

Abstract
During malignant transformation the cells of origin give rise to cancer stem cells which possess the capacity to undergo limitless rounds of self-renewing division, regenerating themselves while producing more tumor cells. Within normal tissues, a limitless self-renewal capacity is unique to the stem cells, which divide asymmetrically to produce more restricted progenitors. Accumulating evidence suggests that misregulation of the self-renewal machinery in stem cell progeny can lead to tumorigenesis, but how it influences the properties of the resulting tumors remains unclear. Studies of the type II neural stem cell (neuroblast) lineages in the Drosophila larval brain have identified a regulatory cascade that promotes commitment to a progenitor cell identity by restricting their response to the self-renewal machinery. Brain tumor (Brat) and Numb initiate this cascade by asymmetrically extinguishing the activity of the self-renewal factors. Subsequently, Earmuff (Erm) and the SWI/SNF complex stably restrict the competence of the progenitor cell to respond to reactivation of self-renewal mechanisms. Together, this cascade programs the progenitor cell to undergo limited rounds of division, generating exclusive differentiated progeny. Here we review how defects in this cascade lead to tumor initiation and how inhibiting the self-renewal mechanisms may be an effective strategy to block CSC expansion.
Introduction
Investigation of the mechanisms by which cancers are formed and regulated may aid in the development of more effective cancer therapies [1, 2]. Accumulating evidence suggests that aberrant activity of stem cell self-renewal pathways can transform progenitor cells into tumor initiating cells [3-11]. In addition, aberrant activity of stem cell self-renewal pathways has also been implicated in the regulation of the cancer stem cell (CSC) types that support long term tumor growth [12-18]. These CSCs are defined by their capacity to self-renew while producing a hierarchical lineage of cells that either differentiate and become non-tumorigenic or form more CSCs to expand the tumor [8, Magee, 2012 #277]. How different oncogenic lesions coerce non-stem cell types into aberrantly responding to the self-renewal machinery and initiating tumor formation, and how this contributes to the regulation of the resulting CSCs remains unclear.
The type II neuroblast lineage in the Drosophila larval brain serves as an exceptional in vivo model to study the regulation of progenitor cells during normal development and tumorigenesis [19-21]. A type II neuroblast undergoes repeated rounds of asymmetric division to self-renew and to produce uncommitted (immature) intermediate neural progenitors (INPs) that are transiently arrested in the cell cycle progression (Figure 1A). [22-24]. Following division, the expression of the self-renewal factors remains on in the type II neuroblast but is asymmetrically extinguished in the immature INP [25-27]. The immature INP then undergoes a series of maturation steps to commit to the functional identity of an INP [26, 28, 29]. Subsequently, INPs reactivate expression of neuroblast self-renewal factors, but their response is severely restricted, ensuring INPs only undergo five-to-six rounds of asymmetric division to exclusively generate ganglion mother cells (GMC) and differentiated cells [30] (Figure 1A). A series of recent studies have demonstrated that defects in restricting the responses to self-renewal factors in immature INPs or INPs allows them to aberrantly reacquire a neuroblast like identity [25, 26, 29, 31-34]. This leads to the formation of massive numbers of supernumerary neuroblasts that act as CSCs, forming metastatic tumors that can be serially propagated upon transplantation into adult hosts [32, 35]. In this review, we will discuss how different mutations lead to aberrant responses to the self-renewal factors, resulting in different cells types acting as the cell of origin and producing CSCs with distinct properties. In addition, we will discuss how by modifying the aberrant responses to self-renewal factors, it may be possible to specifically interfere with the inter-conversion of progenitors to a less restricted stem cell type, thereby preventing tumor growth.
Figure 1. Aberrant expression of self-renewal factors causes uncommitted progenitors to revert to form CSCs.
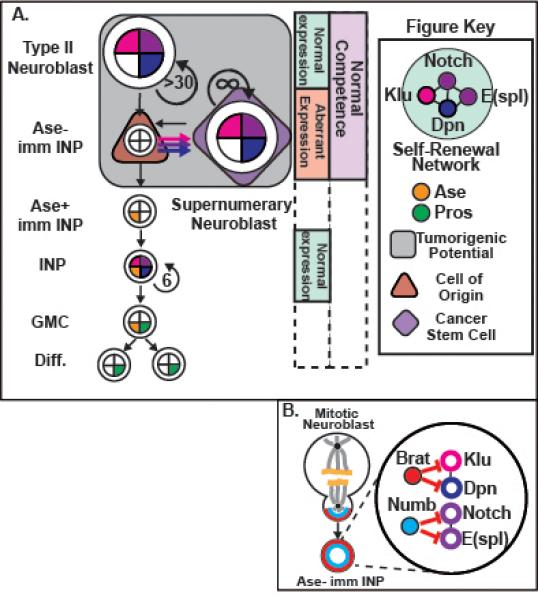
A) Lineage diagram depicting tumorigenesis resulting from overexpression of self-renewal factors or brat or numb mutation. Type II neuroblasts express a self-renewal network (light blue) that includes Notch (purple), E(spl)mγ(purple), Dpn (blue), and Klu (magenta). Ase− immature INPs remain competent (light purple box) to respond to aberrant expression (salmon box) of the self-renewal factors and can revert to form supernumerary neuroblasts. In these tumor types, Ase− immature INPs act as the cell of origin (brown triangle) and supernumerary neuroblasts are likely the CSCs (purple diamond); these cell types retain tumorigenic potential (grey box). In contrast, INPs do not retain tumorigenic potential in these tumor types, and only divide 5-6 times to generate exclusively GMCs and differentiated cells that express nuclear Pros (green). (B) Brat and Numb repress expression of self-renewal factors in Ase− immature INPs. (Left) Schematic depicting the mitotic division of a type II neuroblast showing: Brat (red) and Numb (blue) are basally segregated into the Ase− immature INP; DNA is shown in yellow; spindle in grey; and centrosomes in black. (Right) Once in the Ase− immature INPs, Brat and Numb act in parallel to inhibit aberrant expression of distinct components of the self-renewal network.
Aberrant reversion of uncommitted progenitor cells induced by self-renewal factors leads to the formation of tumor-initiating cells
Studies from several groups have collectively established a network of factors that plays critical roles in promoting the self-renewal of type II neuroblasts (Figure 1A) [26, 27, 32, 36]. Consistent with aberrant responses to self-renewal factors promoting tumor formation, over-expression of components of this self-renewal network triggers formation of massive numbers of supernumerary neuroblasts that are tumorigenic [32]. A highly conserved component of the type II neuroblast self-renewal network is Notch, which encodes a transmembrane protein [37, 38]. Upon proteolytic activation, the Notch intra-cellular domain (NICD) translocates to the nucleus where it complexes with the DNA binding protein Suppressor of Hairless (Su(H)) to activate target gene expression. Notch is indispensable for the maintenance of type II neuroblasts, and over-expression of the NICD in type II neuroblast lineages leads to supernumerary neuroblast formation [26, 39]. Thus, Notch is both necessary and sufficient to promote type II neuroblast self-renewal. Notch promotes the self-renewal of type II neuroblasts in part by directly regulating the expression of Enhancer of split mγ(E(spl)mγ), which encodes a basic helix-loop-helix-Orange transcription factor [27] (Figure 1A). Removing Notch function abrogates the expression of the E(spl)mγ-gfp reporter transgene in all neuroblasts, and loss of the E(spl) locus renders over-expression of the NICD unable to induce supernumerary type II neuroblast formation. Although over-expression of E(spl)mγ in type II neuroblast lineages induces supernumerary neuroblast formation, loss of the E(spl) locus does not affect the maintenance of type II neuroblasts [27, 32]. Thus, E(spl)mγ is only sufficient to promote neuroblast self-renewal, suggesting that additional parallel mechanisms must exist. Similar to E(spl)mγ, over-expression of deadpan (dpn), which also encodes a basic helix-loop-helix-Orange transcription factor, in type II neuroblast lineages also induces supernumerary neuroblast formation, but loss of dpn function does not affect the maintenance of type II neuroblasts [32, 36, 40]. Most importantly, type II neuroblasts lacking both dpn and the E(spl) loci rapidly undergo premature differentiation, indicating that Dpn and E(spl)mγ function cooperatively to maintain the self-renewal of type II neuroblasts [27] (Figure 1A). Despite containing many functional Su(H) binding sites in the regulatory region, the expression of Dpn does not require Notch function, and dpn is dispensable for supernumerary neuroblast formation induced by over-expression of NICD [27]. Therefore, Dpn functions in parallel with E(spl)mγ to regulate the self-renewal of type II neuroblasts possibly by integrating multiple upstream signaling inputs including Notch (Figure 1A).
Klumpfuss (Klu) is another core component of the type II neuroblast self-renewal network (Figure 1A). While type II neuroblasts in klu mutant larval brains undergo premature differentiation, over-expression of klu induces supernumerary neuroblast formation [26, 32]. Thus, Klu is also necessary and sufficient for the self-renewal of type II neuroblasts. In addition, removal of klu function completely compromises the ability of overexpression of the NICD to induce supernumerary neuroblast formation, and over-expression of klu suppresses premature differentiation of type II neuroblasts mutant for Notch function [26]. Thus, klu likely functions downstream of Notch in promoting the self-renewal of type II neuroblasts (Figure 1A).
How does increased function of the self-renewal factors in type II neuroblast lineages cause tumorigenesis? One possibility might be that increased function of the self-renewal factors induces symmetric divisions of type II neuroblasts, and that tumor-initiating cells arise from type II neuroblasts that self-renew excessively. However, type II neuroblasts over-expressing NICD or klu reproducibly undergo asymmetric cell divisions to self-renew and to generate immature INPs [26, 32]. Thus, it is unlikely that tumor-initiating cells arise from type II neuroblasts that excessively self-renew. Alternatively, tumor-initiating cells might arise from the reversion of newly born Ase− immature INPs, which also aberrantly express self-renewal factors following asymmetric cell division. Consistent with this hypothesis, mis-expression of the NICD, klu, dpn, or E(spl)mγ in Ase− immature INPs leads to the formation of supernumerary neuroblasts [26, 34]. Thus, aberrantly reverting Ase− immature INPs serve as the cells of origin in the fly brain tumor models induced by over-expression of self-renewal factors (Figure 1A).
Brat and Numb prevent the formation of tumor-initiating cells by extinguishing the function of type II neuroblast self-renewal factors
Since Ase− immature INPs remain extremely competent to respond to the self-renewal factors, extinguishing aberrant responses to the self-renewal machinery in these cells plays a key role in suppressing tumor initiation (Figure 1A) [26, 34]. Brat and Numb, which uniquely segregate into the newly born Ase− immature INP following asymmetric division of a type II neuroblast [24, 25, 41, 42] (Figure 1B), are excellent candidate proteins to extinguish aberrant responses to the neuroblast self-renewal factors in immature INPs. Several recent studies have collectively indicated that Brat and Numb function to prevent tumorigenesis by preventing Ase− immature INPs from reverting into supernumerary neuroblasts [26, 29]. Consistently, transplantation of brat mutant brains or brains bearing numb mutant clones into adult hosts gives rise to metastatic tumors that can be serially propagated [35].
A brat mutant type II neuroblast always divides asymmetrically to self-renew and to generate a newly born Ase− immature INP [24, 26]. However, a newly born Ase− immature INP lacking brat function does not progress through stereotypical maturation steps to commit to an INP functional identity, but instead, rapidly reverts back into a supernumerary neuroblast. Thus, similar to overexpression of self-renewal factors, Ase− immature INPs are the cell of origin in brat mutant tumors (Figure 1A). Accumulating evidence suggests that Brat prevents newly born Ase− immature INPs from reverting into supernumerary neuroblasts in part by extinguishing the activity of core components of the type II neuroblast self-renewal network (Figure 1B). Consistent with this model, Dpn is aberrantly expressed in brat mutant immature INPs, and removing the function of klu or dpn completely suppresses supernumerary neuroblast formation in the brat mutant genetic backgrounds [26, 34]. Thus, Brat prevents newly born Ase− immature INPs from reverting into supernumerary neuroblasts by preventing the aberrant expression of Klu and Dpn (Figure 1A&B). In addition to regulating core components of the self-renewal machinery, Brat also restricts progenitor potential through context dependent signaling mechanisms. While loss- or gain-of-function of the Wnt signaling pathway has no effect in wild-type brains, increased Wnt signaling further exacerbates supernumerary neuroblast formation in brat mutant brains whereas decreasing Wnt signaling reduces supernumerary neuroblast formation [29]. Thus, Brat also suppresses the reversion of Ase− immature INPs by antagonizing the Wnt signaling pathway. These studies indicate that Brat prevents uncommitted progenitor cells from aberrantly reverting into tumor-initiating cells by suppressing multiple conserved oncogenic pathways.
Similar to brat, a numb mutant type II neuroblast also reproducibly undergoes asymmetric division to generate a newly born Ase− immature INP that reverts back into a supernumerary neuroblast [26]. Thus, newly born Ase− immature INPs also serve as the cell of origin in numb mutant tumors (Figure 1A). Numb functions as an evolutionarily conserved negative regulator of Notch signaling [43, 44], and likely promotes the commitment to a functional INP identity by restricting Notch function (Figure 1A&B). Consistently, over-expression of the NICD in Ase− immature INPs efficiently induces supernumerary neuroblast formation (Xiao and Lee, unpublished observation). How does Numb antagonize Notch signaling in type II neuroblasts? During asymmetric divisions of sensory organ precursor cells, Numb promotes endocytosis of the Notch receptor [43, 45]. While it is possible that Numb functions in a similar manner in immature INPs, Numb may also function through a distinct mechanism [25]. Interestingly, while overexpression of numb normally triggers premature differentiation of type II neuroblasts, overexpression of numb cannot suppress supernumerary neuroblast formation in brat mutants [29]. Thus, the reversion and self-renewal of brat mutant tumors may be particularly dependent on klu and dpn. In addition, overexpression of brat is not sufficient to suppress supernumerary neuroblast formation in numb mutants [29]. Thus, Brat and Numb likely function in parallel to prevent Ase− immature INPs from serving as the cells of origin for tumorigenesis by extinguishing the function of distinct components of the type II neuroblast self-renewal network (Figure 1B).
Lineage trace experiments strongly suggest that the supernumerary neuroblasts that form in brat and numb mutant tumors retain tumorigenic potential and likely act as CSCs (Figure 1A). Consistent with this model, most Ase− immature INPs in hypomorphic brat mutants produce exclusively differentiated cells, but some stochastically revert to form supernumerary neuroblasts [34]. Once reverted, these supernumerary neuroblasts can self-renew indefinitely, expanding the tumor while simultaneously producing differentiated cell types (Figure 1A). Strikingly, removing just dpn or klu alone completely prevents further reversion of Ase− immature INPs in brat mutants [26, 34]. Thus, selectively targeting components of the self-renewal network that are aberrantly expressed in specific tumor types may halt expansion of the CSCs population by causing them to produce exclusively differentiated cell types. Alternatively, reinstating the restriction imposed by proteins asymmetrically inherited by the uncommitted progenitor cells might also slow down the expansion of the CSCs.
Earmuff prevents INPs from reverting into supernumerary neuroblasts by restricting their competence to respond to self-renewal transcription factors
Intriguingly, INPs reactivate the expression of neuroblast self-renewal factors but their proliferative and developmental potential remains restricted, suggesting that during maturation their competence to respond to this self-renewal network is attenuated (Figure 1A). The transcription factor earmuff (erm) is an excellent candidate to stably restrict the response of INPs to the neuroblast self-renewal network. In contrast to brat and numb mutants, lineage trace experiments reveal that aberrantly reverting INPs act as the cell of origin in erm mutant brain tumors [31] (Figure 2A). These experiments suggest that erm mutant INPs revert to form supernumerary neuroblasts that possess unrestrained self-renewal capacity and produce both differentiated cells and more tumorigenic neuroblasts [31, 34]. Thus, erm mutant supernumerary neuroblasts may also function as CSCs. Consistent with erm suppressing the response of INPs to the self-renewal network, co-overexpression of erm can completely suppress the formation of supernumerary neuroblasts induced by over-expression of NICD, dpn, klu, or E(spl)mγ [34]. In addition, removing the function of dpn or klu or knocking down of Notch function strongly suppresses the formation of supernumerary neuroblasts in erm mutants [31, 34]. This indicates that the competence of INPs to respond to the normal reactivation of self-renewal factors is extended in erm mutants, allowing INPs to revert and form supernumerary neuroblasts (Figure 2A).
Figure 2. Failure to restrict the competence of progenitors to respond to self-renewal factors allows them to revert to form CSCs.
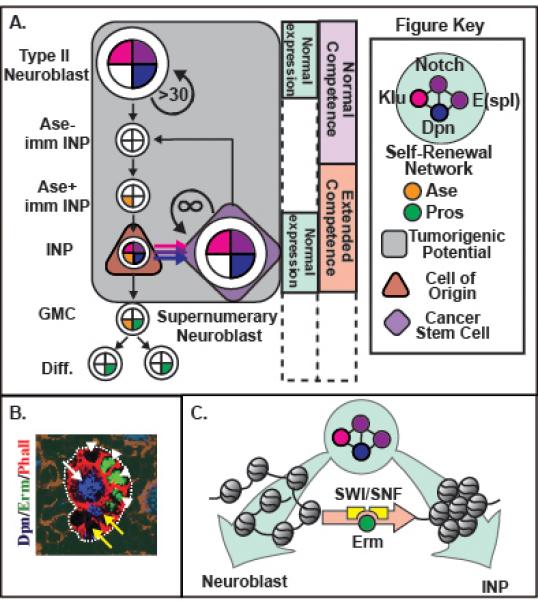
(A) Lineage diagram depicting tumorigenesis when erm or the SWI/SNF complex is mutated. INPs possess extended competence (salmon box) to respond to normal reactivation (light blue box) of self-renewal factors, causing INPs to revert to form supernumerary neuroblasts. Thus, INPs serve as the cell of origin (brown triangle), and revert to form supernumerary neuroblasts that likely act as the CSCs (purple diamond). In erm or SWI/SNF mutants type II neuroblasts, immature INPs, INPs and supernumerary neuroblasts retain tumorigenic potential (grey box) whereas, GMCs, and Diff. cells do not. (B) Confocal image showing Erm expression pattern (green). The following are indicated: type II neuroblast (white arrow); immature INPs (white arrowhead); and INP (yellow arrow). (C) Schematic showing Erm (green) and the SWI/SNF complex (yellow) remodeling the chromatin to restrict the competence to respond to the self-renewal network: nucleasome (grey oval); DNA (black line).
Although erm mutant INPs aberrantly respond to neuroblast self-renewal factors and revert into supernumerary neuroblasts, Erm is specifically expressed in immature INPs, indicating that erm functions during the commitment to an INP functional identity (Figure 2B) [34]. Interestingly, Erm expression is undetectable in brat or numb mutant Ase− immature INPs, indicating that Erm functions temporally after Brat and Numb during the commitment process [34]. These observations raise two intriguing possibilities: (1) the restricted response of progenitors to the self-renewal network is the temporally coordinated effort of asymmetrically segregated proteins and asymmetrically expressed transcription factors, and (2) that by directing activation of Erm, Brat and Numb flip a transcriptional switch. One potential explanation for how Brat and Numb direct Erm activation is by suppressing the self renewal transcription factor activity. Consistent with this model, three of the four core components of the self-renewal network, Klu, Dpn and E(spl)mγ, have been shown to act as transcriptional repressors in other developmental contexts [46-48]. Thus, the premature reactivation of Klu, Dpn or E(spl)mγ in brat or numb mutant immature INPs respectively may transcriptionally repress erm activation. What promotes erm expression in type II neuroblast lineages? The transcription factor pointed (isoform P1) (pntP1) is specifically expressed in type II neuroblasts, and over-expression of PntP1 in other neuroblast lineages is sufficient to trigger the production of ectopic INP-like cells that activate erm-reporter expression [49]. In addition, mutation of pnt genetically interacts with both brat and erm (Janssens, Komori and Lee, unpublished). Thus, the transcriptional networks that distinguish type II neuroblasts from other neuroblasts may prime type II neuroblasts to produce progenitors that asymmetrically express Erm. While, additional genes have been suggested to act in the networks that distinguish type II neuroblasts from differentiated cells or other neuroblasts [32, 50, 51], the potential role of these networks during the regulation of erm expression has not been explored.
How does erm restrict the competence of INPs to respond to the neuroblast self-renewal network? The vertebrate homologues of erm, fez and fezl, are also expressed in neural stem cell lineages [52, 53], and the function of erm is likely evolutionarily conserved [31]. However, fez and fezl have been shown to act in a context dependent manner to both activate and repress transcription [54-57]. Evidence strongly suggests that erm restricts INP potential by acting primarily as a transcriptional repressor [34]. In vertebrates, fez and fezl suppress Notch activity by directly repressing transcription of the homologues of dpn and E(spl)mγ [54]. However, in the Drosophila larval brain dpn and E(spl)mγ are reactivated following erm expression, and co-expression of erm is sufficient to block the ability of dpn or E(spl)mγ to promote supernumerary neuroblast formation [34]. Thus, while erm restricts the function of dpn and E(spl)mγ in Drosophila, they are not likely the direct transcriptional targets of erm. Alternatively, fezl has been shown to activate proneural genes in zebrafish embryos [56]. However, converting erm into an activator has a dominant negative effect in the larval brain [34]. Thus, the described functions of fez and fezl cannot explain the tumor suppressor function of erm, warranting continued study to identify novel erm regulated genes which may serve as new targets for selectively inhibiting tumor growth.
The SWI/SNF complex promotes the directional lineage progression of type II neuroblasts, preventing the reversion of INPs into supernumerary neuroblasts
Alteration of cellular response to networks of transcription factors is dependent on remodeling of the chromatin [58, 59]. The highly conserved SWI/SNF complex is an ATP dependent chromatin remodeling complex that alters nucleosome stability and has diverse functions during stem cell self-renewal and differentiation [60-63]. While members of the SWI/SNF complex are among the most frequently mutated genes in human cancer [64-66], whether their role during the regulation of stem cell fate decisions is connected to their function during tumor suppression remains unclear.
The SWI/SNF complex can be separated into two subtypes, BAP and PBAP based on the presence of the scaffolding protein Osa specifically within the BAP complex [67, 68]. A genome wide RNAi study showed that knocking down critical components of the BAP complex including osa, the catalytic subunit brm, or the assembly and stability factor moira, specifically in type II lineages results in the formation of supernumerary neuroblasts [51]. Upon transplantation into adult hosts these ectopic stem cells form tumors [33]. Thus, the type II neuroblast lineage provides an exceptional model to understand the role of the SWI/SNF complex during the regulation of stem cell lineages and the suppression of tumorigenesis.
Although the SWI/SNF complex is expressed ubiquitously in the fly brain, several lines of evidence suggest that the SWI/SNF complex suppresses tumorigenesis through a specific function during the commitment to an INP functional identity. First, multiple components of the SWI/SNF complex genetically interact with both brat and numb but knock down of brm, mora, or osa does not disrupt neuroblast polarity [34]. This indicates that the SWI/SNF complex likely functions downstream of brat and numb. Consistent with this model, expression of a dominant negative form of brm specifically within Ase− immature INPs can genetically enhance a brat mutant phenotype, and most importantly, knocking down the function of osa specifically within Ase− immature INPs can result in the formation of supernumerary neuroblasts and tumors [33]. Thus the SWI/SNF complex suppresses tumor formation through a specific function during the commitment to an INP functional identity.
Lineage trace experiments reveal that osa mutant supernumerary neuroblasts do not arise from rapidly reverting immature INPs, but instead from INPs that undergo spontaneous reversion [33]. Thus, similar to erm, the SWI/SNF functions in immature INPs to establish the restriction on their developmental potential following the commitment to an INP functional identity (Figure 2A). How does the SWI/SNF complex prevent INPs from reverting into supernumerary neuroblasts? Chromatin remodeling complexes rarely bind to DNA directly but are instead recruited to distinct genomic loci by cell type specific transcription factors. Erm is specifically expressed in immature INPs, and mutation of the SWI/SNF complex phenocopies erm mutants, making it an excellent candidate to direct the function of the SWI/SNF complex during INP maturation. Consistent with this model, brm genetically interacts with erm [34]. Thus, Erm likely functions as an efficient mechanism to direct the SWI/SNF complex to distinct loci to remodel the chromatin, thereby restricting the competence of INPs to respond to the self-renewal network (Figure 2C).
In addition to potentially mediating the repressive effects of Erm, the SWI/SNF complex also activates expression of the transcription factor hamlet (ham) [33]. Ham is uniquely expressed in INPs and directs the birth order in which INPs produce specific types of neurons and glia. Reducing the function of ham causes INPs to undergo excessive rounds of division, suggesting that ham is required to schedule the terminal differentiation of INPs. Consistent with ham promoting the progressive restriction of INPs, over-expression of ham is sufficient to trigger premature differentiation of type II neuroblasts. However, when ham function is reduced INPs continue to divide asymmetrically and do not reacquire a neuroblast like identity. Thus, ham is required to schedule the end of INP self-renewal, but not to prevent INPs from to reverting into supernumerary neuroblasts.
Intriguingly, transplantation of brains in which ham is knocked down into adult hosts does not trigger malignant tumor formation [33]. This suggests that increased self-renewal alone is not sufficient to trigger tumorigenesis, and that regulation of ham expression cannot fully explain the tumor suppressive effects of the SWI/SNF complex. This leaves two potential explanations for how mis-regulation of the stem cell self-renewal machinery contributes to tumorigenesis: (1) CSCs may need to acquire a more plastic stem cell like identity in order to become tumorigenic, or (2) the exponential growth of the CSC pool afforded by inter-conversion of progenitor states may drive tumorigenesis. In fact, evidence supporting both models exists. In human leukemias a stem cell like gene signature is a negative prognostic factor, suggesting that stem cell like CSCs may be more tumorigenic [12 , 13]. In contrast, many leukemias and solid tumors have been described where the CSC pool more closely resembles a restricted progenitor type than a stem cell [69-73]. To unambiguously distinguish between these models using the type II lineage requires examination of a mutant that allows inter-conversion between more restricted GMCs and INPs while preventing reversion to a neuroblast identity. Indeed, mutation of pros specifically within INPs presents such a phenotype; however, the tools to prospectively target pros mutation to INPs while avoiding other clone types have only recently become available [74]. Continued study of type II neuroblast lineages will likely identify the mechanisms by which mis-regulation of self-renewal machinery can lead to the formation and regulation of CSCs.
Inter-conversion between the cells of origin and CSCs fuels tumor growth in type II neuroblast lineages
CSCs often display stem or progenitor cell-like features and are able to self-renew while producing all the other differentiated tumor cell types [75, 76]. While different molecular lesions are thought to affect the cells of origin and the properties of the CSC pool, the relationship between these factors remains poorly understood. The precise lineage information and genetic control of type II neuroblasts provides a unique opportunity to study the formation and regulation of CSCs. Clonal analysis of numb and brat mutants reveals that tumorigenesis is initiated by Ase− immature INPs that revert into supernumerary neuroblasts (Figure 1A). In contrast, when the SWI/SNF complex is impaired or erm is mutated, immature INPs commit to an INP functional identity but spontaneously revert to form supernumerary neuroblasts (Figure 2A). Thus, different molecular lesions result in different cell types becoming misregulated and acting as the cell of origin. Lineage trace experiments performed in these mutant genetic backgrounds suggest that supernumerary neuroblasts serve as CSCs that self-renew extensively and produce both nontumorigenic cells and more supernumerary neuroblasts. However, the gold standard for assessing CSC properties is transplantation. Thus, the development of efficient protocols that allow the transplantation of a small number of genetically homogeneous supernumerary neuroblasts from mutant brains is essential to demonstrate the existence of CSCs in these fly brain tumor models.
The prominence of the CSC model and the parallels between CSCs and normal stem cells has led to the misconception that aberrant self-renewal is the underlying defect that leads to tumor formation. Comparison of knock-down of the SWF/SNF complex with knock-down of ham in type II lineages reveals that while aberrant activity of genes that promote stem cell self-renewal can initiate tumorigenesis, unrestrained self-renewal alone cannot [33]. Importantly, when ham is knocked down, INPs self-renew indefinitely but never reacquire a neuroblast like identity. This strongly suggests that aberrant responses to the self-renewal factors leads to tumorigenesis by promoting inter-conversion between the progenitor cell state and the stem cell state, rather than by promoting uncontrolled self-renewal. Consistent with this model numerous tumors have been shown to contain CSCs that resemble different progenitor types that can interconvert [13, 18]. Thus, aberrant responses to the self-renewal machinery may trigger tumorigenesis by promoting interconversion of progenitor cells into CSCs, allowing exponential amplification of the CSC pool.
Importantly, genetic analysis reveals that reversion of immature INPs and INPs into CSCs may be particularly reliant on distinct components of the self-renewal machinery. While mutation of either dpn or klu alone has only a mild effect on self-renewal, mutating either klu or dpn strongly suppresses reversion in both brat and erm mutant tumors [26, 34]. This corroborates a model where targeting components of the self-renewal network, or perhaps more feasibly, the genes they regulate, may be an effective strategy to halt tumor growth [76]. Interestingly, mutation of klu has no effect on numb mutant tumors, and over-expression of numb is not sufficient to suppress supernumerary neuroblast formation in brat or erm mutants (Xiao and Lee, unpublished observation). Thus, different lesions likely produce CSCs through aberrant activity of distinct components of the self-renewal network. This indicates that treatment of tumors that arise from distinct lesions may require selectively targeting the specific components of the self-renewal pathway that are mis-regulated. Ultimately, a better understanding of how specific tumor types are formed and regulated may lead to more effective therapies.
Highlights (for review).
Defects in the mechanisms that restrict type II neuroblast lineages cause tumors.
Aberrant expression of self-renewal factors in immature INPs can initiate tumors.
Extended competence of INPs to respond to self-renewal factors can initiate tumors.
Inter-conversion between the cell of origin and cancer stem cells fuels tumor growth.
Inhibiting self-renewal pathways may be an effective strategy to halt tumor growth.
Acknowledgements
We thank the members of the Lee lab for the critical reading of this review. This work was supported by a NIH grant R01-GM092818.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Remke M, Ramaswamy V, Taylor MD. Medulloblastoma molecular dissection: the way toward targeted therapy. Curr Opin Oncol. 2013;25(6):674–81. doi: 10.1097/CCO.0000000000000008. [DOI] [PubMed] [Google Scholar]
- 2.Sadanandam A, et al. A colorectal cancer classification system that associates cellular phenotype and responses to therapy. Nat Med. 2013;19(5):619–25. doi: 10.1038/nm.3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Akala OO, et al. Long-term haematopoietic reconstitution by Trp53-/-p16Ink4a-/-p19Arf-/- multipotent progenitors. Nature. 2008;453(7192):228–32. doi: 10.1038/nature06869. [DOI] [PubMed] [Google Scholar]
- 4.Chen W, et al. Malignant transformation initiated by Mll-AF9: gene dosage and critical target cells. Cancer Cell. 2008;13(5):432–40. doi: 10.1016/j.ccr.2008.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McCormack MP, et al. The Lmo2 oncogene initiates leukemia in mice by inducing thymocyte self-renewal. Scienc. 2010;327(5967):879–83. doi: 10.1126/science.1182378. [DOI] [PubMed] [Google Scholar]
- 6.Youssef KK, et al. Identification of the cell lineage at the origin of basal cell carcinoma. Nat Cell Biol. 2010;12(3):299–305. doi: 10.1038/ncb2031. [DOI] [PubMed] [Google Scholar]
- 7.Liu C, et al. Mosaic analysis with double markers reveals tumor cell of origin in glioma. Cell. 2011;146(2):209–21. doi: 10.1016/j.cell.2011.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Visvader JE. Cells of origin in cancer. Nature. 2011;469:314–322. doi: 10.1038/nature09781. 20 January. [DOI] [PubMed] [Google Scholar]
- 9.Chen J, McKay RM, Parada LF. Malignant glioma: lessons from genomics, mouse models, and stem cells. Cell. 2012;149(1):36–47. doi: 10.1016/j.cell.2012.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu C, Zong H. Developmental origins of brain tumors. Curr Opin Neurobiol. 2012;22(5):844–9. doi: 10.1016/j.conb.2012.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schwitalla S, et al. Intestinal Tumorigenesis Initiated by Dedifferentiation and Acquisition of Stem-Cell-like Properties. Cell. 2013;152(1-2):25–38. doi: 10.1016/j.cell.2012.12.012. [DOI] [PubMed] [Google Scholar]
- 12.Gentles AJ, et al. Association of a leukemic stem cell gene expression signature with clinical outcomes in acute myeloid leukemia. JAMA. 2010;304(24):2706–15. doi: 10.1001/jama.2010.1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eppert K, et al. Stem cell gene expression programs influence clinical outcome in human leukemia. Nat Med. 2011;17(9):1086–93. doi: 10.1038/nm.2415. [DOI] [PubMed] [Google Scholar]
- 14.Guryanova OA, et al. Nonreceptor tyrosine kinase BMX maintains self-renewal and tumorigenic potential of glioblastoma stem cells by activating STAT3. Cancer Cell. 2011;19(4):498–511. doi: 10.1016/j.ccr.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shats I, Gatza ML, Chang JT, Mori S, Wang J, Rich J, Nevins JR. Using a stem cell-based signature to guide therapeutic selection in cancer. Cancer Res. 2011;71(5):1772–80. doi: 10.1158/0008-5472.CAN-10-1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Merlos-Suárez A, et al. The intestinal stem cell signature identifies colorectal cancer stem cells and predicts disease relapse. Cell Stem Cell. 2011;8(5):511–24. doi: 10.1016/j.stem.2011.02.020. [DOI] [PubMed] [Google Scholar]
- 17.Wong DJ, et al. Module map of stem cell genes guides creation of epithelial cancer stem cells. Cell Stem Cell. 2008;2(4):333–44. doi: 10.1016/j.stem.2008.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sarry JE, et al. Human acute myelogenous leukemia stem cells are rare and heterogeneous when assayed in NOD/SCID/IL2Rγc-deficient mice. J Clin Invest. 2011;121(1):384–95. doi: 10.1172/JCI41495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boyan GS, Reichert H. Mechanisms for complexity in the brain: generating the insect central complex. Trends Neurosci. 2011;34(5):247–57. doi: 10.1016/j.tins.2011.02.002. [DOI] [PubMed] [Google Scholar]
- 20.Weng M, Lee CY. Keeping neural progenitor cells on a short leash during Drosophila neurogenesis. Curr Opin Neurobiol. 2011;21(1):36–42. doi: 10.1016/j.conb.2010.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Homem CC, Knoblich JA. Drosophila neuroblasts: a model for stem cell biology. Development. 2012;139(23):4297–310. doi: 10.1242/dev.080515. [DOI] [PubMed] [Google Scholar]
- 22.Bello BC, et al. Amplification of neural stem cell proliferation by intermediate progenitor cells in Drosophila brain development. Neural Develop. 2008;3(1):5. doi: 10.1186/1749-8104-3-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Boone JQ, Doe CQ. Identification of Drosophila type II neuroblast lineages containing transit amplifying ganglion mother cells. Dev Neurobiol. 2008;68(9):1185–95. doi: 10.1002/dneu.20648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bowman SK, et al. The Tumor Suppressors Brat and Numb Regulate Transit-Amplifying Neuroblast Lineages in Drosophila. Dev Cell. 2008;14(4):535–46. doi: 10.1016/j.devcel.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Haenfler JM, Kuang C, Lee CY. Cortical aPKC kinase activity distinguishes neural stem cells from progenitor cells by ensuring asymmetric segregation of Numb. Dev Biol. 2012;365(1):219–228. doi: 10.1016/j.ydbio.2012.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xiao Q, Komori H, Lee CY. klumpfuss distinguishes stem cells from progenitor cells during asymmetric neuroblast division. Development. 2012;139(15):2670–80. doi: 10.1242/dev.081687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zacharioudaki E, Magadi SS, Delidakis C. bHLH-O proteins are crucial for Drosophila neuroblast self-renewal and mediate Notch-induced overproliferation. Development. 2012;139(7):1258–69. doi: 10.1242/dev.071779. [DOI] [PubMed] [Google Scholar]
- 28.Bayraktar OA, et al. Drosophila type II neuroblast lineages keep Prospero levels low to generate large clones that contribute to the adult brain central complex. Neural Dev. 2010;5:26. doi: 10.1186/1749-8104-5-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Komori H, et al. Brain tumor specifies intermediate progenitor cell identity by attenuating β-catenin/Armadillo activity. Development. 2014;141(1) doi: 10.1242/dev.099382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Homem CC, et al. Long-Term Live Cell Imaging and Automated 4D Analysis of Drosophila Neuroblast Lineages. PLoS One. 2013;8(11):e79588. doi: 10.1371/journal.pone.0079588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Weng M, Golden KL, Lee CY. dFezf/Earmuff maintains the restricted developmental potential of intermediate neural progenitors in Drosophila. Dev Cell. 2010;18(1):126–35. doi: 10.1016/j.devcel.2009.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Berger C, et al. FACS purification and transcriptome analysis of drosophila neural stem cells reveals a role for Klumpfuss in self-renewal. Cell Rep. 2012;2(2):407–18. doi: 10.1016/j.celrep.2012.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Eroglu E, et al. SWI/SNF complex regulates Prdm protein Hamlet to ensure lineage directionality in Drosopihla neural stem cells. Cell. 2014 In press. [Google Scholar]
- 34.Janssens DH, et al. dFezf/Earmuff restricts progenitor cell potential by attenuating the competence to respond to self renewal factors. Development. 2014 doi: 10.1242/dev.106534. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Caussinus E, Gonzalez C. Induction of tumor growth by altered stem-cell asymmetric division in Drosophila melanogaster. Nat Genet. 2005;37(10):1125–9. doi: 10.1038/ng1632. [DOI] [PubMed] [Google Scholar]
- 36.San-Juán BP, Baonza A. The bHLH factor deadpan is a direct target of Notch signaling and regulates neuroblast self-renewal in Drosophila. Dev Biol. 2011;352(1):70–82. doi: 10.1016/j.ydbio.2011.01.019. [DOI] [PubMed] [Google Scholar]
- 37.Liu J, et al. Notch signaling in the regulation of stem cell self-renewal and differentiation. Curr Top Dev Biol. 2010;92:367–409. doi: 10.1016/S0070-2153(10)92012-7. [DOI] [PubMed] [Google Scholar]
- 38.Hori K, Sen A, Artavanis-Tsakonas S. Notch signaling at a glance. J Cell Sci. 2013;126(10):2135–40. doi: 10.1242/jcs.127308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Song Y, Lu B. Regulation of cell growth by Notch signaling and its differential requirement in normal vs. tumor-forming stem cells in Drosophila. Genes Dev. 2011;25(24):2644–58. doi: 10.1101/gad.171959.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhu S, et al. The bHLH Repressor Deadpan Regulates the Self-renewal and Specification of Drosophila Larval Neural Stem Cells Independently of Notch. PLoS One. 2012;7(10):e46724. doi: 10.1371/journal.pone.0046724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee CY, et al. Brat is a Miranda cargo protein that promotes neuronal differentiation and inhibits neuroblast self-renewal. Dev. Cell. 2006;10(4):441–9. doi: 10.1016/j.devcel.2006.01.017. [DOI] [PubMed] [Google Scholar]
- 42.Betschinger J, Mechtler K, Knoblich JA. Asymmetric segregation of the tumor suppressor brat regulates self-renewal in Drosophila neural stem cells. Cell. 2006;124(6):1241–53. doi: 10.1016/j.cell.2006.01.038. [DOI] [PubMed] [Google Scholar]
- 43.Kandachar V, Roegiers F. Endocytosis and control of Notch signaling. Curr Opin Cell Biol. 2012;24(4):534–40. doi: 10.1016/j.ceb.2012.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Giebel B, Wodarz A. Notch signaling: numb makes the difference. Curr Biol. 2012;22(4):133–5. doi: 10.1016/j.cub.2012.01.006. [DOI] [PubMed] [Google Scholar]
- 45.Couturier L, Vodovar N, Schweisguth F. Endocytosis by Numb breaks Notch symmetry at cytokinesis. Nat Cell Biol. 2012;14(2) doi: 10.1038/ncb2419. [DOI] [PubMed] [Google Scholar]
- 46.Giagtzoglou N, et al. Two modes of recruitment of E(spl) repressors onto target genes. Development. 2003;130(2):259–70. doi: 10.1242/dev.00206. [DOI] [PubMed] [Google Scholar]
- 47.Fischer A, Gessler M. Delta-Notch- and then? Protein interactions and proposed modes of repression by Hes and Hey bHLH factors. Nucleic Acids Res. 2007;35:4583–4596. doi: 10.1093/nar/gkm477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Terriente-Felix A, et al. Notch cooperates with Lozenge/Runx to lock haemocytes into a differentiation programme. Development. 2013;140(4):926–37. doi: 10.1242/dev.086785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhu S, et al. Ets transcription factor Pointed promotes the generation of intermediate neural progenitors in Drosophila larval brains. Proc Natl Acad Sci U S A. 2011;108(51):20615–20620. doi: 10.1073/pnas.1118595109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Carney TD, et al. Functional genomics identifies neural stem cell sub-type expression profiles and genes regulating neuroblast homeostasis. Developmental Biology. 2012;361(1):137–146. doi: 10.1016/j.ydbio.2011.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Neumüller RA, et al. Genome-wide analysis of self-renewal in Drosophila neural stem cells by transgenic RNAi. Cell Stem Cell. 2011;8(5):580–93. doi: 10.1016/j.stem.2011.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nelson BR, et al. Dynamic interactions between intermediate neurogenic progenitors and radial glia in embryonic mouse neocortex: potential role in Dll1-Notch signaling. J Neurosci. 2013;33(21):122–39. doi: 10.1523/JNEUROSCI.0791-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Guo C, et al. Fezf2 Expression Identifies a Multipotent Progenitor for Neocortical Projection Neurons, Astrocytes, and Oligodendrocytes. Neuron. 2013;80(5) doi: 10.1016/j.neuron.2013.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shimizu T, et al. Zinc finger genes Fezf1 and Fezf2 control neuronal differentiation by repressing Hes5 expression in the forebrain. Development. 2010;137:1875–1885. doi: 10.1242/dev.047167. [DOI] [PubMed] [Google Scholar]
- 55.Yang N, Dong Z, Guo S. Fezf2 regulates multilineage neuronal differentiation through activating basic helix-loop-helix and homeodomain genes in the zebrafish ventral forebrain. J Neurosci. 2012;32(32):10940–8. doi: 10.1523/JNEUROSCI.2216-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen L, et al. Genomic Selection Identifies Vertebrate Transcription Factor Fezf2 Binding Sites and Target Genes. J Biol Chem. 2011;286(21):18641–18649. doi: 10.1074/jbc.M111.236471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shu X-S, et al. FEZF2, a novel 3p14 tumor suppressor gene, represses oncogene EZH2 and MDM2 expression and is frequently methylated in nasopharyngeal carcinoma. Carcinogenesis. 2013;0(0):1–10. doi: 10.1093/carcin/bgt165. [DOI] [PubMed] [Google Scholar]
- 58.Orkin S, Hochedlinger K. Chromatin Connections to Pluripotency and Cellular Reprogramming. Cell. 2011;145(6):835–850. doi: 10.1016/j.cell.2011.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Koche RP, et al. Reprogramming Factor Expression Initiates Widespread Targeted Chromatin Remodeling. Cell Stem Cell. 2011;8(1):96–105. doi: 10.1016/j.stem.2010.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kidder BL, Palmer S, Knott JG. SWI/SNF-Brg1 regulates self-renewal and occupies core pluripotency-related genes in embryonic stem cells. Stem Cells. 2009;27(2):317–328. doi: 10.1634/stemcells.2008-0710. [DOI] [PubMed] [Google Scholar]
- 61.de la Serna IL, Ohkawa Y, Imbalzano AN. Chromatin remodelling in mammalian differentiation: lessons from ATP-dependent remodellers. Nat Rev Genet. 2006;7(6):461–473. doi: 10.1038/nrg1882. [DOI] [PubMed] [Google Scholar]
- 62.Ho L, et al. An embryonic stem cell chromatin remodeling complex, esBAF, is an essential component of the core pluripotency transcriptional network. Proc Natl Acad Sci U S A. 2008;106(13):5187–5191. doi: 10.1073/pnas.0812888106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lessard JA, Crabtree GR. Chromatin regulatory mechanisms iin pluripotency. Annu. Rev. Cell Dev. Biol. 2010;26:503–532. doi: 10.1146/annurev-cellbio-051809-102012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Reisman D, Glaros S, Thompson EA. The SWI/SNF complex and cancer. Oncogene. 2009;28(14):1658–68. doi: 10.1038/onc.2009.4. [DOI] [PubMed] [Google Scholar]
- 65.Kadoch C, et al. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat Genet. 2013;45(6):592–601. doi: 10.1038/ng.2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shain AH, Pollack JR. The spectrum of SWI/SNF mutations, ubiquitous in human cancers. PLoS One. 2013;8(1) doi: 10.1371/journal.pone.0055119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Moshkin YM, et al. Functional differentiation of SWI/SNF remodelers in transcription and cell cycle control. Mol Cell Biol. 2007;27(2):651–61. doi: 10.1128/MCB.01257-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Vázquez M, Moore L, Kennison J. The trithorax group gene osa encodes an ARID-domain protein that genetically interacts with the brahma chromatin-remodeling factor to regulate transcription. Development. 1999;126(4):733–42. doi: 10.1242/dev.126.4.733. [DOI] [PubMed] [Google Scholar]
- 69.Goardon N, et al. Coexistence of LMPP-like and GMP-like leukemia stem cells in acute myeloid leukemia. Cancer Cell. 2011;19(1):138–52. doi: 10.1016/j.ccr.2010.12.012. [DOI] [PubMed] [Google Scholar]
- 70.Cozzio A, et al. Similar MLL-associated leukemias arising from self-renewing stem cells and short-lived myeloid progenitors. Genes Dev. 2003;17(24):3029–3035. doi: 10.1101/gad.1143403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Huntly BJ, et al. MOZ-TIF2, but not BCR-ABL, confers properties of leukemic stem cells to committed murine hematopoietic progenitors. Cancer Cell. 2001;6:587–96. doi: 10.1016/j.ccr.2004.10.015. [DOI] [PubMed] [Google Scholar]
- 72.Krivtsov AV, et al. Transformation from committed progenitor to leukaemia stem cell initiated by MLL AF9. Nature. 2006;442(7104):818–22. doi: 10.1038/nature04980. [DOI] [PubMed] [Google Scholar]
- 73.Zhao Z, et al. p53 loss promotes acute myeloid leukemia by enabling aberrant self-renewal. Genes Dev. 2010;24(13):1389–402. doi: 10.1101/gad.1940710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wang Y-C, et al. Drosophila intermediate neural progenitors produce lineage-dependent related series of diverse neurons. Development. 2014;141:1–6. doi: 10.1242/dev.103069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Magee JA, Piskounova E, Morrison SJ. Cancer stem cells: impact, heterogeneity, and uncertainty. Cancer Cell. 2012;21(3):283–96. doi: 10.1016/j.ccr.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Visvader JE, Lindeman GJ. Cancer stem cells: current status and evolving complexities. Cell Stem Cell. 2012;10(6):717–28. doi: 10.1016/j.stem.2012.05.007. [DOI] [PubMed] [Google Scholar]