

Abstract
Vaccinia virus (VACV) strain MVA is a highly attenuated vector for vaccines that is being explored in clinical trials. We compared the CD8+ T cell immunogenicity of MVA with that of a virulent laboratory strain of VACV (strain WR) in BALB/c mice by examining epitope-specific responses as well as estimating the total number of activated CD8+ T cells, irrespective of specificity. We found that MVA elicited total CD8+ T cell responses that were reduced by at least 20-fold compared with strain WR in BALB/c mice. In C57Bl/6 mice we also found a substantial difference in immunogenicity between these VACV strains, but it was more modest at around 5-fold. Of note, the size of responses to the virulent WR virus were similar in both strains of mice suggesting that BALB/c mice can mount robust CD8+ T cell responses to VACV. While the data for total responses clearly showed that MVA overall is poorly immunogenic in BALB/c mice, we found one epitope for which strong responses were made irrespective of virus strain. Therefore in the context of a vaccine, some recombinant epitopes may have similar immunogenicity when expressed from MVA and other strains of VACV, but we would expect these to be exceptions. These data show clearly the substantial difference in immunogenicity between MVA and virulent VACV strains and suggest that the impact of host genetics on responses to attenuated vaccine vectors like MVA requires more consideration.
Keywords: Poxvirus, Vaccine, mouse strain, CD8+ T cells, Modified Vaccinia Ankara
Introduction
Since its use to eradicate smallpox, vaccinia virus (VACV) has attracted considerable interest as a recombinant vaccine vector, having a large capacity for foreign genes and inherent immunogenicity.1, 2 As a result, vaccines based on highly attenuated strains of VACV, such as Modified Vaccinia Ankara (MVA), have entered clinical trials.3, 4 Mouse models are invaluable for developing vaccines, but are frequently limited to a single inbred strain of mice. Further, it is assumed that data from virulent VACV strains such as Western Reserve (WR) can be directly extrapolated to attenuated vectors, even though virulence and antigen dose are linked to immunogenicity. In this context, the CD8+ T cell (TCD8+) response to VACV has been most extensively investigated using C57Bl/6 mice and for VACV strain WR.5, 6 Here we ask whether this data can be reasonably generalized to other strains of VACV, including MVA and to BALB/c mice, a commonly used inbred strain preferred for some infectious models.7, 8
Results and Discussion
First, we measured TCD8+ responses in BALB/c mice 7 days after immunization with VACV strains MVA, Copenhagen and WR using peptides published as H-2d-presented epitopes 9, 10 to restimulate splenocytes prior to intracellular staining for IFN-γ (IFN-γ ICS).11 This set of native VACV epitopes was used rather than a single native (or recombinant) epitope to obtain representative results and because previous studies have shown that changes in immunodominance can be misinterpreted as differences in immunogenicity.12 Immunization with WR resulted in larger TCD8+ responses than with Copenhagen and especially MVA for almost all peptides (Fig. 1A). The only exception was E3140, which elicits a similar sized TCD8+ response for all strains of VACV. Comparisons between the virus strains were similar in mice >30 days after immunization (not shown).
Figure 1. Peptide-specific TCD8+ responses to different strains of VACV in BLAB/c mice and impact of sequence variation in the immunodominant F226 peptide.

Groups of BALB/c mice were immunized with 1×106 PFU of VACV i.p. and 7d later peptide-specific TCD8+ cell responses were measured using IFN-γ ICS. (A) Average percentages (and SEMs) of TCD8+ cells from BALB/c mice infected with VACV WR, Copenhagen (Cop) or MVA that produce IFN-γ in ex vivo stimulations with the indicated peptides. (B) Average percentages (and SEMs) of TCD8+ cells from BALB/c mice infected with VACV WR, WR F2G, MVA or MVA F2Y that produce IFN-γ in ex vivo stimulations with the indicated peptides. (C) Sum of TCD8+ cell responses to the 7 epitopes conserved across all strains shown in B (including only the appropriate F226 variant). Data are means and SEM from two independent experiments, each with n = 5 – 10 (*p < 0.01, Mann Whitney test).
Lack of conservation of key epitopes across the VACV strains complicates the experiment above. In particular, the most immunogenic epitope in WR (F226(Y); SPYAAGYDL) exists as a variant (F226(G); SPGAAGYDL) in Copenhagen and MVA and in these viruses anti-F226 responses are far less prominent. A5275 and B249 are absent in MVA and C674 exists as two alternate variants (GFIRSLQTI in WR and SFIRSLQNI in Copenhagen and MVA). Both versions of this peptide were used in all IFN-γ ICS assays, but for all the virus strains produced the same result and for simplicity we show data from homologous variants for each virus.
As noted above, the F226(G) variant of the immunodominant F226(Y) epitope in WR elicits relatively weak TCD8+responses from Copenhagen and even more so from MVA, but it is not clear to what extent this is due to the epitope variant or the virus strain background. To distinguish between these possibilities and to allow matching of a known immunodominant epitope between WR and MVA, we made recombinants of both, engineered such that they expressed the alternate variant of the F226 epitope. WR F2G had the usual dominant WR F226(Y) epitope replaced with F226(G) and MVA F2Y had the reverse replacement. After immunizing mice with these viruses, we found that replacement of the F226(Y) epitope of WR with F226(G) in WR F2G substantially reduced F226-specific responses, but other specificities were not altered (Fig. 1B). Conversely, the introduction of F226(Y) to MVA, while improving F226–specific TCD8+ responses compared with MVA, produced a response that was less than a quarter of that elicited by WR. Indeed WR F2G, induced a significantly higher F226-specific response than MVA F2Y, even though they expressed subdominant and immunodominant variants, respectively (p=0.0256). Summing up the responses to the conserved epitopes tested in this experiment (including only the appropriate variant of F226) showed that MVA F2Y was much less immunogenic than WR even though it shared the immunodominant F226(Y) (Fig. 1C). Likewise, WR F2G remained substantially more immunogenic than MVA, despite both viruses containing the subdominant F226(G) epitope. These data suggest that MVA is inherently poorly immunogenic in BALB/c mice compared with WR.
To ensure that the results above were not unwittingly biased by the set of epitopes that we had available, we used an epitope-independent method of measuring TCD8+ responses. Upon activation of TCD8+ during VACV immunization in vivo, CD62L and granzyme B (GzmB) are down- and up-regulated respectively. Therefore the expression of intracellular GzmB and surface CD62L on uncultured TCD8+ can be used as a surrogate measure of the total acute anti-VACV response in the acute phase.13 Using this method demonstrated that while WR induced a large response of around 60% of TCD8+ in BALB/c, Copenhagen was less immunogenic and MVA elicited responses around 8-fold lower (Figure 2A–B left). When the data were analyzed as the total number of activated TCD8+ cells per spleen, the difference between WR and MVA was even more striking at >30-fold (Figure 2B right). We were concerned that this method might be missing TCD8+ primed by MVA if GzmB expression or CD62L down regulation were not as marked on these cells such that they fell outside the gate, which was set on the basis of samples from WR-infected mice. To address this we used MVA F2Y that had poor immunogenicity like MVA but allowed the use of F226(Y)-loaded DimerX (similar to peptide-MHC-tetramer) reagents to identify a population of virus-specific TCD8+ that could be compared with similar cells from WR-infected mice (Figure 2C). After MVA F2Y and WR immunization, 61.4% and 93.5% of F226(Y)-specific TCD8+ were able to be identified as activated according to their CD62L and GzmB profile, respectively (Figure 2C). This suggests our method underestimates numbers of TCD8+ primed by both viruses in BALB/c mice, but more so for MVA than WR. Taking these different levels of underestimation into account, the difference in total response elicited by these viruses was ~20-fold. Less virulent strains of VACV have been found previously to induce lower responses in C57Bl/6 mice, but MVA was not tested and the differences were not as large as we show here in BALB/c mice.14 To examine this, we used CD62L and GzmB to compare responses to immunization with VACV strains in C57Bl/6 mice (Figure 2D). Around 60% of TCD8+ responded to WR in these mice, similar to WR in BALB/c mice and a previous study.13 Copenhagen and MVA were less immunogenic than WR, but the difference was not as great as was seen for BALB/c mice. In C57Bl/6 mice, MVA induced responses that were 2.5-fold lower than WR when analyzed as a percent of TCD8+ and 5-fold when total numbers of activated cells in the spleen were compared.
Figure 2. Total TCD8+ cell response to VACV strains in BALB/c and C57Bl/6 mice.
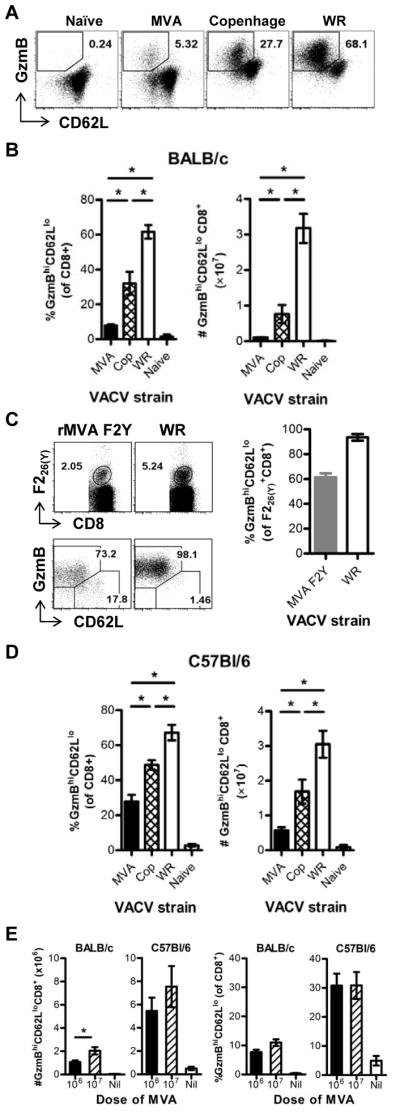
Groups mice were immunized i.p. with 1×106 or 1×107 PFU of VACV strains as shown or were control (naïve) injected with 200 μl PBS, i.p.. After 7 days splenocytes were analyzed for surface CD62L and intracellular GzmB (A, B, D and E) or F226(Y)-loaded DimerX-binding, CD62L and GzmB (C). (A) Representative flow cytometry plots (gated on CD8+ events) for each VACV strain and naïve control in BALB/c mice. (B) Averages (and SEM) of CD62Llo GzmBhi events in BALB/c mice analyzed as a % of CD8+ events (left) and total numbers of CD8+, CD62Llo GzmBhi events (right). Data are from 2 for uninfected mice, 6 Copenhagen-, 6 MVA- and 12 WR-infected mice across two experiments. (C) Representative flow cytometry plots gated on CD8+ (top row), and on F226(Y)-DimerX+, as shown on top plots (bottom row) of splenocytes from mice immunized with MVA F2Y or WR in BALB/c mice. Graph on right shows average percent (and SEM) of CD62LloGzmBhi amongst F226(Y)-specific TCD8+. (D) Same as B, but using C57Bl/6 mice. (E) Comparison of two doses of MVA (1×106 and 1×107 PFU in BALB/c and C57Bl/6 mice as shown on graphs. Total numbers of activated TCD8+ per spleen are on the left and the percent of activated cells amongst CD8+ events on the right. (D and E) Data are from 2 uninfected mice and 6 mice infected with each VACV strains across two experiments. (*p < 0.05, Mann Whitney test).
These data show that BALB/c and C57Bl/6 mice mount similar sized TCD8+ responses to WR, so the former are not necessarily compromised in their ability to respond to VACV in general. However, we did note that BALB/c mice made poorer responses to strain Copenhagen than did C57Bl/6 mice (around 2-fold less). It is possible that BALB/c mice need relatively higher levels of infection to generate the same TCD8+ response compared with C57Bl/6 mice and the high virulence of WR masks this defect. Understanding differences in immune responses to virulent viruses across mouse strains is complicated because it is not possible to dissect the roles of virus burden, antigen loads and inflammation, all of which will vary with virus replication and spread. However for a non-replicating virus such as MVA, which causes no pathology and has limited spread15–17, these complications are not a problem. Therefore the simplest explanation of these data is that BALB/c mice have a simple defect in priming TCD8+ responses to MVA compared with C57Bl/6 mice. To determine if the difference in anti-MVA TCD8+ numbers in the two mouse strains could be resolved at a higher dose, we compared responses in mice immunized with the standard 1×106 PFU with a 10-fold higher dose (Figure 2E). The number, but not percent of TCD8+ was significantly increased in BALB/c mice given 1×107 PFU but the response remained around a third of that elicited by the standard dose in C57Bl/6 mice. In C57Bl/6 mice, no significant increase in response was seen for the higher dose, supporting the notion that TCD8+ responses in BALB/c mice might be more dependent on high doses of VACV antigen. They also underscore the defect of BALB/c mice in making TCD8+ responses to MVA.
When examining individual epitopes, the poor immunogenicity we describe here was not apparent for one epitope. Responses to E3140 were comparable in all the strains of VACV tested and this suggests that for at least one epitope, MVA can prime good TCD8+ responses. This most likely explains the apparent superiority of a recombinant MVA over a thymidine kinase negative WR in priming TCD8+ to an encoded β-galactosidase epitope when the opposite result was found for VACV-specific responses.16 The interpretation in that case was that the induction of modest anti-vector and good anti-recombinant antigen responses is a desirable general characteristic of recombinant MVA vaccines16. However in the light of the data here, perhaps the previous result reflects a fortuitous choice of foreign antigen rather than a general principle. Looking across the native epitopes we tested (e.g. Figure 1B), it is clear that E3140 is an exception rather than the rule and there is no reason to expect that recombinant antigens in vaccine would follow a different pattern.
In summary, MVA elicits many fewer TCD8+ than WR in BALB/c and C57Bl/6 mice, but this was especially notable in BALB/c. The contrast between these mouse strains highlights the importance of host genetic background in responses to attenuated vaccine vectors such as MVA.
Methods
Viruses
The unmodified VACV used were low passage stocks grown in BHK-21. WR refers to WR (NIH TC-adapted), ATCC VR-1354, sequence accession AY243312.1. MVA refers to ‘clone 1’ characterized extensively in17 that was derived at NIH from an original isolate harvested after 572 passages in primary chick cells in 1974. Both were provided by Bernard Moss (NIH).
Mice and infections
Female C57Bl/6 and BALB/c mice greater than 8 weeks of age were housed and experiments were done according to ethical requirements and under an approval from the Australian National University Animal Ethics and Experimentation Committee. Mice were injected via the intraperitoneal (i.p.) route with 1×106 PFU of virus in 200 μl PBS except in Figure 2E where some mice received 1×107 PFU.
TCD8+ assays
Mice were euthanized 7 days after infection and single cell suspensions of spleens used in the following assays12: A) Splenocytes were cultured with synthetic peptides at 0.1 μM in the presence of brefeldin A for 4 hours before surface staining for CD8 (clone 53-6.7; BioLegend), fixation with 1% paraformaldehyde and intracellular staining for IFN-γ (clone XMG1.2; BioLegend). Peptide sequences used were: F226(Y), SPYAAGYDL; F226(G), SPGAAGYDL; A5275, KYGRLFNEI; E3140, VGPSNSPTF; C674(G), GFIRSLQTI; C674(S), SFIRSLQTI; I890, LPNPAFIHI; I8511, QYIYSEHTI; A3190, IYSPSNHHI; B249, KYMWCYSQV; D1797, KYEGPFTTT; (Mimotopes or Genscript).9, 10 The use of this method to make quantitative comparisons of TCD8+ responses after VACV infection has been validated rigorously11. B) splenocytes were stained for surface CD8 (clone 53-6.7) and CD62L (clone 53-6.7, BioLegend) before fixing and staining for intracellular GzmB (clone GB12; Invitrogen, supplied by Caltag).13 C) Splenocytes were stained for CD8 (clone 53-6.7) and with F226(Y)-loaded H-2Ld:Ig fusion protein (DimerX; BD Biosciences) pre-bound with α-mouse IgG1-PE (clone A85-1, BD Biosciences).18 In some instances surface CD62L and intracellular GzmB were also assessed as above and as published.13 For all assays data were acquired using an LSR II flow cytometer (BD Biosciences) and analysis was done with the aid of with Flowjo software (Treestar).
Generation of VACVs with the alternate F226 peptide
Recombinant VACV WR expressing the alternate F226 peptide SPGAAGYDL and recombinant VACV MVA expressing the alternate F226 SPYAAGYDL peptide were engineered using a transient dominant method in which unstable intermediates were enriched and identified through the use of a fusion protein between enhanced green fluorescent protein (GFP) and BsdR (confers resistance to blasticidin).19 The sequences used to encode the F226 epitope were altered to include identifying restriction enzyme sites and some additional changes to allow each recombinant to be distinguished from its parent by PCR, but the only amino acid changes were the desired glycine and tyrosine substitutions.
Acknowledgments
We thank Bernard Moss (NIH, Bethesda) for VACV WR and MVA, Geoffrey Smith (University of Cambridge) for VACV Copenhagen and RSB animal services for mouse husbandry. This work was funded by grants from the NIH (R01AI067401), NHMRC (APP1023141) and an ARC Future Fellowship (FT110100310) to DCT.
References
- 1.Smith GL, Mackett M, Moss B. Infectious vaccinia virus recombinants that express hepatitis B virus surface antigen. Nature. 1983;302:490–5. doi: 10.1038/302490a0. [DOI] [PubMed] [Google Scholar]
- 2.Frey SE, Newman FK, Cruz J, Brian Shelton W, Tennant JM, Polach T, et al. Dose-related effects of smallpox vaccine. N Engl J Med. 2002;346:1275–80. doi: 10.1056/NEJMoa013431. [DOI] [PubMed] [Google Scholar]
- 3.Kantoff PW, Schuetz TJ, Blumenstein BA, Glode LM, Bilhartz DL, Wyand M, et al. Overall Survival Analysis of a Phase II Randomized Controlled Trial of a Poxviral-Based PSA-Targeted Immunotherapy in Metastatic Castration-Resistant Prostate Cancer. J Clin Oncol. 2010;28:1099–105. doi: 10.1200/JCO.2009.25.0597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Goepfert PA, Elizaga ML, Sato A, Qin L, Cardinali M, Hay CM, et al. Phase 1 Safety and Immunogenicity Testing of DNA and Recombinant Modified Vaccinia Ankara Vaccines Expressing HIV-1 Virus-like Particles. J Infect Dis. 2011;203:610–9. doi: 10.1093/infdis/jiq105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moutaftsi M, Peters B, Pasquetto V, Tscharke DC, Sidney J, Bui HH, et al. A consensus epitope prediction approach identifies the breadth of murine TCD8+-cell responses to vaccinia virus. Nat Biotechnol. 2006;24:817–9. doi: 10.1038/nbt1215. [DOI] [PubMed] [Google Scholar]
- 6.Remakus S, Sigal LJ. Gamma interferon and perforin control the strength, but not the hierarchy, of immunodominance of an antiviral CD8+ T cell response. J Virol. 2011;85:12578–84. doi: 10.1128/JVI.05334-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Graham BS, Perkins MD, Wright PF, Karzon DT. Primary respiratory syncytial virus infection in mice. J Med Virol. 1988;26:153–62. doi: 10.1002/jmv.1890260207. [DOI] [PubMed] [Google Scholar]
- 8.Vladimirov V, Badalová J, Svobodová M, Havelková H, Hart AAM, Blažková H, et al. Different genetic control of cutaneous and visceral disease after Leishmania major infection in mice. Infect Immun. 2003;71:2041–6. doi: 10.1128/IAI.71.4.2041-2046.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tscharke DC, Woo WP, Sakala IG, Sidney J, Sette A, Moss DJ, et al. Poxvirus CD8+ T-cell determinants and cross-reactivity in Balb/c mice. J Virol. 2006;80:6318–23. doi: 10.1128/JVI.00427-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Oseroff C, Peters B, Pasquetto V, Moutaftsi M, Sidney J, Panchanathan V, et al. Dissociation between epitope hierarchy and immunoprevalence in CD8+ responses to vaccinia virus Western Reserve. J Immunol. 2008;180:7193–202. doi: 10.4049/jimmunol.180.11.7193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Flesch IEA, Hollett NA, Wong YC, Tscharke DC. Linear fidelity in quantification of anti-viral CD8+ T cells. PLoS ONE. 2012;7:e39533. doi: 10.1371/journal.pone.0039533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lin LCW, Flesch IEA, Tscharke DC. Immunodomination during peripheral vaccinia virus infection. PLoS Pathogens. 2013;9:e1003329. doi: 10.1371/journal.ppat.1003329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yuen TJ, Flesch IEA, Hollett NA, Dobson BM, Russell TA, Fahrer AM, et al. Analysis of A47, an immunoprevalent protein of vaccinia virus, leads to a reevaluation of the total antiviral CD8+ T cell response. J Virol. 2010;84:10220–9. doi: 10.1128/JVI.01281-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Salek-Ardakani S, Flynn R, Arens R, Yagita H, Smith GL, Borst J, et al. The TNFR family members OX40 and CD27 link viral virulence to protective T cell vaccines in mice. J Clin Invest. 2011;121:296–307. doi: 10.1172/JCI42056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carroll MW, Moss B. Host range and cytopathogenicity of the highly attenuated MVA strain of vaccinia virus: Propagation and generation of recombinant viruses in a nonhuman mammalian cell line. Virol. 1997;238:198–211. doi: 10.1006/viro.1997.8845. [DOI] [PubMed] [Google Scholar]
- 16.Ramirez JC, Gherardi MM, Esteban M. Biology of attenuated modified vaccinia virus Ankara recombinant vector in mice: Virus fate and activation of B- and T-cell immune responses in comparison with the western reserve strain and advantages as a vaccine. J Virol. 2000;74:923–33. doi: 10.1128/jvi.74.2.923-933.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wyatt LS, Earl PL, Eller LA, Moss B. Highly attenuated smallpox vaccine protects mice with and without immune deficiencies against pathogenic vaccinia virus challenge. PNAS. 2004;101:4590–5. doi: 10.1073/pnas.0401165101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Flesch IE, Woo WP, Wang Y, Panchanathan V, Wong YC, La Gruta NL, et al. Altered CD8+ T cell immunodominance after vaccinia virus infection and the naive repertoire in inbred and F1 mice. Journal of immunology (Baltimore, Md : 1950) 2010;184:45–55. doi: 10.4049/jimmunol.0900999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wong YC, Lin LCW, Melo-Silva CR, Smith SA, Tscharke DC. Engineering recombinant poxviruses using a compact GFP–blasticidin resistance fusion gene for selection. J Virol Met. 2011;171:295–8. doi: 10.1016/j.jviromet.2010.11.003. [DOI] [PubMed] [Google Scholar]