to 11 and 13.
- 23.a) Cha JK, Christ WJ, Kishi Y. Tetrahedron Lett. 1983;24:3943–3946. [Google Scholar]; b) Christ WJ, Cha JK, Kishi Y. Tetrahedron Lett. 1983;24:3947–3950. [Google Scholar]
- 24.a) Schmidt RR, Gohl A. Chem Ber. 1979;112:1689–1704. [Google Scholar]; b) Wender PA, Bi FC, Buschmann N, Gosselin F, Kan C, Kee JM, Ohmura H. Org Lett. 2006;8:5373–5376. doi: 10.1021/ol062234e. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.