

Abstract
Anaplasma phagocytophilum, an obligate intracellular bacterium, modifies functions of its in vivo host, the neutrophil. The challenges of using neutrophils ex vivo necessitate cell line models. However, cell line infections do not currently mimic ex vivo neutrophil infection characteristics. To understand these discrepancies, we compared infection of cell lines to ex vivo human neutrophils and differentiated hematopoietic stem cells with regard to infection capacity, oxidative burst, host defense gene expression, and differentiation. Using established methods, marked ex vivo neutrophil infection heterogeneity was observed at 24-48 h necessitating cell sorting to obtain homogeneously-infected cells at levels observed in vivo. Moreover, gene expression of infected cell lines differed markedly from the prior standard of unsorted infected neutrophils. Differentiated HL-60 cells sustained similar infection levels to neutrophils in vivo and closely mimicked functional and transcriptional changes of sorted infected neutrophils. Thus, care must be exercised using ex vivo neutrophils for A. phagocytophilum infection studies since a major determinant of transcriptional and functional changes among all cells was the intracellular bacteria quantity. Furthermore, comparisons of ex vivo neutrophils and the surrogate HL-60 cell model allowed the determination that specific cellular functions and transcriptional programs are targeted by the bacterium without significantly modifying differentiation.
Introduction
The obligate intracellular pathogen, Anaplasma phagocytophilum, survives and propagates primarily within neutrophils by reprogramming critical granulocyte functions. This reprogramming includes delayed neutrophil apoptosis that allows time for bacterial replication (Choi et al., 2005; Ge and Rikihisa, 2006; Yoshiie et al., 2000), increased recruitment and clustering of neutrophils which promotes bacterial dissemination and inflammatory response (Akkoyunlu et al., 2001; Klein et al., 2000; Scorpio et al., 2004), and impaired host defenses, such as reduced NADPH oxidase superoxide anion production that permits intracellular survival (Banerjee et al., 2000; Carlyon et al., 2004; Choi and Dumler, 2003; IJdo and Mueller, 2004; Wang et al., 2002). These modifications occur with active intracellular replication and with changes in host gene transcription. For example, reduced NADPH oxidase activation is in part attributed to decreased granulocyte CYBB transcription (Banerjee et al., 2000; Garcia-Garcia et al., 2009a; Thomas et al., 2005). The A. phagocytophilum nucleomodulin AnkA, binds to the CYBB promoter and downregulates its expression (Garcia-Garcia et al., 2009b). A. phagocytophilum infection also leads to downregulation of host granulocyte defense genes including catalase (CAT), cathepsin G (CTSG), defensin alpha 4 (DEFA4), elastase 2 (ELA2), myeloperoxidase (MPO), proteinase 3 (PRTN3), and bactericidal/permeability-increasing protein (BPI), among others (Carlyon et al., 2002; Garcia-Garcia et al., 2009a). Moreover, delayed apoptosis is in part maintained by upregulated transcription of BCL2 family genes, whereas neutrophil recruitment is enhanced by upregulated chemokine gene transcription, especially IL8 (Borjesson et al., 2005; de la Fuente et al., 2005; Lee et al., 2008; Pedra et al., 2005; Sukumaran et al., 2005). Complex and coordinated functional changes such as reduced adhesion of infected neutrophils to endothelial cells, their transmigration through endothelium, enhanced degranulation, and impaired phagocytosis are phenotypic expressions that resemble neutrophil progenitors more than terminally differentiated neutrophils (Choi et al., 2003; Choi et al., 2004; Garyu et al., 2005). Yet, the coordinated subversion of each function provides a significant fitness advantage for intracellular survival in neutrophils and subsequent acquisition by tick blood meal. Understanding the genome-wide basis for transcriptional and epigenetic subversion of complex phenotypic functions by A. phagocytophilum will require infections in ex vivo neutrophils or other adequate tractable surrogate cell models.
Investigation of functional alterations owing to A. phagocytophilum infection is most relevant in the natural mammalian target cell, the neutrophil. However, neutrophils present difficult challenges for experimental studies ex vivo, specifically with regard to pre-programmed apoptosis and short ex vivo life span, inability to manipulate transcription, and difficulty with transfection for expression of exogenous proteins or silencing of gene expression. As a result, investigation of the functional effects of A. phagocytophilum infection is most often conducted in granulocyte cell line models including HL-60, THP-1, and NB4 cells (Carlyon et al., 2002; Garcia-Garcia et al., 2009a; Pedra et al., 2005). Although cell lines have substantially contributed to studies of the functional effects of A. phagocytophilum infection, each cell model has deficits for study of neutrophil differentiation or function. Moreover, ex vivo neutrophil transcriptional responses with A. phagocytophilum infection do not yield the same results as observed in granulocyte cell lines (Borjesson et al., 2005; de la Fuente et al., 2005; Lee et al., 2008; Pedra et al., 2005; Sukumaran et al., 2005). No study has examined why such discrepancies exist or which cell line(s) most closely mimic responses and behavior of infected neutrophils. Additionally, the PLB-985 human myelomonoblastic cell line and differentiated human hematopoietic stem cells (HSCs) have yet to be investigated as in vitro models of infection. HSCs hold promise for modeling A. phagocytophilum infection because they are primary cells that lack neoplastic mutations and can be transfected to express exogenous proteins or silence endogenous gene expression. To determine which cell line models could accurately be used to study genome-wide modulation of transcriptional programs that underlie neutrophil reprogramming with A. phagocytophilum infection, we sought to test: i) why neutrophil transcriptional responses differ from those in granulocyte cell models, ii) how accurately these cell line models reflect the differentiation, functional, and transcriptional effects of infection in neutrophils, and iii) whether these models can be used to determine if transcriptional and functional changes are the result of induced de-differentiation.
Materials and Methods
Cell lines and cell culture
Human neutrophils were obtained from EDTA anti-coagulated peripheral blood under a protocol approved by the Johns Hopkins Medical Institutions IRB. Dextran-sedimented, leukocyte-rich plasma was centrifuged through Ficoll-Paque density gradients, mononuclear cells were discarded, and residual erythrocytes were lysed in hypotonic buffer to obtain purified neutrophils. The promyelocytic HL-60 (ATCC CCL-240) and acute monoblastic/myelomonocytic leukemia THP-1 (ATCC TIB-202) cell lines were purchased from American Type Culture Collection (Manassas, VA). The acute promyelocytic leukemia NB4 and myelomonoblastic leukemia PLB-985 cell lines were gifts from Dr. Alan Friedman (The Johns Hopkins University, Baltimore, MD, USA) and Dr. Frank DeLeo (Rocky Mountain Laboratories, NIAID, Hamilton, MT, USA), respectively. The neutrophils were maintained and cell lines were grown in RPMI 1640 medium (Hyclone, Thermo Fisher Scientific, Waltham, MA) supplemented with 10% fetal bovine serum (Thermo Fisher Scientific, Waltham, MA) and Glutamax (Life Technologies, Carlsbad, CA). HL-60, NB4, and THP-1 cells were differentiated 5 days prior to infection with 1 μM all-trans retinoic acid (ATRA) (Breitman et al., 1980; Drach et al., 1993; Idres et al., 2001) while PLB-985 cells were differentiated with 1.25% dimethylsulfoxide (DMSO) (Tucker et al., 1987). HSCs purchased from Promocell GmbH (Heidelberg, Germany) were expanded in and differentiated for 14 days prior to infection in hematopoietic progenitor cell expansion medium DXF supplemented with cytokine E mix (Promocell GmbH, Heidelberg, Germany). All cells were grown in a humidified incubator at 37°C with 5% CO2 . Cell density was kept <106 cells/mL by diluting with fresh medium.
A. phagocytophilum isolation and infection
High passage A. phagocytophilum Webster strain was re-activated for virulence by equine passage and re-isolated in HL-60 cells (Davies et al., 2011). Briefly, two A. phagocytophilum seronegative and PCR-negative horses were inoculated with 106 cell-free bacteria. Eight days post inoculation both horses were febrile (103°F and 104°F) and blood containing approximately 106 infected cells was used to inoculate a second set of horses. By day 7 the horses were febrile and blood was obtained for isolation in HL-60 cell culture. These experiments were conducted under an IACUC-approved protocol at the University of California, Davis.
The re-activated A. phagocytophilum was maintained in HL-60 cells as previously described (Goodman et al., 1996). For all experiments A. phagocytophilum was passed less than 10 times in vitro. The GFP- and mCherry-transgenic A. phagocytophilum HGE1 strains (a gift from Dr. Ulrike Munderloh, University of Minnesota, St. Paul, MN, USA) were maintained in HL-60 cells supplemented with 100 μg/mL spectinomycin (Felsheim et al., 2006). To obtain cell-free A. phagocytophilum, 90-95% infected HL-60 cells were centrifuged at 500 g for 5 min. Cells were suspended in 1X PBS and sonicated two times using output 4 of a Branson Sonifier 250 (Branson Ultrasonics, Danbury, CT) for 15 sec. Cell debris was removed by centrifugation at 1,000 g for 10 min. The bacteria-containing supernatant was then centrifuged at 13,000 g for 30 min and bacteria were suspended in RPMI 1640 medium.
A multiplicity of infection (MOI) of 25 bacteria per cell was used for infection following methods modified from Borjesson et al. and Goodman et al. (Borjesson et al., 2005; Lee et al., 2008). Infection levels were determined by microscopic examination of Wright-Giemsa-stained (Protocol HEMA3, Thermo Fisher Scientific, Waltham, MA) cytocentrifuged cells and by real time quantitative PCR (qPCR) at 72 h post-infection for cell lines and HSCs, and 24 h post-infection for neutrophils. qPCR was performed with DNeasy Blood and Tissue Kit (Qiagen, Valencia, CA) extracted DNA and an msp2/ACTB 5’ nuclease assay (Scorpio et al., 2004). A standard curve based on cloned msp2 was generated and used to calculate the number of bacteria per cell. There are >100 copies of msp2 per A. phagocytophilum genome.
Fluorescence activated cell sorting
Prior transcriptional studies in A. phagocytophilum-infected neutrophils used intervals up to 24 h during which bacterial replication (12-24 h doubling time) was likely to be minimal (de la Fuente et al., 2005; Lee et al., 2008; Sukumaran et al., 2005). Despite using conditions applied by others to study differential neutrophil transcription with A. phagocytophilum infection (Borjesson et al., 2005; Lee et al., 2008), morphologic and qPCR methods showed a highly heterogeneous population comprised of small numbers of infected cells and large numbers of apparently uninfected cells. To preclude the confounding variable introduced by using heterogeneously-infected neutrophil cultures, after 24 h infection, 107 neutrophils infected with GFP or mCherry transgenic A. phagocytophilum were washed in RPMI 1640 medium containing 1% FBS, examined by flow cytometry and a homogeneously-infected population was isolated by fluorescence activated cell sorting (FACS) into cell lysis buffer (RNeasy Mini Prep, Qiagen, Valencia, CA) using a MoFlo sorter (Beckman Coulter, Brea, CA). RNA extraction, cDNA synthesis RT-PCR and qPCR were performed as described below. Non-viable cells were excluded by gating and among a population of 5 × 106 viable neutrophils, 104 (0.2%) of the most fluorescent cells were sorted for analysis of transcription (Fig. S2). As a control, unsorted infected neutrophil cultures were also examined.
RNA isolation, Reverse Transcriptase-PCR, and quantitative PCR
RNA was extracted using RNeasy RNA Extraction Kit (Qiagen, Valencia, CA) and cDNA was synthesized using iScript cDNA synthesis kit (Bio-Rad, Hercules, CA) according to manufacturer’s protocols. Reverse transcriptase PCR (RT-PCR) was performed with VeriQuest SYBR Green qPCR Master Mix (Affymetrix, Santa Clara, CA) in a CFX384 Multicycler (Bio-Rad, Hercules, CA) (Table S1). The thermal cycling conditions included 95°C for 3 min followed by 40 cycles of amplification at 95°C for 10 sec and 55°C for 30 sec. A melt curve analysis was performed to confirm amplification of a single product for each primer pair. A panel of 12 reference genes (Table S1) was tested for each cell line to determine which were stably expressed (geNorm M score ≤0.5, Qbase PLUS, Biogazelle, Zwijnaarde, Belgium) with infection and differentiation. Fold change expression of defense genes was calculated using the ΔΔCt method (Livak and Schmittgen, 2001) normalized to selected reference genes and relative to uninfected cells.
Because FACS separated more heavily infected neutrophils directly into RNA cell lysis buffer, we tested DNA and RNA from 24 h A. phagocytophilum-infected HL-60 cells by qPCR and RT-qPCR in the msp2/ACTB 5’ nuclease assay (Scorpio et al., 2004) to determine the ratio of msp2 transcripts to msp2 genes per bacterium. This ratio was then used to quantify the number of bacteria per neutrophil based on msp2 transcript quantitation before and after flow cytometric sorting of infected neutrophils.
Oxidative burst
Production of superoxide anion was measured by incubating 106 cells with 0.25 mM 2′,7′-dichlorohydrofluorescein diacetate (DCFH-DA) in PBS for 30 min at room temperature. 105 cells were then either stimulated with 1 μg/mL phorbol 12-myristate 13-acetate (PMA) or not stimulated and fluorescence was measured every 2 min with a Victor3 1420 Multilabel Counter (PerkinElmer, Norwalk, CT). The relative light units at 2 h in three independent experiments were averaged and compared using a two-sided Student's t-test, α 0.05.
Immunophenotyping of myeloid differentiation
Expression of cell surface markers was used to characterize the differentiation of cell lines when exposed to ATRA, DMSO, and A. phagocytophilum. 105 cells were incubated in 2% FBS in PBS with fluorescently-tagged antibodies (Table S2) for 20 min and then fixed with BD FACS Lysing solution (Becton Dickinson, Franklin Lakes, NJ) for 10 min. Cells were washed and resuspended in 2% FBS in PBS. 10,000 events were counted on a BD Fortessa flow cytometer (Becton Dickinson, Franklin Lakes, NJ). Cells were first gated and separated by viability as determined by Live/Dead staining (Invitrogen, Grand Island, NY).The fluorescence minus one (FMO) method was used to determine standard gates for each marker. Expression of surface markers CD34 (hematopoietic progenitor cell antigen, expressed on myeloblasts through promyelocytes), CD66b (CEACAM8, expressed on promyelocytes through neutrophils), CD11b (ITGAM, expressed on myelocytes through neutrophils), CD16 (FCGR3, expressed on metamyelocytes through neutrophils), CD35 (complement receptor [CR] 1, expressed on band neutrophils through neutrophils), and CD10 (neprilysin, expressed on mature neutrophils) were examined to determine the degree of differentiation (Fig. 4A). CD162 (selectin P ligand) was also investigated because of its role as attachment ligand and in invasion of A. phagocytophilum (Goodman et al., 1999; Herron et al., 2000).
Figure 4. A. phagocytophilum alters cellular immunophenotype.

(A) Schematic diagram of the range over which granulocyte immunophenotypic surface markers are expressed with differentiation. In this panel, green represents less differentiation and red represents greater differentiation. (B) The expression of selected cellular markers (CD34, CD66b, CD11b, CD16, CD35 and CD10) of myeloid differentiation were measured using flow cytometry to determine differentiation state of uninfected and infected cellular models relative to ex vivo peripheral blood human neutrophils. The geometric mean of three independent experiments was averaged for each immunophenotypic marker. The total fluorescence for a marker within the individual cell lines was calculated and used to determine the proportional expression of each marker for a particular cell line. The proportional expression was then weighted (−1 for CD34 , +1 for CD66b, +2 for CD11b, +3 for CD16, +4 CD35, and +5 for CD10). The overall differentiation status for each cell line and condition was determined by the total of the weighted scores for each marker. Undiff = undifferentiated, Diff = differentiated; Aph – A. phagocytophilum infected, mock – mock infected. The weighted scores were ranked and the Wilcoxon Rank-Sum test performed. (C) The CD162 geometric mean fluorescence of three independent experiments was averaged for each condition (cell line, differentiation status and time point). The proportion of CD162 expression was calculated within a cell line and ranked. Green bars – PLB-985 cells; blue bars – THP-1 cells; orange bars – HL-60 cells; brown bars – HSCs; pink/red bars – neutrophils (PMNs or polymorphonuclear neutrophils).
The geometric mean of three independent experiments was calculated and averaged for each marker per cell type, differentiation status, and time point. Mean fluorescence intensity was normalized across each cell type for each surface marker to allow comparisons within cell lines. For example, the geometric mean fluorescence of CD34 for all PMN samples was summed, and the proportion of CD34 expression for each condition (differentiated, not differentiated, infected, uninfected at 3, 24, and 48 h) was calculated based on the total sum fluorescence of that marker in PMNs. This was repeated for each marker within each cell line. The proportion of expression for each marker was then weighted based on the specific differentiation marker, such that those markers reflecting a less differentiated phenotype received a negative weight (-1 for CD34) or no weight (+1 for CD66b), and markers associated with an increasingly neutrophil-like differentiation were weighted more heavily (+2 for CD11b, +3 for CD16, +4 for CD35, and +5 for CD10). The weighted scores were summed for each cell type to score the relative degree of differentiation across the variables (differentiated, not differentiated, infected, uninfected, and time post-infection if applicable). Since the final scores were not normally distributed, they were ranked for nonparametric analysis. The most differentiated cells received the highest rank (22 out of 22) and the least differentiated the lowest rank (1 out of 22). CD162 surface expression was analyzed separately from the differentiation markers. Similarly, the proportion of CD162 expression for each condition was ranked and cells with highest proportion received the highest rank. Ranked scores were compared using a two-sided Mann-Whitney test, with α=0.05.
Results
Anaplasma phagocytophilum infection level differs among granulocyte models
Peripheral blood neutrophils infected by A. phagocytophilum in vivo, such as in humans, horses, or mouse models, generally develop single morulae containing approximately 10 bacterial profiles (Popov et al., 1998). Thus, the degree to which each cell line became infected with A. phagocytophilum was first determined. Anaplasma phagocytophilum re-activated for virulence by passage in horses was used for in vitro infection to preclude the effects of bacterial adaption to HL-60 cells with prolonged passage. Neutrophils as well as undifferentiated and differentiated cell lines were infected with an MOI of 25 bacteria per cell. At 24 and 48 h post-infection for neutrophils and 72 h post-infection for cell lines, cells were stained with Wright-Giemsa and qPCR was performed on DNA from 105 cells. Neutrophil infection was assessed at 3, 24 and 48 h because the cells became apoptotic after 48 h post-infection; uninfected neutrophils were apoptotic at 24 h. Cell lines were assessed at 72 h to obtain homogeneous infections. Despite using methods for infection as described in other neutrophil transcription studies, we found that by 48 h, neutrophils were infected with an average of less than 1 bacterium per cell (Fig. 1) as detected by qPCR. Wright-Giemsa stains showed a heterogeneous mixture that included only a very small number of infected but viable cells and a large proportion of uninfected apoptotic neutrophils (Fig. S1A), in line with our and other previous reports (Choi et al., 2005; Ge and Rikihisa, 2006; Lee and Goodman, 2006; Scaife et al., 2003; Yoshiie et al., 2000). After sorting the most fluorescent 0.2% of cells, the mean was 40.4 (± 6.0 s.e.m.) A. phagocytophilum/neutrophil.
Figure 1. A. phagocytophilum infects granulocytes at differing efficiencies.
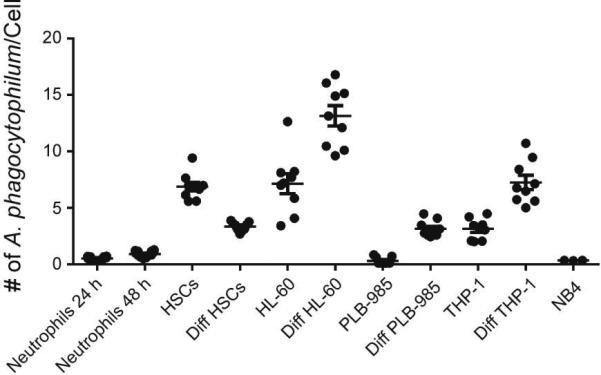
DNA was extracted from cells at 24 and 48 h for PMNs and 72 h for other cells. The DNA was analyzed by qPCR for the presence of A. phagocytophilum infection with msp2/B-actin 5’ nuclease assay and the number of A. phagocytophilum per cell was determined. Infection was performed in quadruplicate for neutrophils and triplicate for remaining cells. The mean ± SEM for 24 and 48 h infected neutrophils and 72 h time point for all other cell lines is displayed.
All cell lines differentiated with ATRA or DMSO and HSCs differentiated as per protocol developed morphology that was more neutrophil-like with multi-lobed nuclei and increased cytoplasmic granules as compared to undifferentiated cells (Fig. S1B). Differentiated HL-60 cells had the highest infection burden with approximately 12 bacteria per cell (Fig. 1 and S1B), similar to that anticipated with infection in vivo. Undifferentiated PLB-985 and NB4 cells had the lowest number of bacteria per cell, and in some cases, infection was undetectable at 72 h (Fig. 1 and S1B). Because infection could not be consistently detected in NB4 cells, analysis of this cell line was not pursued.
Transcription of cellular defense genes is altered with infection
Anaplasma phagocytophilum infection of granulocytes and neutrophils results in differential downregulation and upregulation of host genes. A number of defense genes are downregulated in A. phagocytophilum-infected THP-1 and HL-60 cells (Carlyon et al., 2002; Garcia-Garcia et al., 2009a) and neutrophils (Borjesson et al., 2005; Lee et al., 2008). In addition, chemokine gene expression is increased, and we used IL8 as a monitor for upregulated gene expression. A panel of these genes was analyzed for expression in infected neutrophils, HSCs and the 3 cell lines (Fig. 2) to determine similarities of responses in cell lines vs. infected neutrophils. After fluorescence sorting to examine homogeneously-infected neutrophils, the defense gene expression profile was different than observed in the unsorted neutrophils, but consistent with that observed with differentiated HL-60 and HSCs (Fig. 2 B-D). Unsorted, heterogeneously-infected neutrophils, as observed in other studies, showed increased expression of CYBB (gp91phox), ELA (elastase) and MPO (myeloperoxidase) (Fig. 2A), consistent with cellular activation of host defense pathways. In contrast, all 7 defense genes downregulated in granulocyte cell models of A. phagocytophilum infection were found repressed in the sorted homogeneously-infected population of neutrophils (Fig. 2B).
Figure 2. Defense gene transcription is altered with infection.
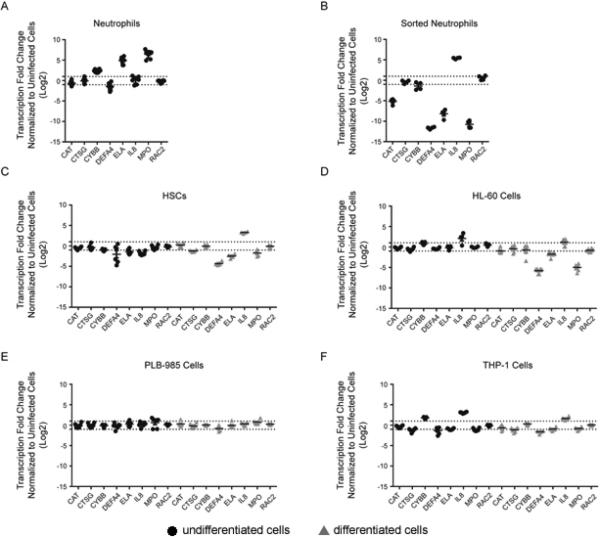
Transcription of defense genes CAT, CTSG, CYBB, DEFA4, ELA2, IL8, MPO, and RAC2 was assessed in infected cells relative to uninfected cells (fold change shown on y-axis). Infected neutrophils (A) and sorted mCherry-expressing A. phagocytophilum-infected neutrophils (B) were normalized to the reference genes B2M, GNB2LI, SDHA and fold change in transcription at 24 h (●) post-infection was determined relative to 3 h uninfected neutrophils (since uninfected neutrophils are apoptotic at 24 h ex vivo). HSCs (C) were normalized using GNB2LI and RPL32 as reference genes. HL-60 cells (D) were normalized with GNB2LI and RPL13A. PLB-985 cells (E) were normalized with the reference genes HMBS and HPRT1. THP-1 cells (F) were normalized using ACTB and SDHA. 72 h time points are displayed for the HSC, HL-60, PLB-985 and THP-1 cells for both undifferentiated (●) and differentiated (▲) cells. Infections were repeated in triplicate with technical duplicate readings for RT-PCR. Mean ± SEM is plotted. The dotted lines delimit the upper and lower two-fold changes in gene expression.
Of the cell lines, differentiated HL-60 cells, which were the most heavily and homogeneously infected without sorting, had the greatest number of defense genes downregulated (6 of 7) (Fig. 2D). Differentiated HSCs showed a similar pattern to HL-60 cells and neutrophils (Fig. 2C). Interestingly, not all genes previously shown to be downregulated in THP-1 cells had decreased transcript levels (Fig. 2D), possibly due to heterogeneity of infection or variations in transcriptional responses that accrued with continued passage of this cell line. For this study, we selected examination at 72 h post-infection for all cells lines and not when >95% of cells were infected, which could have biased toward HL-60 cells that are typically used for propagation, despite the use of low passage bacteria. Undifferentiated and differentiated PLB-985 cells had no significant changes in gene expression with infection (Fig. 2E), in accordance with the low or absent bacterial loads.
A. phagocytophilum decreases intracellular oxidative burst
Suppression of superoxide production is critical for A. phagocytophilum survival in HL-60 cells after the first 3-5 hours of infection (Banerjee et al., 2000; Carlyon et al., 2002), and is a hallmark of infected human and murine neutrophils in vivo (Wang et al., 2002). Intracellular levels of superoxide anion were measured in response to the strong stimulant PMA in the presence and absence of A. phagocytophilum (Fig. 3). With differentiation, all cells increased production of superoxide anion in response to PMA except PLB-985 cells (Fig.3B). Neutrophils, HSCs, and undifferentiated and differentiated HL-60 cells all decreased production of superoxide anion when infected with A. phagocytophilum. Infection did not decrease superoxide anion generation in differentiated HSCs, or in undifferentiated and differentiated PLB-985 and THP-1 cells (Fig. 3).
Figure 3. ranulocyte oxidative burst is decreased with A. phagocytophilum infection.

Intracellular levels of reactive oxygen species in response to the stimulant PMA were detected using DCFH-DA 72 h post-infection for cell lines and HSCs and 24 h post-infection for neutrophils. The relative fluorescence at the 2 h time point was plotted for undifferentiated (A) and differentiated (B) cells with (Aph) and without infection. Infections were repeated in triplicate and each sample was read in duplicate. Mean ± SEM is plotted.
A. phagocytophilum infection does not affect cellular differentiation
Because the functions of A. phagocytophilum-infected neutrophils resemble those of less differentiated myeloid cells (e.g. decreased i] oxidative burst, ii] apoptosis, iii] margination, iv] emigration, and v] phagocytosis) (Choi et al., 2004; Garyu et al., 2005; Mott et al., 2002; Scaife et al., 2003), we examined the degree and effect of infection on differentiation by immunophenotype (Fig. 4B). As expected, neutrophils were most differentiated and varied considerably from all cell lines tested (p<0.001; Mann-Whitney test). Anaplasma phagocytophilum-infected cells did not show a significant change in differentiation compared to uninfected cells (p=0.27) suggesting that the bacterium does not target cellular differentiation or de-differentiation programs but is directed at specific host cell programs that impact functional changes. However, of 11 pairs of infected and uninfected cells, 9 infected samples were more differentiated than their uninfected counterparts. Of the infected/uninfected pairs, HSCs were markedly more differentiated when infected with A. phagocytophilum and only undifferentiated PLB-985 and undifferentiated HL-60 cells appeared less differentiated after infection.
Anaplasma phagocytophilum binds to CD162 to facilitate uptake (Goodman et al., 1999; Herron et al., 2000) and infection-induced degranulation leads to shedding of surface CD162 (Choi et al., 2003). Differentiated HL-60 and HSC cells had the highest proportional increases in cells expressing CD162 with differentiation and HL-60 cells also demonstrated modest CD162 loss with infection (Fig. 4C). However, no overall significant differences in CD162 expression were detected when comparing infected and uninfected cells or neutrophils (p=0.224; Wilcoxon test). As anticipated, ex vivo neutrophils rapidly lost surface CD162 expression over 48 h, greater for uninfected cells probably because of apoptosis.
Discussion
Anaplasma phagocytophilum is a unique bacterium, in part because of its small genome and its obligate intracellular niche within the chief mammalian host defense cell, the neutrophil. In addition, mounting evidence shows that A. phagocytophilum evolved multiple mechanisms to subvert host cell functions as fitness advantages for survival and transmission. This includes the delivery of type IV secretion system effectors that not only mediate direct protein-protein disturbances of signaling but also bind host DNA, alter chromatin structure, and modify transcriptional programs governing host cell functions (Garcia-Garcia et al., 2009a; Lin et al., 2007). Advances in understanding the mechanisms by which A. phagocytophilum alters functional programs via signaling or transcriptional regulation is best accomplished using their natural mammalian target, neutrophils. Since experimental manipulation of neutrophils ex vivo is currently challenging, cell models that closely parallel neutrophil functions and that are validated to share underlying transcriptional and signaling programs would be beneficial. Currently, some accepted transcriptional responses proven important for A. phagocytophilum survival and transmission in cell lines have not been replicated in neutrophil transcriptome studies (Borjesson et al., 2005; Lee et al., 2008). Because of these discrepancies, we sought to compare several existing and potentially useful granulocyte cell line models to determine which most closely mimics the functional, phenotypic, and transcriptional changes observed in neutrophils infected with A. phagocytophilum.
Higher levels of infection in all cell models including neutrophils, was clearly the major determinant of transcriptional and functional changes observed with infection. This was especially evident in differentiated HL-60 cells which were the most heavily and homogeneously infected and concurrently had the most dramatic reductions in defense gene transcription and oxidative burst. This is in agreement with our prior studies and the results of other investigators (Banerjee et al., 2000; de la Fuente et al., 2005; Garcia-Garcia et al., 2009a). In contrast, PLB-985 and THP-1 cells had low levels of infection and minimal change in function and defense gene expression, while NB4 cells never became sufficiently infected and were not further studied. In concert with these observations, the infection level in neutrophils also clearly influenced transcriptional changes as we found defense gene transcription was dramatically different in a homogeneously-infected neutrophil population with an average of 40 bacteria/cell as compared to unsorted heterogeneously-infected group with <3 bacteria/cell on average. In the heterogeneous neutrophil populations, the response of the more highly infected neutrophils was masked by the much larger population of uninfected neutrophils precluding the ability to observe the true response of ex vivo neutrophils to A. phagocytophilum. This observation in particular has important implications for studies of A. phagocytophilum conducted by infecting peripheral blood neutrophils ex vivo when responses are analyzed within 12 or 24 hours post-infection. Hypothetically, the data suggest that a higher intracellular bacterial load results in the delivery of more effector molecules into the host cell to impact specific signaling pathways or transcriptional programs governing neutrophil functions.
Among the A. phagocytophilum infected cell models with homogeneous and high level infections, the most dramatic functional changes in antimicrobial responses, such as the lack of superoxide generation, are phenotypic features of immature progenitor cells. Given the relationship between cellular bacterial load and transcriptional and functional alterations, we investigated the effect of differentiation on bacterial load, transcription, and respiratory burst for each cell line and HSCs to determine if A. phagocytophilum de-differentiates cells or reprograms specific cellular functions. The cellular immunophenotypes selected generally reflect maturation of neutrophil antimicrobial response and expression of ligands to detect microbe-associated molecules. The findings here suggest that infection is either neutral or minimally accelerates differentiation and maturation in cell lines and neutrophils. Surprisingly, PLB-985 cells were the least differentiated, and in addition to low bacterial load, this could also explain the absence of transcriptional and functional changes with infection. Furthermore, differentiated HL-60 and HSCs, like neutrophils, had the highest proportions of surface CD162, a key granulocyte adhesion molecule for A. phagocytophilum binding and invasion (Goodman et al., 1999; Herron et al., 2000), suggesting that its sustained expression allows a higher proportion of cells to become infected. Thus, the mechanism(s) that A. phagocytophilum uses to reprogram granulocyte function are not analogous to de-differentiation as might be observed with reprogramming of differentiated cells into induced pluripotent stem cells (iPSCs). Instead, these data indicate neutrophil/granulocyte reprogramming that results in differentially-regulated gene expression patterns evolved to target key cellular functions that improve microbial survival, propagation, and transmission.
A single cell in vivo that loses antimicrobial activity is unlikely to have much impact on disease. However, the loss of antimicrobial activity and the delayed apoptosis that occur with A. phagocytophilum could permit the infected cell to survive for several rounds of bacterial replication before disseminating to other susceptible granulocytes, which could be acquired in a subsequent tick blood meal. The proinflammatory response, including IL-8 and other upregulated chemokine/cytokine genes, clearly benefits bacterial population expansion by recruiting new host cells (Scorpio et al., 2004). Along with neutrophil degranulation, the proinflammatory response could have significant ancillary impacts on tissue injury and disease since these responses are amplified locally, as seen with the inflammatory lesions in tissues of mice, horses and humans (Lepidi et al., 2000). In fact, disease with A. phagocytophilum infection is more closely linked to the overall host inflammatory and immune response to the bacterium than to the direct impact of infection on neutrophil function (Choi et al., 2007; Martin et al., 2001). The cumulative effect whereby a median of 1% of circulating human neutrophils are infected, or about 1.7 × 108cells (1.7 × 109 bacteria) in a 70 kg human, could be dramatic.
How A. phagocytophilum alters antimicrobial responses, promotes self-survival and replication to be transmitted from reservoir hosts and to ultimately cause disease in humans will facilitate an improved understanding of microbial control of infected host cells, mechanisms to reduce disease severity, or could expand knowledge of how to control or contain undesired granulocyte responses in inflammatory diseases. Until better tractability of ex vivo neutrophils is achieved, susceptible, flexible, robust cell models that closely mimic neutrophils in function and transcriptional program responses are needed. The data here provide firm evidence that A. phagocytophilum infection studies designed to interrogate cellular functions in ex vivo neutrophils are flawed without approaches that achieve homogeneous infection at levels similar to those observed in vivo. These data also demonstrate that the well-described HL-60 cell infection model mimics and provides many desirable attributes by comparison. Moreover, it is now clear that the complex functional changes that occur in neutrophils or granulocyte cells infected by A. phagocytophilum are not the result of a de-differentiation program as seen with iPSCs. Rather, these changes occur from targeted reprogramming of functions that affect specific transcriptional and/or signaling pathways. This ultimately benefits microbial survival and transmission, but can concurrently generate inflammatory injury and disease.
Supplementary Material
Acknowledgments
This research was supported by research grant R01-AI44102 from the National Institute of Allergy and Infectious Diseases to J.S.D. We thank Dennis J. Grab, The Johns Hopkins University School of Medicine, for critical discussions and reading of the manuscript. Alan Friedman, The Johns Hopkins University School of Medicine, and Frank DeLeo, Rocky Mountain Laboratories, NIAID, kindly provided the NB4 and PLB-985 cell lines, respectively. Ulrike Munderloh of the University of Minnesota provided mCherry and GFP transgenic A. phagocytophilum. Hao Zhang and Joseph Margolick at the Johns Hopkins Bloomberg School of Public Health provided fluorescence activated cell sorting services. Janet Foley and John Madigan at the University of California, Davis for re-activating A. phagocytophilum Webster strain virulence by passage in horses. Cindy Chen, The Johns Hopkins University School of Medicine, assisted with oxidative burst assays.
Footnotes
Author Contributions
K.E.R.-B. designed and did most of the experiments and prepared the manuscript. S.H.S performed immunophenotype experiments. M.A.L. assisted with culturing and RT-PCR analysis. J.S.D. helped analyze data and interpret results and supervised all studies. All authors contributed to discussions and manuscript preparation.
References
- Akkoyunlu M, Malawista SE, Anguita J, Fikrig E. Exploitation of interleukin-8-induced neutrophil chemotaxis by the agent of human granulocytic ehrlichiosis. Infect. Immun. 2001;69:5577–5588. doi: 10.1128/IAI.69.9.5577-5588.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee R, Anguita J, Roos D, Fikrig E. Cutting edge: infection by the agent of human granulocytic ehrlichiosis prevents the respiratory burst by down-regulating gp91phox. J. Immunol. 2000;164:3946–3949. doi: 10.4049/jimmunol.164.8.3946. [DOI] [PubMed] [Google Scholar]
- Borjesson DL, Kobayashi SD, Whitney AR, Voyich JM, Argue CM, Deleo FR. Insights into pathogen immune evasion mechanisms: Anaplasma phagocytophilum fails to induce an apoptosis differentiation program in human neutrophils. J. Immunol. 2005;174:6364–6372. doi: 10.4049/jimmunol.174.10.6364. [DOI] [PubMed] [Google Scholar]
- Breitman TR, Selonick SE, Collins SJ. Induction of differentiation of the human promyelocytic leukemia cell line (HL-60) by retinoic acid. Proc. Natl. Acad. Sci. U. S. A. 1980;77:2936–2940. doi: 10.1073/pnas.77.5.2936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlyon JA, Abdel-Latif D, Pypaert M, Lacy P, Fikrig E. Anaplasma phagocytophilum utilizes multiple host evasion mechanisms to thwart NADPH oxidase-mediated killing during neutrophil infection. Infect. Immun. 2004;72:4772–4783. doi: 10.1128/IAI.72.8.4772-4783.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlyon JA, Chan WT, Galan J, Roos D, Fikrig E. Repression of RAC2 mRNA expression by Anaplasma phagocytophila is essential to the inhibition of superoxide production and bacterial proliferation. J. Immunol. 2002;169:7009–7018. doi: 10.4049/jimmunol.169.12.7009. [DOI] [PubMed] [Google Scholar]
- Choi KS, Dumler JS. Early induction and late abrogation of respiratory burst in A. phagocytophilum-infected neutrophils. Ann. N. Y. Acad. Sci. 2003;990:488–493. doi: 10.1111/j.1749-6632.2003.tb07415.x. [DOI] [PubMed] [Google Scholar]
- Choi KS, Garyu J, Park J, Dumler JS. Diminished adhesion of Anaplasma phagocytophilum-infected neutrophils to endothelial cells is associated with reduced expression of leukocyte surface selectin. Infect. Immun. 2003;71:4586–4594. doi: 10.1128/IAI.71.8.4586-4594.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi KS, Grab DJ, Dumler JS. Anaplasma phagocytophilum infection induces protracted neutrophil degranulation. Infect. Immun. 2004;72:3680–3683. doi: 10.1128/IAI.72.6.3680-3683.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi KS, Park JT, Dumler JS. Anaplasma phagocytophilum delay of neutrophil apoptosis through the p38 mitogen-activated protein kinase signal pathway. Infect. Immun. 2005;73:8209–8218. doi: 10.1128/IAI.73.12.8209-8218.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi KS, Webb T, Oelke M, Scorpio DG, Dumler JS. Differential innate immune cell activation and proinflammatory response in Anaplasma phagocytophilum infection. Infect. Immun. 2007;75:3124–3130. doi: 10.1128/IAI.00098-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies RS, Madigan JE, Hodzic E, Borjesson DL, Dumler JS. Dexamethasone-induced cytokine changes associated with diminished disease severity in horses infected with Anaplasma phagocytophilum. Clin. Vaccine Immunol. 2011;18:1962–1968. doi: 10.1128/CVI.05034-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Fuente J, Ayoubi P, Blouin EF, Almazan C, Naranjo V, Kocan KM. Gene expression profiling of human promyelocytic cells in response to infection with Anaplasma phagocytophilum. Cell. Microbiol. 2005;7:549–559. doi: 10.1111/j.1462-5822.2004.00485.x. [DOI] [PubMed] [Google Scholar]
- Drach J, Lopez-Berestein G, McQueen T, Andreeff M, Mehta K. Induction of differentiation in myeloid leukemia cell lines and acute promyelocytic leukemia cells by liposomal all-trans-retinoic acid. Cancer Res. 1993;53:2100–2104. [PubMed] [Google Scholar]
- Felsheim RF, Herron MJ, Nelson CM, Burkhardt NY, Barbet AF, Kurtti TJ, Munderloh UG. Transformation of Anaplasma phagocytophilum. BMC Biotechnol. 2006;6:42. doi: 10.1186/1472-6750-6-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Garcia JC, Barat NC, Trembley SJ, Dumler JS. Epigenetic silencing of host cell defense genes enhances intracellular survival of the rickettsial pathogen Anaplasma phagocytophilum. PLoS Pathog. 2009a;5:e1000488. doi: 10.1371/journal.ppat.1000488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Garcia JC, Rennoll-Bankert KE, Pelly S, Milstone AM, Dumler JS. Silencing of host cell CYBB gene expression by the nuclear effector AnkA of the intracellular pathogen Anaplasma phagocytophilum. Infect. Immun. 2009b;77:2385–2391. doi: 10.1128/IAI.00023-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garyu JW, Choi KS, Grab DJ, Dumler JS. Defective phagocytosis in Anaplasma phagocytophilum-infected neutrophils. Infect. Immun. 2005;73:1187–1190. doi: 10.1128/IAI.73.2.1187-1190.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge Y, Rikihisa Y. Anaplasma phagocytophilum delays spontaneous human neutrophil apoptosis by modulation of multiple apoptotic pathways. Cell. Microbiol. 2006;8:1406–1416. doi: 10.1111/j.1462-5822.2006.00720.x. [DOI] [PubMed] [Google Scholar]
- Goodman JL, Nelson C, Vitale B, Madigan JE, Dumler JS, Kurtti TJ, Munderloh UG. Direct cultivation of the causative agent of human granulocytic ehrlichiosis. N. Engl. J. Med. 1996;334:209–215. doi: 10.1056/NEJM199601253340401. [DOI] [PubMed] [Google Scholar]
- Goodman JL, Nelson CM, Klein MB, Hayes SF, Weston BW. Leukocyte infection by the granulocytic ehrlichiosis agent is linked to expression of a selectin ligand. J. Clin. Invest. 1999;103:407–412. doi: 10.1172/JCI4230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herron MJ, Nelson CM, Larson J, Snapp KR, Kansas GS, Goodman JL. Intracellular parasitism by the human granulocytic ehrlichiosis bacterium through the P-selectin ligand, PSGL-1. Science. 2000;288:1653–1656. doi: 10.1126/science.288.5471.1653. [DOI] [PubMed] [Google Scholar]
- Idres N, Benoit G, Flexor MA, Lanotte M, Chabot GG. Granulocytic differentiation of human NB4 promyelocytic leukemia cells induced by all-trans retinoic acid metabolites. Cancer Res. 2001;61:700–705. [PubMed] [Google Scholar]
- IJdo JW, Mueller AC. Neutrophil NADPH oxidase is reduced at the Anaplasma phagocytophilum phagosome. Infect. Immun. 2004;72:5392–5401. doi: 10.1128/IAI.72.9.5392-5401.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein MB, Hu S, Chao CC, Goodman JL. The agent of human granulocytic ehrlichiosis induces the production of myelosuppressing chemokines without induction of proinflammatory cytokines. J. Infect. Dis. 2000;182:200–205. doi: 10.1086/315641. [DOI] [PubMed] [Google Scholar]
- Lee HC, Goodman JL. Anaplasma phagocytophilum causes global induction of antiapoptosis in human neutrophils. Genomics. 2006;88:496–503. doi: 10.1016/j.ygeno.2006.06.002. [DOI] [PubMed] [Google Scholar]
- Lee HC, Kioi M, Han J, Puri RK, Goodman JL. Anaplasma phagocytophilum-induced gene expression in both human neutrophils and HL-60 cells. Genomics. 2008;92:144–151. doi: 10.1016/j.ygeno.2008.05.005. [DOI] [PubMed] [Google Scholar]
- Lepidi H, Bunnell JE, Martin ME, Madigan JE, Stuen S, Dumler JS. Comparative pathology, and immunohistology associated with clinical illness after Ehrlichia phagocytophila-group infections. Am. J. Trop. Med. Hyg. 2000;62:29–37. doi: 10.4269/ajtmh.2000.62.29. [DOI] [PubMed] [Google Scholar]
- Lin M, den Dulk-Ras A, Hooykaas PJ, Rikihisa Y. Anaplasma phagocytophilum AnkA secreted by type IV secretion system is tyrosine phosphorylated by Abl-1 to facilitate infection. Cell. Microbiol. 2007;9:2644–2657. doi: 10.1111/j.1462-5822.2007.00985.x. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Martin ME, Caspersen K, Dumler JS. Immunopathology and ehrlichial propagation are regulated by interferon-gamma and interleukin-10 in a murine model of human granulocytic ehrlichiosis. Am. J. Pathol. 2001;158:1881–1888. doi: 10.1016/s0002-9440(10)64145-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mott J, Rikihisa Y, Tsunawaki S. Effects of Anaplasma phagocytophila on NADPH oxidase components in human neutrophils and HL-60 cells. Infect. Immun. 2002;70:1359–1366. doi: 10.1128/IAI.70.3.1359-1366.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedra JH, Sukumaran B, Carlyon JA, Berliner N, Fikrig E. Modulation of NB4 promyelocytic leukemic cell machinery by Anaplasma phagocytophilum. Genomics. 2005;86:365–377. doi: 10.1016/j.ygeno.2005.05.008. [DOI] [PubMed] [Google Scholar]
- Popov VL, Han VC, Chen SM, Dumler JS, Feng HM, Andreadis TG, Tesh RB, Walker DH. Ultrastructural differentiation of the genogroups in the genus Ehrlichia. J. Med. Microbiol. 1998;47:235–251. doi: 10.1099/00222615-47-3-235. [DOI] [PubMed] [Google Scholar]
- Scaife H, Woldehiwet Z, Hart CA, Edwards SW. Anaplasma phagocytophilum reduces neutrophil apoptosis in vivo. Infect. Immun. 2003;71:1995–2001. doi: 10.1128/IAI.71.4.1995-2001.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scorpio DG, Akkoyunlu M, Fikrig E, Dumler JS. CXCR2 blockade influences Anaplasma phagocytophilum propagation but not histopathology in the mouse model of human granulocytic anaplasmosis. Clin. Diagn. Lab. Immunol. 2004;11:963–968. doi: 10.1128/CDLI.11.5.963-968.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukumaran B, Carlyon JA, Cai JL, Berliner N, Fikrig E. Early transcriptional response of human neutrophils to Anaplasma phagocytophilum infection. Infect. Immun. 2005;73:8089–8099. doi: 10.1128/IAI.73.12.8089-8099.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas V, Samanta S, Wu C, Berliner N, Fikrig E. Anaplasma phagocytophilum modulates gp91phox gene expression through altered interferon regulatory factor 1 and PU.1 levels and binding of CCAAT displacement protein. Infect. Immun. 2005;73:208–218. doi: 10.1128/IAI.73.1.208-218.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker KA, Lilly MB, Heck L, Jr, Rado TA. Characterization of a new human diploid myeloid leukemia cell line (PLB-985) with granulocytic and monocytic differentiating capacity. Blood. 1987;70:372–378. [PubMed] [Google Scholar]
- Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002;3:RESEARCH0034. doi: 10.1186/gb-2002-3-7-research0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Malawista SE, Pal U, Grey M, Meek J, Akkoyunlu M, Thomas V, Fikrig E. Superoxide anion production during Anaplasma phagocytophila infection. J. Infect. Dis. 2002;186:274–280. doi: 10.1086/341451. [DOI] [PubMed] [Google Scholar]
- Yoshiie K, Kim HY, Mott J, Rikihisa Y. Intracellular infection by the human granulocytic ehrlichiosis agent inhibits human neutrophil apoptosis. Infect. Immun. 2000;68:1125–1133. doi: 10.1128/iai.68.3.1125-1133.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Ding L, Sandford AJ. Selection of reference genes for gene expression studies in human neutrophils by real-time PCR. BMC Mol. Biol. 2005;6:4. doi: 10.1186/1471-2199-6-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.