

Abstract
Endothelial cell (EC) dedifferentiation in relation to neovascularization is a poorly understood process. In this report we addressed the role of Wnt signaling in the mechanisms of neovascularization in adult tissues. Here, we show that a low-dose of 6-bromoindirubin-3′-oxime (BIO), a competitive inhibitor of Glycogen Synthase Kinase (GSK)-3β, induced the stabilization of β-catenin and its subsequent direct interaction with the transcription factor NANOG in the nucleus of ECs. This event induced loss of VE-cadherin from the adherens junctions, increased EC proliferation accompanied by asymmetric cell division (ACD), and formed cellular aggregates in a hanging drop assays indicating the acquisition of a dedifferentiated state. In a chromatin immunoprecipitation assay, nuclear NANOG protein bound to the NANOG- and VEGFR2-promoters in ECs, and the addition of BIO activated the NANOG-promoter-luciferase reporter system in a cell-based assay. Consequently, NANOG-knockdown decreased BIO-induced NOTCH-1 expression, thereby decreasing cell proliferation, ACD and neovascularization. In a Matrigel plug assay, BIO induced increased neovascularization, secondary to the presence of VEGF. Moreover, in a mouse model of hind limb ischemia, BIO augmented neovascularization that was coupled with increased expression of NOTCH-1 in ECs and increased smooth muscle α-actin (SMA)+ cell recruitment around the neovessels. Thus, these results show the ability of a low-dose of BIO to augment neovascularization secondary to VEGF, a process that was accompanied by a partial dedifferentiation of ECs via β-catenin and the NANOG signaling pathway.
Keywords: BIO, Dedifferentiation, Endothelial cells, Hind limb ischemia, Neovascularization
INTRODUCTION
In adults, most cells exit from the cell cycle to undergo differentiation; however, cardiomyocytes and ECs have long been thought to undergo terminal differentiation [1–3]. Contrary to this long held view, a current model posits that in response to tissue injury, resident cells surrounding the injured area migrate and rapidly re-enter the cell cycle to induce tissue regeneration [4–6]. Accumulating experimental evidence suggests the presence of an intrinsic mechanism of in situ tissue regeneration [7–15], however, the underlying mechanisms are not well known.
Transcription factors that regulate the reprogramming of somatic cells into iPS cells include the embryonic genes Nanog, Oct4, Sox2, and Klf4 [16]. In particular, transcription factor Nanog is known for its ability to convert somatic cells into a pluripotent stem cell state [17–20], while Wnt signaling mediates the expression of NANOG [21]. The expression of Nanog is highly enriched in vivo in sprouting ECs, including those found in the capillaries, dorsal aorta, and intersomitic vessels [21]. ECs express several Wnts, Wnt receptors and co-receptors [24–27]. Wnt signaling is known not only to regulate stem cell self-renewal [28–30], but also to upregulate expression of NANOG in ECs [21]. These observations led us to consider if the activation of canonical Wnt signaling induces dedifferentiation of mature ECs into an immature phenotype by activating Nanog. Loss of cell-cell adhesion, disappearance of VE-/E-cadherins from adherens junctions, increased proliferation, formation of cellular aggregates, asymmetric cell division (ACD), and acquisition of migratory phenotypes are considered hallmarks of cellular dedifferentiation [4,5,7,8,22–23]. These events are considered crucial for wound healing and tissue regeneration in adults [7–15].
Activation of the canonical Wnt pathway inhibits GSK-3β from phosphorylating β-catenin, resulting in the accumulation of active β-catenin polypeptide species that translocate into the nucleus to activate Wnt gene targets, including NANOG [21,28–30]. Small molecule inhibitors of GSK-3β, BIO and CHIRON99021, have been shown to be effective at nanomolar concentrations [31–33]. Interestingly, BIO promotes and enhances the reprogramming of somatic cells into induced pluripotent stem cells via the induction of Nanog and Oct4 [34–36]. Since BIO induced dedifferentiation of cardiomyocytes [34] and rescued the angiogenic phenotype in R-spondin-1-deficient zebrafish [37], we addressed the hypothesis that BIO-mediated activation of NANOG in ECs can induce partial dedifferentiation of these cells secondary to presence of VEGF, thereby augmenting neovascularization in vivo. Accordingly, we demonstrated that a low-dose of BIO has the ability to induce an interaction between β-catenin and NANOG in the nucleus of HUVECs and HSaVECs, which controls neovascularization via the upregulation of NANOG gene networks.
Materials and Methods
Additional Methods are available in the online-only Supplement
Antibodies and reagents
Anti-VEGFR2/FLK1 (C-1158), anti-VEGFR2/FLK1 (N-931), anti-human β-catenin (E-5), anti-human β-catenin (H-102), anti-human NANOG (J29), anti-human Glut-1 (C-20), anti-human GAPDH (4G5), anti-human JAM-A (1H2A9), and small interfering RNAs (siRNAs; modified 25-mer duplexes) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-NANOG polyclonal antibody was purchased from Cell Signaling (Beverly, MA). Anti-human NANOG monoclonal antibody was purchased from Novus Biologicals (Littleton, CO). Anti-mouse CD31 was purchased from BD Biosciences (San Jose, CA). Anti-NOTCH-1 monoclonal antibody was purchased from Affinity BioReagents (Golden, CO). Anti-mouse α-smooth muscle actin (αSMA; clone A2547) was purchased from Sigma Aldrich (St. Louis, MO). Anti-mouse von Willebrand Factor (vWF) was bought from Millipore (Billerica, MA). Growth Factor reduced Matrigel was purchased from R&D Systems (Minneapolis, MN). 6-bromoindirubin-3′-oxime (BIO) was purchased from Stemgent (San Diego, CA). Stock solution of BIO (10 mM) was prepared in Dimethy sulphoxide (DMSO) and stored in aliquots in −20°C freezers in a dark sealed container.
Cell Culture and siRNA transfection
Human umbilical vein endothelial cells (HUVECs), human pulmonary arterial endothelial cells (HPAECs), and Human saphenous vein endothelial cells (HSaVECs) were cultured in EndoGRO-VEGF (vascular endothelial growth factor) Complete Media Kit (Millipore, Billerica, MA). HUVECs were purchased from Millipore, HPAECs from Lonza (Walkersville, MD), and HSaVECs were purchased from PromoCell (Heidelberg, Germany). Knockdown experiments were performed as described previously [21,43].
Western blot analysis
HUVECs or HSaVECs were solubilized using 1X TNT [20mM Tris (pH 7.5), 150mM NaCl, 1% Triton X-100, 0.1% SDS, 0.25% NP-40, 1mM EDTA, and freshly added protease inhibitors] cell extraction buffer as described previously [21,43]. All Western blot analyses were carried out as described previously [21,43].
RNA extraction, RT-PCR, qRT-PCR, 5′-bromo-2′-deoxyuridine (BrdU) incorporation assay
Total RNA extraction, cDNA synthesis using SuperScript II reverse transcriptase (RT; Invitrogen), RT-PCR and q-RT-PCR methods have been described previously[21,43]. Quantitative RT-PCR was performed using Power SYBR Green RNA-to-CT TM 1-Step Kit (Applied Biosystems) as described previously [21,43]. Primers were purchased from IDT DNA Technologies (Skokie, IL): NANOG (NM_024865.2), forward primer 5′-CCTGAAGACGTGTGAAGATGAG-3′, and reverse primer 5′-CCAGTGTCCAGACTGAAATTGA-3′ (product size 59 bp); OCT4 (NM_002701.4), forward primer 5′-GGAGATATGCAAAGCAGAAACC-3′, and reverse primer 5′-CCTCTCACTCGGTTCTCGATAC-3′ (product size 74 bp); FLK1 (NM_002253.2), forward primer 5′-GCTACCAGTCCGGATATCACTC-3′, and reverse primer 5′-TCTGCTTCCTCACTGGAGTACA-3′ (product size 64 bp); VE-Cadherin (NM_001795.3), forward primer 5′-GCTGTACTGAGCACTGAACCAC-3′, and reverse primer 5′-CTGTCACTCCTGATCTCCACTG-3′ (product size 100 bp); GAPDH (NM_002046.3), forward primer 5′-TTGCCATCAATGACCCCTTCA-3′, and reverse primer 5′-CGCCCCACTTGATTTTGGA-3′ (product size 174 bp); BRACHYURY (NM_080646.1), forward primer 5′-AAGGACAAGGAAGTGAAAGCTG-3′, and reverse primer 5′-GCTCCACTTCTCTCTCTGGTGT-3′ (product size 58 bp); CD133 (NM_001145852.1), forward primer 5′-TTGGAGTGCAGCTAACATGAGT-3′, and reverse primer 5′-TGCTGGACACCAGATCTAAGAA-3′ (product size 100 bp); β-Catenin (NM_001098209.1), forward primer 5′-ACAAATGGATTTTGGGAGTGAC-3′, and reverse primer 5′-CTTGTGATCCATTCTTGTGAC-3′ (product size 58 bp); CD31 (NM_000442.4), forward primer 5′-AGCCCTAGAAGCCAATTAGTCC-3′, and reverse primer 5′-GCAATTCTTAGGGGACAGTGAC-3′ (product size 57 bp); von Willebrand Factor (vWF) (NM_000552.3), forward primer 5′-AGGAGGAGTGCAAAAGAGTGTC-3′, and reverse primer 5′-TACTCATCACAGCACTGGGTCT-3′ (product size 85 bp). BrdU incorporation was performed using 5-Bromo-2′-deoxy-uridine Labeling and Detection Kit II (Roche, Branchburg, NJ) assay, quantification and imaging were performed as previously described [21,43].
RESULTS
BIO Regulates the Stabilization of β-catenin and its Association with NANOG in the Nucleus, and Mediates NANOG-promoter Activity
We used HPAECs, HUVECs, and HSaVECs for this report. First we established 0.2 μM BIO as an optimal concentration that elicited a proliferative response in HUVECs in presence of VEGF (50ng/ml) (Fig. S1&S2). This concentration of VEGF was necessary for optimal proliferation of these cells and included for all in vitro experiments (excluding ELISA and co-IP), as BIO alone was not highly effective without VEGF. Analysis of ECs stained with anti-β-catenin and anti-NANOG antibodies showed basal NANOG expression in untreated control HUVECs (Fig. 1A). BIO-treated (6 hrs) ECs showed increased colocalization of β-catenin with NANOG in the nucleus (Fig. 1B). In contrast, immunostaining with anti-VE-cadherin and anti-NANOG showed no colocalization of these two proteins in the nucleus, although intense staining of NANOG in the nucleus was clearly evident after BIO treatment (Fig. 1C). After treatment of the ECs with 0.2 μM BIO for 36 hrs, VE-Cadherin (green) became less apparent, while the accumulation of NANOG (red) in the nucleus clearly increased (Fig. 1D–F). We also confirmed that Wnt3a induced NANOG accumulation in HUVECs (Fig. S3). Thus, the staining and microscopy analyses indicated the loss of VE-cadherin from adherens junctions in ECs that were treated with BIO (0.2μM) or with Wnt3a (Fig. S3D–F). The phosphorylation of GSK-3β at Ser9, thereby increase in non-phosphorylated β-catenin and NANOG in was confirmed by Western blot (Fig. 1G). To address whether β-catenin interacts with NANOG, HUVECs were growth factor starved overnight, thereafter nuclear extracts were subjected a co-immunoprecipitation (co-IP) assay. Reciprocal co-IP showed a minimal basal interaction of β-catenin with NANOG in control ECs (Fig. 1H), while the interaction of β-catenin with NANOG increased in response to BIO treatment (16 hrs). When nuclear extracts were subjected to immunoprecipitation with anti-NANOG antibody, the β-catenin polypeptide level was always higher. In contrast, anti-β-catenin did not co-IP equivalent level of NANOG polypeptide. However, this association was highly enriched in response to BIO treatment of ECs (Fig. 1H). To examine whether their interaction is direct, we carried-out far-Western assays (also called ligand blotting). Far-Western analysis showed that NANOG protein was bound to the 85 kDa β-catenin polypeptide species but not to Glut-1 (Fig. 1I, top panel, negative control). The identity of the β-catenin polypeptide was confirmed by reprobing the membranes with anti-β-catenin antibody (Fig. 1I, bottom panel). These data indicate that β-catenin binds to NANOG directly, which may be responsible for the BIO-mediated (0.2 μM) increased number of ECs observed in Fig. S2.
Figure 1. BIO mediates interaction of β-catenin with NANOG, and NANOG binds to the NANOG and BRACHYURY promoters.

Sparsely plated HUVECs treated without or with BIO were subjected to staining and microscopy. (A) Anti-VE-cadherin (green), (B) Anti-human β-catenin (green) and anti-human NANOG (red); (C) Anti-human VE-cadherin (green) and anti-human NANOG (red) staining. (D) Reduced VE-cadherin (green) staining in ECs treated with BIO. (E) Nuclear accumulation of NANOG (red). (F) NANOG, red; DAPI, blue (merge). Scale bar is 100 μm. (G) EC extracts were analyzed by immunoblotting with the indicated antibodies. Note: increased β-catenin and NANOG in nuclear extracts. GAPDH represents equal loading. (H) Reciprocal co-IP of NANOG with β-catenin. (I) Far-Western showing the interaction is direct: EC extracts were immunoprecipitated with indicated antibodies, membrane incubated (ligand blotting) with human recombinant NANOG protein (2.0 μg/ml), then analyzed by immunoblotting with anti-human NANOG antibody (top). This blot was stripped then reprobed with anti-β-catenin antibody (bottom). Results are representative of 3 independent experiments. (J) Chromatins IP prepared from HPAECs and HUVECs were analyzed for the presence of indicated promoters. Compared with control cells, anti-NANOG showed enrichment of NANOG, OCT4, BRACHYURY, CD133, and VEGFR-2 promoters after BIO stimulation. In contrast, there was no amplification in anti-Glut-1 ChIP (negative control). (K) Schematic of the NANOG-promoter/enhancer region showing putative NANOG (asterisks, direct strand; diamond, reverse strand) binding consensus sites. The position is relative to the TSS. (L) ECs transiently transfected with pGL4.84 (control) or with pGL4.84-(−2.1-NANOG) promoter were treated with BIO (0.2 μM) for 6 hrs. NANOG-promoter Renilla luciferase activity after BIO treatment is presented as the fold induction of RLU (relative luciferase unit) versus control. Results represent the mean of 3 independent experiments ± S.E.M. *, P < 0.05.
Analyses of the human NANOG-promoter/enhancer from −2298 to +1 relative to the transcription start site (TSS) identified 18 NANOG binding sites (ATTA), while the BRACHYURY-promoter showed 15 sites (ATTA) within the −3.8 kb upstream of the TSS (Fig. S4&S5). Supplemental Table 1 shows the primers used to detect NANOG binding sequences flanking the putative NANOG-binding sites on the NANOG-, OCT4-, BRACHYURY-, CD133- and VEGFR-2-promoters and to amplify expected PCR products prepared from the anti-Glut-1 (control) and anti-NANOG antibody chromatin IPs. To test the hypothesis that NANOG binds to the NANOG-promoter and that NANOG can also bind to the OCT4-, BRACHYURY-, CD133- and VEGFR-2-promoters in HPAECs and HUVECs in response to BIO stimulation, we subjected these cells to ChIP experiments. Accordingly, NANOG, OCT4, BRACHYURY, CD133 and VEGFR-2 promoters were enriched only in the cells receiving BIO and not the control ECs (Fig. 1J). These data establish that NANOG not only auto-regulates itself but can also bind the endogenous OCT4-, BRACHYURY-, CD133- and VEGFR-2-promoters in two different types of ECs in response to BIO treatment.
A −2.1 kb-NANOG promoter/enhancer genomic fragment was subcloned into pGL4.84 in a promoterless vector to drive the luciferase gene, generating pGL4.84-(−2.1 kb-NANOG) (Fig. 1K). To test if stimulation of these cells with BIO leads NANOG to bind to and activate the NANOG promoter and to drive expression of the luciferase reporter gene, ECs were transiently transfected with the pGL4.84-(−2.1kb-NANOG) construct along with β-galactosidase as a tracer. Using the NANOG-promoter/enhancer luciferase reporter gene that signifies the activation state of NANOG-sensitive transcription, we treated transfected ECs with the vehicle alone (control) or with BIO (0.2 μM) and assayed for optimal luciferase activity 6 hrs post-treatment (Fig. 1L). There was a generalized increase in luciferase activity (5-fold) after 6 hrs even without BIO treatment in ECs transfected the NANOG-sensitive pGL4.84-(−2.1 kb-NANOG) construct; however, BIO treatment induced a ≥17-fold increase in luciferase activity. The ability of BIO to increase the accumulation of β-catenin in the nucleus, where it binds NANOG, as well as to activate the NANOG reporter gene is, therefore, a key mechanism related to the function of the β-catenin-NANOG pathway in BIO’s action.
BIO Promotes Aggregation of ECs in a Hanging Drop Assay and Concomitantly Induces Expression of Pluripotency-Associated Genes in presence of VEGF
BIO induced cellular aggregates in a hanging drop assay (Fig. S6). The formation of cellular aggregates is henceforth considered a dedifferentiated phenotype (an immature cell state). To address the proposed mechanisms underlying the phenotypic switch, mRNAs and cell extracts were prepared for q-RT-PCR and Western blot analyses. As shown in Fig. 2A, we observed increased levels of transcripts for β-catenin, NANOG, BRACHYURY, OCT4, CD133, and FLK1 in ECs treated with BIO. In contrast, the levels of mature EC markers CD31 and vWF transcripts decreased. Immunostaining followed by microscopic analyses revealed increased β-catenin and NANOG but decreased VE-cadherin and vWF in BIO-treated ECs (Fig. 2B–I). Immunoblotting of cell extracts showed increased non-phosphorylated β-catenin (nuclear), NANOG (nuclear), NOTCH-1 (280 kDa), the NOTCH intracellular cleaved domain (NICD), DLL4, and NUMB, while VE-cadherin decreased and GAPDH remained unchanged (Fig. 2J). These data demonstrate the potential of BIO to induce expression of stemness genes and proteins in ECs.
Figure 2. Acquisition of a dedifferentiated phenotype by a subset of ECs.

(A) Q-RT-PCR showing an increased expression of NANOG, β-catenin, OCT4, BRACHYURY, CD133, and FLK1 after BIO treatment, while the level of vWF and CD31 decreased. The baseline value was calculated as 1 fold. Experiments were repeated at least 5 times. Results represent the mean of 3 independent experiments ± S.E.M. *, P < 0.01. (B–I) Control or BIO (0.2 μM for 6 hrs) treated HUVECs were fixed and stained with the indicated antibodies. (B) β-catenin (green) is mostly distributed in the plasma-membrane, while NANOG (red) is undetectable. (C) Increased accumulation of β-catenin and NANOG in the nucleus in response to BIO stimulation, concomitantly inducing formation of cellular aggregrates. (D) Anti-VE-cadherin (green) staining reveals zipper-like adherens junctions, while (E) BIO induces phenotypic alterations. (F) Anti-vWF staining reveals normal EC characteristics, while (G) BIO down-regulates expression of vWF. Scale bar is 100 μm. Original magnification, 20X. (H) Control HUVECs stained with anti-human NOTCH-1. (I) BIO-treated HUVECs stained with anti-NOTCH-1. (J) Cell extracts prepared from control or BIO treated HUVECs were subjected to Western blotting with indicated antibodies. The numerical values presented below each western blot panels indicate signal intensities in arbitrary units, control signal value was considered 1. Experiments were repeated at least 3 times with replicates.
BIO Augments EC proliferation and Asymmetric Cell Division (ACD) in the presence of VEGF
To evaluate BIO-induced proliferative activities, we monitored BrdU uptake as a measure of HUVEC entry into the S-phase of the cell cycle. The timeline of the BrdU experiment is as shown (Fig. 3A). Without VEGF and BIO the basal proliferation of ECs were close to 7%, while addition of BIO (0.2 μM) alone, with no VEGF increased proliferation of these cells to >35% (Fig. 3B–D). The proliferation of ECs in presence of VEGF (50ng/ml), without BIO was 60% (Fig. 3B, E, G). However, the proportion of BrdU-positive cells was significantly higher (>80%) in ECs receiving BIO (0.2 μM) + VEGF (50ng/ml) in the culture compared to untreated control ECs (Fig. 3B, F, H). The BrdU assays showed morphologically distinguishable symmetric cell division (SCD) in control cells, whereas cells receiving BIO showed both SCD and ACD subpopulations (Fig. 3G and H). Next, FACS assay of HUVECs receiving either VEGF (50ng/ml) or BIO (0.2 μM), or together were monitored by annexin-v and propidium iodide (PI) staining. Interestingly, both VEGF and BIO either alone or in combination reduced basal apoptosis and necrosis of HUVECs, thereby favoring cell survival (Fig. S7A&B). Accordingly, Western Blot analysis indicated that this increase in proliferative activity was accompanied by increased Cyclin-D1 expression and decreased levels of p21 and p53 proteins (Fig. 3I).
Figure 3. BIO increases proliferation of HUVECs.
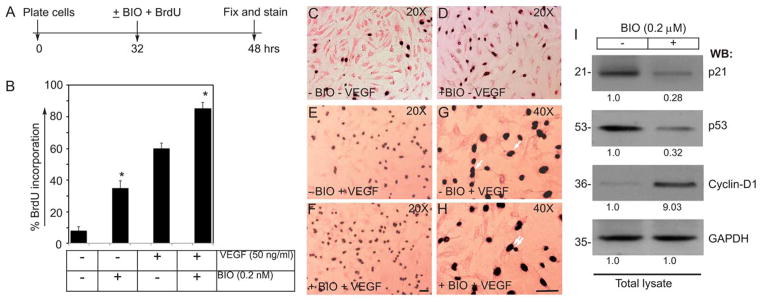
(A) Timeline of BrdU incorporation assay. (B) BIO (0.2 μM) stimulation promotes cell cycle progression in primary HUVECs. A greater percentage of HUVECs that were stimulated with BIO showed higher incorporation of BrdU. (C–H) Representative images of BrdU incorporation of control and BIO-treated HUVECs, in presence or in absence of VEGF (50ng/ml). Single and double white arrows indicate SCD and ACD, respectively. Scale bar, 200 μm (I) Representative images of the Western blot analyses of the total cellular proteins prepared from control or BIO treated HUVECs. The numerical values presented below each western blot panels indicate signal intensities in arbitrary units, control signal value was considered 1. Experiments were repeated > 3 times. Results represent the mean of 3 independent experiments ± S.E.M. *, P < 0.01 vs. control.
To examine BIO-induced ACD and the contribution of NANOG to this process, we performed BrdU experiments using 0.2 μM BIO to stimulate HUVECs in culture in the presence of VEGF. ACD was quantified as a percentage of the total number of dividing cells. Control HUVECs showed a limited pool of asymmetrically dividing cells (7%) (Fig. 4A). As expected, the addition of BIO increased the ACD population from 7.5 to 27.8% (Fig. 4A and Fig. S7). To test the importance of NANOG in BIO-induced ACD in ECs, we knocked down NANOG in BIO-treated HUVECs. Supporting the importance of NANOG in ACD in ECs, we found that NANOG knockdown significantly decreased the ACD/BrdU+ EC subpopulation close to basal levels, from 27.8% ± 1.18% for the BIO-treated cells to 10.5% ± 1.22% for the BIO-treated NANOG-knockdown cells (Fig. 4A). Efficiency of NANOG-knockdown cells was confirmed by Western blot (Fig. 4B). These data indicate that BIO can induce both SCD and ACD in ECs and that NANOG knockdown decreases the ACD subpopulation significantly. Representative images of SCD and ACD are shown in Fig. 4C–L. CD133 and NOTCH-1 staining of control ECs revealed very little or no signal (Fig. 4G and I), yet staining with DAPI (a nuclear marker) showed morphologically distinguishable symmetric nuclei (white arrows, Fig. 4I). However, in a subpopulation of ECs receiving BIO, the intensity of anti-CD133 (Fig. 4H) and anti-NOTCH-1 (Fig. 4J) staining was stronger, and the nuclear content was morphologically unequal (Fig. 4E, F, H, J, white arrowheads). As NOTCH-1 is known to regulate cell fate determination by binding to DLL4 on the cell surface, we used DLL4 to examine ACD and SCD. Thus, NOTCH-1 and DLL4 staining provided evidence that NOTCH-1 protein was distributed unevenly in the ACD subpopulation (Fig. 4J, M and N). In addition, we observed increased coexpression of NUMB with low-level NOTCH-1 in smaller daughter cells (Fig. 4N). These data show that BIO induces ACD in a subset of ECs by upregulating NANOG.
Figure 4. BIO mediated NANOG expression plays a role in ACD in ECs.

(A) Quantification of the percent of ACD of the total dividing cell population using the BrdU assay. Results represent the mean of 3 independent experiments ± S.E.M. *, P < 0.05 vs. control. (B) Western blot analyses for the total proteins prepared from control or BIO treated HUVECs with the indicated antibodies. (C–F) Representative images of BrdU incorporation in vehicle control and BIO-treated HUVECs. (G–I) Representative images of the immunofluorescent staining of control HUVECs with anti-CD133 (red) and anti-NOTCH-1 (green). (H–J) Representative images of BIO treated HUVECs stained with anti-CD133 (red) and anti-NOTCH-1 (green). Representative images of control or BIO treated HUVECs stained with: (K) NOTCH-1 (green) and DLL4 (red); (L) NOTCH-1 (red) and NUMB (green); (M) NOTCH-1 (green) and DLL4 (red); (N) and NOTCH-1 (red) and NUMB (green); DAPI, nucleus (blue). The arrows and arrowheads indicate morphologically distinguishable SCD and ACD, respectively. Experiments were repeated 3 (n=3) times. Scale bar, 150 μm.
BIO induces the Secretion of Angiogenic Factors and Angiogenic Activities of ECs in vitro
To monitor whether BIO can induce the migration of HUVECs, we performed Boyden chamber and wound-healing scratch assays. Timeline of the migration assay is shown (Fig. 5A). Fig. 5B shows the migration of the ECs to the lower surface of Boyden chamber transfilters in response to increasing concentrations of BIO (0, 0.05, 0.2, and 0.5 μM). In the absence of BIO (control), ECs showed basal migration. However, ECs responded significantly to BIO treatment, and this increased response was concentration-dependent (0.2 and 0.5 μM), with the peak response greater than 16-fold over the control ECs. Thus, the optimal concentration of BIO that is needed to induce migration over 6 hrs was 0.2 μM. Representative images of the Boyden chamber filter assay are shown in Fig. 5C–F. Next, to test whether BIO can induce wound closure, we performed a scratch assay on a confluent EC monolayer. Over a period of 6 hrs, there was ~15% wound closure in the absence of BIO (control) (Fig. 5G–I). In contrast, the EC monolayer receiving BIO showed significant cell movement toward the open scratched area, which induced 60% wound closure (Fig. 5J–L). To test whether ECs can secrete angiogenic factors, growth factor- and serum-starved ECs were stimulated with BIO (in serum-free media) for 6 hrs. Media collected at 0, 3, and 6 hrs was then subjected to ELISA. Fig. 5N shows the ability of BIO to induce the secretion of Ang-2, bFGF (low level), IL-8 (low level), and TIMP-1 over a period of 3 and 6 hrs. VEGF was not detectable (data not shown). Together, these data show that BIO not only induces cell migration and wound healing but also induces the expression of angiogenic factors.
Figure 5. BIO induces migration of HUVECs and the secretion of angiogenic factors and augments branching point structures in Matrigel.

(A) Timeline of cell migration assay. (B) Quantification of the cell migration through chemotactic Boyden chamber. (C–F) Representative images from the Boyden chamber filters with increasing amounts of BIO. (G–I) Representative images of the control HUVECs with wound introduction at the indicated time points. (J–L) Representative images of the BIO treated HUVECs with wound closure at the indicated time points. Scale bar, 300 μm. (M) Quantification of %wound closure. (N) HUVECs were growth factor and serum starved for 2.5 hrs, washed with 1XPBS, pH 7.4 then stimulated with DMEM (no serum or growth factor) containing BIO (0.2 μM) for indicated period of time. Cell culture supernatants were then subjected to ELISA assay for the indicated angiogenic factors. Experiments were repeated 3 times (n=3) with triplicates. (O) Timeline of Matrigel experiment. (P) HUVECs (2 × 105) were plated onto 12 well dishes coated with growth factor reduced Matrigel supplemented with bFGF (20 ng/ml), VEGF (50 ng/ml), and BIO (0.1 and 0.2 μM). ECs interconnect to form a vascular plexus (branching) like structures, were counted after 18 hrs. Results represent the mean of 3 independent experiments ± S.E.M. *, P < 0.05 vs. control. (Q–S) Representative images of branching points. Black arrows indicate branching points. Experiments were repeated at least 3 times with triplicates.
To examine the effect of BIO on this angiogenic response in vitro, we used growth-factor-reduced Matrigel supplemented with bFGF (20 ng/ml) and VEGF165 (50 ng/ml). The timeline of an in vitro branching point structure assay is shown in Fig. 5O. As control HUVECs plated on Matrigel elongate, interconnect and assemble into vascular-plexus-like structures, we observed secondary sprouting with fine branching points. After 18 hrs, we counted the branching point structures. Thus, stimulation of ECs with 0.1 and 0.2 μM BIO increased branching points in Matrigel in vitro (Fig. 5P). Representative images of branching points are shown in Fig. 5Q–S. These data show that BIO increases angiogenic activities of ECs in Matrigel in vitro.
BIO Augments Neovascularization in Matrigel plugs and in a Mouse Model of Hind Limb Ischemia (HLI)
Next, we evaluated the capacity of BIO to induce neovascularization in Matrigel plugs in two groups of nude mice. In the first group, mice received Matrigel+BIO (0.2 μM), while in the second group, mice received Matrigel containing control HUVECs or HUVECs that were pre-treated with BIO (0.2 μM). Fig. 6A shows the timeline of the Matrigel plug assay. Fig. 6B–E shows the representative Matrigel plugs retrieved at day 7 from the nude mice. Fig. 6F shows the quantification of H&E stained venous structures. The presence of red blood cells in the H&E stained sections indicated the formation of neovessels in the Matrigel plugs, as shown in Figure 6G–J. Fig. 6F&H show a significant increase in neovascularization after 7 days in the nude mice receiving the Matrigel + BIO (0.2 μM), whereas the control Matrigel alone elicited minimal venous formation (Fig. 6F&G). In the next set of experiments, HUVECs were left untreated or treated with BIO prior to mixing with Matrigel (Fig. 6I–J&N–S). To differentiate HUVECs (human cells) from mouse ECs, sections were stained with anti-human vWF (red) and anti-mouse CD31 (green) antibodies. Quantification of anti-SMA+ vascular structures indicated increased neovessel formation in BIO treated group compared to control (Fig. 6K). As shown in Fig. 6L–O, the representative images of vWF− and CD31-stained Matrigel plugs consistently revealed neovessels in BIO treated groups, and the presence of highly autofluorescent red blood cells indicated functional veins (Fig. 6O–Q) green autofluorescent erythrocytes and leukocytes). Similarly, the quantification of anti-vWF and anti-NOTCH-1 staining revealed increased neovessels in the BIO-treated group, compared with the control (Fig. 6R&S). The presence of green autofluorescent erythrocytes and leukocytes demonstrated functional neovessels. We also used anti-Ephrin-B2 and Hey-2 antibodies to label ECs, however, it remained unclear if venous ECs acquired arterial phenotype or not (data not shown).
Figure 6. BIO augments neovascularization of Matrigel implants.


(A) Timeline of the Matrigel plug assay. (B–E) Representative images of the Matrigel implants removed from nude mice at day 7, (B and C) are out of focus. (F) Quantification of the vascular structures per 10X field in the H&E stained sections. (G–J) Representative images of the H&E stained section of the Matrigel plugs. Scale bar, 150 μm. (K) Quantification of SMA+ vascular structures per 40X field. Representative images of Matrigel plug sections control or BIO treated HUVECs stained with, (L–O) Anti-mouse CD31 (green) and anti-human vWF (red). Scale bar, 200 μm; (P&Q) Magnified images of O showing vWF positivity of the vascular structure; (R&S) Quantification of the vWF+ and NOTCH-1+ vascular structures per 40X field in the Matrigel loaded with control ECs (-BIO) or ECs pre-treated with BIO (0.2 μM). Panels below are representative images of Matrigel sections with indicated ECs, receiving no BIO (-BIO) or with BIO (0.2 μM) were stained with indicated antibodies. Autofluorescent erythrocytes and leukocytes (green).
Next, to address the efficacy of BIO in neovascularization, we used a mouse model of HLI. The experimental strategy and timeline are shown in Fig. 7A. Mice (n=3 in each group) were subjected to unilateral HLI. All mice subjected to surgery appeared healthy during and after the operative period. There was no sign of toxicity or death associated with BIO treatment among the groups. H&E and anti-vWF staining experiments followed by microscopy of control and BIO-treated lower limb muscle sections showed that the normal vascular and overall tissue architecture was preserved, indicating no obvious vascular pathology such as edema or hemorrhage (Fig. S9). Importantly, BIO induced Nanog expression at levels greater than 1000-fold, whereas there was no change in the expression of Gapdh in the lower limb muscles (Fig. 7B). At day 28, lower limb tibialis anterior (TA) muscles were collected, fixed, and subjected to immunohistochemistry using anti-mouse vWF anti-mouse α-SMA antibodies. There were more vWF-positive vascular structures in ischemic muscles from all mice after 28 days relative to controls (Fig. 7C–E). Accordingly, the quantification of α-SMA+ neovessels showed increased recruitment of α-SMA+ cells after BIO treatment (Fig. 7F–H).
Figure 7. BIO augments neovessel formation in a mouse model of HLI.

(A) Timeline of the HLI model and regimen of BIO treatment. (B) Q-RT-PCR analysis of Nanog and Gapdh expression in mice receiving PBS (control group) and BIO. (C) Quantification of vWF+ vascular structures in ischemic tibialis anterior (TA) muscles per 20X field. (D&E) Representative images of PBS and BIO-treated ischemic TA muscles stained with anti-vWF (red) and DAPI (blue). (F) Quantification of α-SMA+ vascular structures in ischemic TA muscles per 20X field. (G&H) Representative images of PBS and BIO-treated ischemic TA muscles stained with α-SMA (green) and DAPI (blue). Results represent the mean of 3 independent experiments ± S.E.M. *, P < 0.05. Scale bar, 300 μm.
DISCUSSION
Here, we showed that the stimulation of ECs with BIO in the presence of VEGF (i) induced an interaction of β-catenin with NANOG; (ii) increased the expression of transcriptional networks centering around the NANOG gene in ECs which induced partial dedifferentiation of these cells; (iii) augmented neovascularization in Matrigel plugs and (iv) in ischemic hind limb TA muscles.
We observed increased proliferation at 0.2 to 0.5 μM concentrations, but concentrations of 1 μM and above reduced the proliferation of HUVECs. This is the first report of the ability of BIO to induce proliferation of ECs at 0.2 μM and 0.5 μM concentrations. In Zebrafish, BIO (0.5 mM) rescued Rspo1 angiogenesis defects with no toxic effects [37]. By contrast, persistent death of tumor cells in culture and orthotopic tumor implant assays required doses of BIO above 100 μM [38]. The observation that BIO increased proliferation, migration, and neovascularization clearly indicates the ability of low dose BIO to induce positive effects in these processes. Thus, sensitivity to BIO is likely to differ among cells of different origins. The additive effect of BIO was secondary to the presence of VEGF in all of these processes. Since VEGF is a known EC-specific mitogen and a survival factor, we did not address the role of VEGF in this report.
Our staining and microscopy analyses indicated the ability of BIO to disrupt the cobblestone morphology of endothelial cells. If fact, BIO treatment induced the loss of VE-cadherin from adherens junctions in a consistent manner while increasing the accumulation of β-catenin in the nucleus. The loss of VE-cadherin from adherens junctions induced loss of cell-cell adhesion, and this phenotype is a hallmark of cellular dedifferentiation [4,5,22,23]. Increased colocalization of β-catenin and NANOG in the nucleus was evident in ECs stimulated with BIO. Although we were able to demonstrate an increased binding of NANOG to β-catenin through co-IP experiments, anti-NANOG consistently co-precipitated a higher amount of β-catenin polypeptide. However, anti-β-catenin co-IPs did not pull down the NANOG polypeptide at an equivalent level. These data suggest that nuclear β-catenin may not only bind to NANOG but may also bind to another molecule(s). For example, nuclear β-catenin is known to displace TCF3 [39]. We surmise that local concentrations of β-catenin and NANOG polypeptides in the nuclei of ECs are not equivalent. If these assumptions are correct, then it could explain why we always observed higher levels of β-catenin in anti-NANOG IPs. Alternatively, the anti-β-catenin antibody epitope is close to the NANOG-β-catenin interaction site. For this reason, we performed a far Western experiment to assess if their interaction is direct or indirect. In this experiment, purified NANOG protein bound to the β-catenin polypeptide in the nitrocellulose filter directly. The binding of β-catenin to NANOG has also been observed in the human glioblastoma U87 cell line in response to BIO (data not shown). Thus, these data suggest that NANOG plays a permissive role in ECs.
To address the mechanism of NANOG activation and the relationship between NANOG and the NANOG and BRACHYURY promoters, we selected promoter elements containing putative NANOG binding sites [21]. Accordingly, the results from the ChIP assay showed that NANOG binds to the NANOG and BRACHYURY promoters. The luciferase reporter assay also confirmed the critical role of NANOG in stimulating the NANOG promoter and the requirement of BIO in this response. The NANOG promoter is also reactive to cell surface receptor agonist stimulation by proteins such as BMP4 and TGF-β [17,18]; therefore, it is likely that the NANOG promoter is not only under the singular transcriptional control of NANOG but also of KLF4, OCT4, and SOX2. These experiments established that BIO mediates the stabilization of β-catenin, which then translocates into the nucleus to bind NANOG. Importantly, BIO induced the aggregation of ECs in a hanging drop analysis, i.e., induced EnMT or dedifferentiation. In contrast, there was no such cellular aggregation in untreated control ECs. Because these cellular aggregates that were grown suspended upside down resembled EnMT, we subjected these cells to a q-RT-PCR assay for Wnt/β-catenin target genes including NANOG. Thus, we observed an increased expression of a transcriptional network centered on NANOG, including NANOG itself, OCT4, BRACHYURY, CD133, and FLK1, but decreased expression of vWF and CD31. Bona fide markers of immature EC lineage BRACHYURY, CD133, and FLK1 were upregulated, while the downregulation of VE-cadherin, CD31 and vWF proteins strongly support the hypothesis that BIO mediates a phenotypic switch from a mature EC toward an immature EC state, and that this phenotypic switch is accompanied by the increased expression of the NANOG transcriptional gene network.
Rapid re-entry into the cell cycle is considered a characteristic of dedifferentiation and neovascularization [4–9,38,40–42]. BrdU assays indicated a significant increase in cell proliferation after the addition of low-dose BIO to ECs (0.2 and 0.5 μM; P<0.05). Because ECs were asynchronously growing in presence of serum and VEGF, some of the cells may have either escaped from S-phase or did not enter S-phase. However, it is clear that BIO not only induced BrdU uptake but also induced a decrease in p21 and p53, while it increased Cyclin-D1 protein levels. Upon closer examination of BrdU-labeled cells, it became apparently clear that there was a subpopulation of BrdU+ cells whose nuclear and cytoplasmic contents were distributed unequally between the two daughter cells. This morphologically distinguishable unequal distribution of cellular and nuclear contents resembled ACD. These observations, together with the ability of BIO to induce the formation of cellular aggregates and the expression of a NANOG transcriptional network, suggest that during neovascularization ECs likely undergo a phase of dedifferentiation. Recent studies from our laboratory and those of others have raised the possibility that mature ECs may have the ability to dedifferentiate in response to Wnt3a stimulation, giving rise to highly proliferative cells [31,43–46].
ACD, a conserved biological process in which the parent cell divides unequally and gives rise to two daughter cells with different fates, is a process that generates cellular diversity and stratification and is also a hallmark of stem cell self-renewal [22,23]. One daughter cell undergoes differentiation (e.g., high Notch-1 content, low Numb), while the other daughter cell (e.g., low Notch-1, high Numb) remains connected to a stem cell niche and continues the self-renewal process [22,23]. As ACD can be tracked using the cell markers NOTCH-1, NUMB, and CD133, we used these markers to examine BIO induction of ACD in ECs. It was not surprising to find that the distributions of CD133, NOTCH-1 protein, and DLL4 were unequal among BIO-treated ACD cells. Because NOTCH-1 and DLL4 are highly enriched in the stalk and the tip cells, respectively, during neovascularization, the ability of BIO to induce expression of these two critical molecules further strengthens our overall hypothesis. Increased expression of NUMB in ECs (the daughter cell with a smaller nucleus) also suggests that BIO induces the expression of an endogenous inhibitor of NOTCH-1 signaling in the daughter cells. As it is currently understood, the cell with the smaller nucleus (high NUMB, but low NOTCH) represents a progenitor compartment, while the larger nucleus with higher levels of NOTCH and low NUMB signals a differentiated phenotype. Since NOTCH is known to activate rapid reentry into the cell cycle and progression in quiescent cardiomyocytes [44–46], our data suggest that ECs undergo partial dedifferentiation after BIO stimulation. However, detection, identification and characterization of ACD in vivo have proven to be a very difficult task. Nonetheless, we show that NANOG knockdown of BIO-treated ECs decreased ACD in vitro, indicating the critical role of NANOG in the acquisition of the ACD phenotype.
Based on the above results, we also postulate that BIO not only induces increased cell proliferation but might also induce a migratory phenotype of ECs. Accordingly, our data demonstrates the ability of BIO to both induce chemotactic migration across the Boyden chamber and to close a wound in a scratch assay, which raised the clear potential of BIO’s ability to augment neovascularization. Additionally, ELISA provided evidence that BIO induced secretion of the potent angiogenic factors Ang-2, bFGF (low level), IL-8 (low level), and TIMP-1, although VEGF was not detectable. Ang-2, by binding to its cell surface receptor Tie-2, signals not only for neovessel maturation and stability but also exerts anti-leak and survival activities. Basic FGF is a potent angiogenic factor. These findings suggest that the observed chemotactic and migratory activities of ECs following stimulation with BIO are likely due to the combined actions of these secreted angiogenic factors. Thus, these findings provided us with the impetus to carry out subsequent in vitro and in vivo angiogenesis assays.
Quantification of the functional endpoints strongly indicated that BIO can induce neovascularization in vitro and in vivo at 0.1 and 0.2 μM doses, and these processes involve the contribution of NANOG transcriptional networks, as NANOG knockdown abolished Wnt3a-mediated angiogenesis [21]. We used several markers to identify and quantify neovessels including anti-SMA. Matrigel plugs loaded with BIO showed increased vessel density, compared with control. This trend was similar in ECs that were pretreated with BIO. In Matrigel plugs, we attempted to distinguish venous, arterial and lymphatic ECs using several different markers including Notch-1, Ephrin-B2, Hey-2, anti-alpha-SMA, Prox-1 and LYVE antibodies. In plugs containing HUVECs or HSaVECs or no cells, the host ECs recruited into the Matrigel plugs were heterogeneous including venous, arterial and lymphatic markers. Thus, the origin of ECs recruited into the Matrigel plugs are likely heterogeneous. Although BIO decreased vWF expression in vitro (Fig 2F, G), the vWF+ vascular structures increased in vivo. As vWF is best known for its important role in hemostasis, it has been used as a marker for vessel injury or inflammation. Pro-inflammatory agent such as TNFα is known to induce the expression of vWF. Thus, we speculate that the increase in vWF in vivo may have be due to increased inflammatory action of factor(s) such as TNFα. In a HLI model, mice undergoing HLI received one dose of BIO injection (intramuscular) directly into the lower limbs; thereafter, BIO was given in drinking water at 0.2, 0.4, or 0.6 μM concentration for 28 days. At the end of 28 days, there was no apparent sign of toxicity in these mice. Interestingly, BIO induced Nanog expression in the TA muscles and also in the liver. Although the effect of BIO is likely to be pleiotropic in vivo, we can at least infer that BIO is able to induce Nanog expression in vivo and to exert no visible toxicity. In the HLI model, it remains unclear if the effect of BIO in neovascularization were solely mediated by Nanog. Because both neovascularization and arteriogenesis are crucial for adaptation and regeneration following pathological events such as tissue ischemia or injury [47,48], we used FAL to induce HLI and to model aspects of human occlusive artery disease to investigate vascular regeneration and to test the efficacy of BIO. In vivo, when BIO is given in drinking water it could act on several different cell types including venous, arterial and lymphatic vessels. However, without injury or ischemia BIO did not induce appreciable dedifferentiation of normal quiescent cells or tissues.
Summary
In summary, we have demonstrated that BIO-induced neovascularization is associated with the dedifferentiation of venous to arterial ECs and that this process is NANOG specific. Although we propose a simplistic view of dedifferentiation (Fig. S10), unbridled conversion of venous to arterial ECs (or more immature cells) could trigger pathological conditions such as tumor formation or cardiovascular events. However, further studies will be required to address these possibilities.
Supplementary Material
Figure S1. Endothelial characteristics of HUVECs. Endothelial cell characteristics of HUVECs were determined by staining with, (A) anti-human VE-cadherin (green) and (B) anti-human vWF (red) antibodies. Blue, DAPI. Original magnification is as shown. Scale bar, 100 μm.
Figure S2. Dose-dependent effect of BIO in cultured HUVECs. (A) Timeline of plating the cells, BIO treatment, fixing, staining and quantification. (B) HUVECs (2 × 105) were plated in complete media for the indicated time and were treated with an increasing concentration of BIO (0, 0.2, 0.5, 1.0, 1.5, and 2.0 μM). The number of ECs was counted after 24 hrs of stimulation. Quantification of the number of cells per 20X field, mean ± S.E.M. (C–H) Representative images of H&E stained HUVECs receiving vehicle alone or the indicated concentrations of BIO. The experiment was repeated 3 times with triplicates. Asterisks indicate P<0.05 versus the control vehicle group.
Figure S3. Wnt3a induced NANOG expression in HUVECs. Control HUVECs and HUVECs treated with Wnt3a (20ng/ml) for 4 hours on coverslips and fixed with 4% PFA, then subjected to immunostaining with, (A&D) anti-human VE-cadherin (green), (B&E) anti-NANOG (red) antibodies, and (C&F) Merge. Original magnification is as shown. Scale bar, 100 μm. (G) Quantification of image pixel intensity.
Figure S4. Nucleotide sequence of human NANOG promoter/enhancer (−2.2 kb). Analysis of human NANOG promoter/enhancer from −2240 to +1 relative to the ATG (red) TSS identified 19 NANOG binding sites. NANOG binding nucleotide sequence (ATTA) in direct (+) strand are shown in bold characters, while in reverse (−) strand in bold underlined (TAAT).
Figure S5. Nucleotide sequence of human BRACHYURY promoter/enhancer (−3.8 kb). The BRACHYURY promoter showed 15 sites (ATTA) within the −3.8 kb upstream of the ATG (red) TSS. Putative NANOG binding nucleotide sequence (ATTA) in direct (+) strand are shown in bold characters, while in reverse (−) strand in bold underlined (TAAT).
Figure S6. BIO induces the formation cellular aggregates in a hanging drop assay. (A) Timeline of the hanging drop assay. HUVECs at passage 3–4 were plated in EndoGRO-VEGF in Complete Media Kit + BIO (0.2 μM) for 24 hours in 10 cm dishes. Dishes were inverted and filled with 6 ml sterile 1X PBS, pH 7.4. After 7 or 14 days the dishes were turned up-right, and 6 ml EndoGRO-VEGF Complete Media was placed on cells for a few minutes. Media was aspirated, fixed with 4% PFA and ECs/hanging drops were subjected to H&E staining. (B) Quantification of the number of colonies per 10X field, mean ± S.E.M. Asterisks indicate P<0.05. (C–F) Representative images of hanging drop colonies. Scale bar, 200 μm. (G&H) Unstained live cellular aggregates (colonies) of HUVECs receiving BIO at 1 and 2 weeks. Experiments were repeated at least 3 times.
Figure S7. BIO does not induce apoptosis or necrosis of HUVECs. HUVECs were cultured overnight without VEGF or bFGF or BIO. Next day, (A) HUVECs were either left untreated (control) or treated with VEGF (50ng/ml) in absence or presence of BIO (0.2 μM) for 6 hours. The contribution of apoptosis and necrosis were measured by FACS analyses of Annexin-V and propidium iodide (PI) staining. (B) Quantification of HUVECs death. Experiments were repeated 3 times. *P < 0.05 vs. control.
Figure S8. Representative images of SCD and ACD in HUVECs. (A&B) Images of BrdU incorporation in control and BIO-treated HUVECs. The arrows and arrowheads indicate SCD and ACD, respectively. Scale bar, 200 μm.
Figure S9. Representative images of adductor muscle tissue architecture. Top panel, images of (A&C) H&E stained or (B&D) vWF (red) stained adductor muscle tissue sections prepared from control (unligated) and ligated lower limbs receiving PBS or BIO. Boxed areas from the top panels are magnified in the bottom panels. No obvious changes in overall tissue architecture or necrosis in these mice were detected. Pre-existing vessels showed no pathology as lumens (arrows) were filled with erythrocytes and leukocytes, with no visible hemorrhage or edematous events. (E) Quantification of vWF+ vascular structures. Experiments were carried out at least 3 times. Data represent mean ± S.E.M. Asterisks indicate P<0.05.
Figure S10. Proposed model of dedifferentiation of venous to arterial ECs. In adult, venous ECs are dormant and do not express detectable level of NANOG or NOTCH-1. Following ischemia or injury, BIO or Wnt3a stabilize β-catenin. This event induces β-catenin/NANOG signaling resulting in increased expression of NANOG, Brachyury, FLK1/VEGFR2, NOTCH-1 accompanied by down-regulation of VE-cadherin, CD31 and vWF proteins. As NANOG levels increase in the nucleus, both SCD and ACD occur as characterized by increased NOTCH-1 and NUMB expression. We posit that this event induces a phenotypic switch from venous to an arterial phenotype, thereby contributing to increased neovascularization. However, only a subset of venous ECs undergoes such phenotypic switch. SMC, smooth muscle cells; DLL4, Delta-like ligand-4.
Table 1.
Oligonucleotides used for the amplification of the NANOG-, OCT4-, BRACHYURY-, CD133-, VEGFR2-promoters using the anti-Glut-1 (control) and anti-NANOG chromatins.
| Name | Oligonucleotides | Product size (kb) |
|---|---|---|
| NANOG |
FOR 5′-AATTAGCTAGGCATGGTGGTGTGTG-3′ REV 5′-ATGTTAGTATAGAGGAAGAGGAGGA-3′ |
2.2 |
| BRACHYURY |
FOR 5′-CTCTGAATGGCTGGGCTTGCCAAGGAG-3′ REV 5′-AGGGGAGCTCATCCTCCCGTCCGGCTC-3′ |
3.4 |
| OCT4 |
FOR 5′-CAGTTAAAGGCCGAGAAGTGAAC-3′ REV 5′-AGGGACTACTCAACCCCTCTCTC-3′ |
2.3 |
| CD133 |
FOR 5′-TGCTTCACCCCTTTCTACTGATAC-3′ REV 5′-GTCCCTTACTTAGAATGCAGCTAC C-3′ |
1.4 |
| FLK1/VEGFR2 |
FOR 5′-AGTAACAGGTTACATTATATTTCAG-3′ REV 5′-GTACTCGGTAACGGGCGCTGAGCAAC-3′ |
1.0 |
Acknowledgments
We thank Drs. Asrar B. Malik and Dolly Mehta for their suggestions. These studies were supported by AHA (GRNT4520014) and NIH (HL079356) and by the University of Illinois at Chicago (UIC) Center for Clinical and Translational Science (CCTS) Award Number UL1RR029879 from the National Center for Research Resources and UIC Cancer Center supplemental grant to K.K.W. M.U.F. was supported by R01HL116976 and R21HL112293 grants and E.E.K. was supported by a NIH training grant and an AHA pre-doctoral fellowship.
Footnotes
Author Contribution Summary
E.E.K. conceived idea and designed experiments, performed the biochemical and cell biology, knockdown, western blot and expression analyses, Matrigel plug assays, BrdU and ACD experiments, staining and microscopy, and reporter assays, collected data, generated histograms, and prepared the manuscript. J.B. performed western blotting and staining ACD; I.C. performed biochemical experiments; N.U., T.F., and M.U.F. were responsible for FAL and HLI methodologies, data collection, and interpretation and discussion of results. K.K.W. conceived idea, designed experiments, analyzed and interpreted the data, and prepared the manuscript.
Conflict of Interests
The authors declare no competing financial interests.
References
- 1.Kajstura J, Gurusamy N, Ogórek B, et al. Myocyte turnover in the aging human heart. CIRC RES. 2010;107:1374–1386. doi: 10.1161/CIRCRESAHA.110.231498. [DOI] [PubMed] [Google Scholar]
- 2.Frati C, Savi M, Graiani G, et al. Resident cardiac stem cells. CURR PHARM DES. 2011;17:3252–3257. doi: 10.2174/138161211797904181. [DOI] [PubMed] [Google Scholar]
- 3.Leri A, Kajstura J, Anversa P. Mechanisms of myocardial regeneration. TRENDS CARDIOVASC MED. 2011;21:52–58. doi: 10.1016/j.tcm.2012.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jopling C, Sleep E, Raya M, et al. Zebrafish heart regeneration occurs by cardiomyocyte dedifferentiation and proliferation. NATURE. 2010;464:606–609. doi: 10.1038/nature08899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jopling C, Boue S, Izpisua Belmonte JC. Dedifferentiation, transdifferentiation and reprogramming: three routes to regeneration. NAT REV MOL CELL BIOL. 2011;12:79–89. doi: 10.1038/nrm3043. [DOI] [PubMed] [Google Scholar]
- 6.Urbanek K, Rota M, Cascapera S, et al. Cardiac stem cells possess growth factor-receptor systems that after activation regenerate the infarcted myocardium, improving ventricular function and long-term survival. CIRC RES. 2005;97:663–673. doi: 10.1161/01.RES.0000183733.53101.11. [DOI] [PubMed] [Google Scholar]
- 7.Odelberg SJ, Kollhoff A, Keating MT. Dedifferentiation of mammalian myotubes induced by msx1. CELL. 2000;103:1099–1109. doi: 10.1016/s0092-8674(00)00212-9. [DOI] [PubMed] [Google Scholar]
- 8.Rudnicki MA, Le Grand F, McKinnell I, et al. The molecular regulation of muscle stem cell function. COLD SPRING HARB SYMP QUANT BIOL. 2008;73:323–331. doi: 10.1101/sqb.2008.73.064. [DOI] [PubMed] [Google Scholar]
- 9.Bergmann O, Bhardwaj RD, Bernard S, et al. Evidence for cardiomyocyte renewal in humans. SCIENCE. 2009;324:98–102. doi: 10.1126/science.1164680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Red-Horse K, Ueno H, Weissman IL, et al. Coronary arteries form by developmental reprogramming of venous cells. NATURE. 2010;464:549–553. doi: 10.1038/nature08873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zamora M, Männer J, Ruiz-Lozano P. Epicardium-derived progenitor cells require beta-catenin for coronary artery formation. PROC NATL ACAD SCI USA. 2007;104:18109–18114. doi: 10.1073/pnas.0702415104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Limana F, Zacheo A, Mocini D, et al. Identification of myocardial and vascular precursor cells in human and mouse epicardium. CIRC RES. 2007;101:1255–1265. doi: 10.1161/CIRCRESAHA.107.150755. [DOI] [PubMed] [Google Scholar]
- 13.Zhou B, von Gise A, Ma Q, et al. Genetic fate mapping demonstrates contribution of epicardium-derived cells to the annulus fibrosis of the mammalian heart. DEV BIOL. 2010;338:251–261. doi: 10.1016/j.ydbio.2009.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Smart N, Bollini S, Dubé KN, et al. De novo cardiomyocytes from within the activated adult heart after injury. NATURE. 2011;474:640–644. doi: 10.1038/nature10188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Riley PR, Smart N. Vascularizing the heart. CARDIOVASC RES. 2011;91:260–268. doi: 10.1093/cvr/cvr035. [DOI] [PubMed] [Google Scholar]
- 16.Okita K, Ichisaka T, Yamanaka S. Generation of germline-competent induced pluripotent stem cells. NATURE. 2007;448:313–317. doi: 10.1038/nature05934. [DOI] [PubMed] [Google Scholar]
- 17.Mitsui K, Tokuzawa Y, Itoh H, et al. The homeoprotein Nanog is required for maintenance of pluripotency in mouse epiblast and ES cells. CELL. 2003;113:631–642. doi: 10.1016/s0092-8674(03)00393-3. [DOI] [PubMed] [Google Scholar]
- 18.Hart AH, Hartley L, Ibrahim M, et al. Identification, cloning and expression analysis of the pluripotency promoting Nanog genes in mouse and human. DEV DYN. 2004;230:187–198. doi: 10.1002/dvdy.20034. [DOI] [PubMed] [Google Scholar]
- 19.Yamaguchi S, Kimura H, Tada M, et al. Nanog expression in mouse germ cell development. GENE EXPR PATTERNS. 2005;5:639–646. doi: 10.1016/j.modgep.2005.03.001. [DOI] [PubMed] [Google Scholar]
- 20.Forte A, Schettino MT, Finicelli M, et al. Expression pattern of stemness-related genes in human endometrial and endometriotic tissues. MOL MED. 2009;15:392–401. doi: 10.2119/molmed.2009.00068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kohler EE, Cowan CE, Chatterjee I, et al. NANOG induction of Fetal Liver Kinase-1 (FLK1) transcription regulates endothelial cell proliferation and angiogenesis. BLOOD. 2011;117:1761–1769. doi: 10.1182/blood-2010-07-295261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Horvitz H, Herskowitz I. Mechanisms of asymmetric cell division: two Bs or not two Bs, that is the question. CELL. 1992;68:237–255. doi: 10.1016/0092-8674(92)90468-r. [DOI] [PubMed] [Google Scholar]
- 23.Knoblich JA. Mechanisms of asymmetric stem cell division. CELL. 2008;132:583–597. doi: 10.1016/j.cell.2008.02.007. [DOI] [PubMed] [Google Scholar]
- 24.Wang H, Charles PC, Wu Y, et al. Gene expression profile signatures indicate a role for Wnt signaling in endothelial commitment from embryonic stem cells. CIRC RES. 2006;98:1331–1339. doi: 10.1161/01.RES.0000220650.26555.1d. [DOI] [PubMed] [Google Scholar]
- 25.Ishikawa T, Tamai Y, Zorn AM, et al. Mouse Wnt receptor gene Fzd5 is essential for yolk sac and placental angiogenesis. DEVELOPMENT. 2001;128:25–33. doi: 10.1242/dev.128.1.25. [DOI] [PubMed] [Google Scholar]
- 26.Goodwin AM, Sullivan KM, D’Amore PA. Cultured endothelial cells display endogenous activation of the canonical Wnt signaling pathway and express multiple ligands, receptors, and secreted modulators of Wnt signaling. DEV DYN. 2006;235:3110–3120. doi: 10.1002/dvdy.20939. [DOI] [PubMed] [Google Scholar]
- 27.Wang XY, Lan Y, He WY, et al. Identification of mesenchymal stem cells in aorta-gonad-mesonephros and yolk sac of human embryos. BLOOD. 2008;111:2436–2443. doi: 10.1182/blood-2007-07-099333. [DOI] [PubMed] [Google Scholar]
- 28.Goessling W, North TE, Loewer S, et al. Genetic interaction of PGE2 and Wnt signaling regulates developmental specification of stem cells and regeneration. CELL. 2009;136:1136–1147. doi: 10.1016/j.cell.2009.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chien AJ, Conrad WH, Moon RT. A Wnt survival guide: from flies to human disease. J INVEST DERMATOL. 2009;129:1614–1627. doi: 10.1038/jid.2008.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Merrill BJ. Develop-WNTs in somatic cell reprogramming. CELL STEM CELL. 2008;3:465–466. doi: 10.1016/j.stem.2008.10.011. [DOI] [PubMed] [Google Scholar]
- 31.Dajani R. Crystal structure of glycogen synthase kinase 3 beta: structural basis for phosphate-primed substrate specificity and autoinhibition. CELL. 2001;105:721–732. doi: 10.1016/s0092-8674(01)00374-9. [DOI] [PubMed] [Google Scholar]
- 32.Bertrand JA, Thieffine S, Vulpetti A, et al. Structural characterization of the GSK-3beta active site using selective and non-selective ATP-mimetic inhibitors. J MOL BIOL. 2003;333:393–407. doi: 10.1016/j.jmb.2003.08.031. [DOI] [PubMed] [Google Scholar]
- 33.Meijer L, Skaltsounis AL, Magiatis P, et al. GSK-3-selective inhibitors derived from Tyrian purple indirubins. CHEM BIOL. 2003;10:1255–1266. doi: 10.1016/j.chembiol.2003.11.010. [DOI] [PubMed] [Google Scholar]
- 34.Sato N, Meijer L, Skaltsounis L, et al. Maintenance of pluripotency in human and mouse embryonic stem cells through activation of Wnt signaling by a pharmacological GSK-3-specific inhibitor. NAT MED. 2004;10:55–63. doi: 10.1038/nm979. [DOI] [PubMed] [Google Scholar]
- 35.Tseng AS, Engel FB, Keating MT. The GSK-3 inhibitor BIO promotes proliferation in mammalian cardiomyocytes. CHEM BIOL. 2006;13:957–963. doi: 10.1016/j.chembiol.2006.08.004. [DOI] [PubMed] [Google Scholar]
- 36.Feng B, Ng JH, Heng JC, et al. Molecules that promote or enhance reprogramming of somatic cells to induced pluripotent stem cells. Cell Stem Cell. 2009;4:301–312. doi: 10.1016/j.stem.2009.03.005. [DOI] [PubMed] [Google Scholar]
- 37.Gore AV, Swift MR, Cha YR, et al. Rspo1/Wnt signaling promotes angiogenesis via Vegfc/Vegfr3. DEVELOPMENT. 2011;138:4875–4886. doi: 10.1242/dev.068460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu L, Nam S, Tian Y, Polychronopoulos P, Magiatis P, Skaltsounis L, Jove R, et al. 6-Bromoindirubin-3′-oxime inhibits JAK/STAT3 signaling and induces apoptosis of human melanoma cells. CANCER RES. 2010;71:3972–3979. doi: 10.1158/0008-5472.CAN-10-3852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yi F, Pereira L, Hoffman JA, et al. Opposing effects of Tcf3 and Tcf1 control Wnt stimulation of embryonic stem cell self-renewal. NAT CELL BIOL. 2011;13:762–770. doi: 10.1038/ncb2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gershengorn MC, Hardikar AA, Wei C, et al. Epithelial-to-mesenchymal transition generates proliferative human islet precursor cells. SCIENCE. 2004;306:2261–2264. doi: 10.1126/science.1101968. [DOI] [PubMed] [Google Scholar]
- 41.Timmins NE, Nielsen LK. Generation of multicellular tumor spheroids by the hanging-drop method. METHODS MOL MED. 2007;140:141–151. doi: 10.1007/978-1-59745-443-8_8. [DOI] [PubMed] [Google Scholar]
- 42.Campa VM, Gutiérrez-Lanza R, Cerignoli F, et al. Notch activates cell cycle reentry and progression in quiescent cardiomyocytes. J CELL BIOL. 2008;183:129–141. doi: 10.1083/jcb.200806104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cowan CE, Kohler EE, Dugan TA, et al. Kruppel-like factor-4 transcriptionally regulates VE-cadherin expression and endothelial barrier function. CIRC RES. 2010;107:959–966. doi: 10.1161/CIRCRESAHA.110.219592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kwon C, Cheng P, King IN, et al. Notch post-translationally regulates β-catenin protein in stem and progenitor cells. NAT CELL BIOL. 2011;13:1244–1251. doi: 10.1038/ncb2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lacorre DA, Baekkevold ES, Garrido I, et al. Plasticity of endothelial cells: rapid dedifferentiation of freshly isolated high endothelial venule endothelial cells outside the lymphoid tissue microenvironment. BLOOD. 2004;103:4164–4172. doi: 10.1182/blood-2003-10-3537. [DOI] [PubMed] [Google Scholar]
- 46.Crespo I, Del Sol A. A General Strategy for Cellular Reprogramming: The Importance of Transcription Factor Cross-Repression. STEM CELLS. 2013 doi: 10.1002/stem.1473. (in press) [DOI] [PubMed] [Google Scholar]
- 47.Urao N, Inomata H, Razvi M, et al. Role of nox2-based NADPH oxidase in bone marrow and progenitor cell function involved in neovascularization induced by hindlimb ischemia. CIRC RES. 2008;103:212–220. doi: 10.1161/CIRCRESAHA.108.176230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Urao N, Razvi M, Oshikawa J, et al. IQGAP1 is involved in post-ischemic neovascularization by regulating angiogenesis and macrophage infiltration. PLOS ONE. 2010;5:e13440. doi: 10.1371/journal.pone.0013440. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Endothelial characteristics of HUVECs. Endothelial cell characteristics of HUVECs were determined by staining with, (A) anti-human VE-cadherin (green) and (B) anti-human vWF (red) antibodies. Blue, DAPI. Original magnification is as shown. Scale bar, 100 μm.
Figure S2. Dose-dependent effect of BIO in cultured HUVECs. (A) Timeline of plating the cells, BIO treatment, fixing, staining and quantification. (B) HUVECs (2 × 105) were plated in complete media for the indicated time and were treated with an increasing concentration of BIO (0, 0.2, 0.5, 1.0, 1.5, and 2.0 μM). The number of ECs was counted after 24 hrs of stimulation. Quantification of the number of cells per 20X field, mean ± S.E.M. (C–H) Representative images of H&E stained HUVECs receiving vehicle alone or the indicated concentrations of BIO. The experiment was repeated 3 times with triplicates. Asterisks indicate P<0.05 versus the control vehicle group.
Figure S3. Wnt3a induced NANOG expression in HUVECs. Control HUVECs and HUVECs treated with Wnt3a (20ng/ml) for 4 hours on coverslips and fixed with 4% PFA, then subjected to immunostaining with, (A&D) anti-human VE-cadherin (green), (B&E) anti-NANOG (red) antibodies, and (C&F) Merge. Original magnification is as shown. Scale bar, 100 μm. (G) Quantification of image pixel intensity.
Figure S4. Nucleotide sequence of human NANOG promoter/enhancer (−2.2 kb). Analysis of human NANOG promoter/enhancer from −2240 to +1 relative to the ATG (red) TSS identified 19 NANOG binding sites. NANOG binding nucleotide sequence (ATTA) in direct (+) strand are shown in bold characters, while in reverse (−) strand in bold underlined (TAAT).
Figure S5. Nucleotide sequence of human BRACHYURY promoter/enhancer (−3.8 kb). The BRACHYURY promoter showed 15 sites (ATTA) within the −3.8 kb upstream of the ATG (red) TSS. Putative NANOG binding nucleotide sequence (ATTA) in direct (+) strand are shown in bold characters, while in reverse (−) strand in bold underlined (TAAT).
Figure S6. BIO induces the formation cellular aggregates in a hanging drop assay. (A) Timeline of the hanging drop assay. HUVECs at passage 3–4 were plated in EndoGRO-VEGF in Complete Media Kit + BIO (0.2 μM) for 24 hours in 10 cm dishes. Dishes were inverted and filled with 6 ml sterile 1X PBS, pH 7.4. After 7 or 14 days the dishes were turned up-right, and 6 ml EndoGRO-VEGF Complete Media was placed on cells for a few minutes. Media was aspirated, fixed with 4% PFA and ECs/hanging drops were subjected to H&E staining. (B) Quantification of the number of colonies per 10X field, mean ± S.E.M. Asterisks indicate P<0.05. (C–F) Representative images of hanging drop colonies. Scale bar, 200 μm. (G&H) Unstained live cellular aggregates (colonies) of HUVECs receiving BIO at 1 and 2 weeks. Experiments were repeated at least 3 times.
Figure S7. BIO does not induce apoptosis or necrosis of HUVECs. HUVECs were cultured overnight without VEGF or bFGF or BIO. Next day, (A) HUVECs were either left untreated (control) or treated with VEGF (50ng/ml) in absence or presence of BIO (0.2 μM) for 6 hours. The contribution of apoptosis and necrosis were measured by FACS analyses of Annexin-V and propidium iodide (PI) staining. (B) Quantification of HUVECs death. Experiments were repeated 3 times. *P < 0.05 vs. control.
Figure S8. Representative images of SCD and ACD in HUVECs. (A&B) Images of BrdU incorporation in control and BIO-treated HUVECs. The arrows and arrowheads indicate SCD and ACD, respectively. Scale bar, 200 μm.
Figure S9. Representative images of adductor muscle tissue architecture. Top panel, images of (A&C) H&E stained or (B&D) vWF (red) stained adductor muscle tissue sections prepared from control (unligated) and ligated lower limbs receiving PBS or BIO. Boxed areas from the top panels are magnified in the bottom panels. No obvious changes in overall tissue architecture or necrosis in these mice were detected. Pre-existing vessels showed no pathology as lumens (arrows) were filled with erythrocytes and leukocytes, with no visible hemorrhage or edematous events. (E) Quantification of vWF+ vascular structures. Experiments were carried out at least 3 times. Data represent mean ± S.E.M. Asterisks indicate P<0.05.
Figure S10. Proposed model of dedifferentiation of venous to arterial ECs. In adult, venous ECs are dormant and do not express detectable level of NANOG or NOTCH-1. Following ischemia or injury, BIO or Wnt3a stabilize β-catenin. This event induces β-catenin/NANOG signaling resulting in increased expression of NANOG, Brachyury, FLK1/VEGFR2, NOTCH-1 accompanied by down-regulation of VE-cadherin, CD31 and vWF proteins. As NANOG levels increase in the nucleus, both SCD and ACD occur as characterized by increased NOTCH-1 and NUMB expression. We posit that this event induces a phenotypic switch from venous to an arterial phenotype, thereby contributing to increased neovascularization. However, only a subset of venous ECs undergoes such phenotypic switch. SMC, smooth muscle cells; DLL4, Delta-like ligand-4.