

Abstract
It has been recognized for some time that the Ca2+-dependent slow afterhyperpolarization (sAHP) is larger in hippocampal neurons of aged compared with young animals. In addition, extensive studies since have shown that other Ca2+-mediated electrophysiological responses are increased in hippocampus with aging, including Ca2+ transients, L-type voltage-gated Ca2+ channel activity, Ca2+ spike duration and action potential accommodation. Elevated Ca2+-induced Ca2+ release from ryanodine receptors (RyRs) appears to drive amplification of the Ca2+ responses. Components of this Ca2+ dysregulation phenotype correlate with deficits in cognitive function and plasticity, indicating they may play critical roles in aging-related impairment of brain function. However, the molecular mechanisms underlying aging-related Ca2+ dysregulation are not well understood.
FK506-binding proteins 1a and 1b (FKBP1a/1b, also known as FKBP12/12.6) are immunophilin proteins that bind the immunosuppressant drugs FK506 and rapamycin. In muscle cells, FKBP1a/1b also bind RyRs and inhibits Ca2+-induced Ca2+ release, but it is not clear whether FKBPs act similarly in brain cells. Recently, we found that selectively disrupting hippocampal FKBP1b function in young rats, either by microinjecting adeno-associated viral vectors containing siRNA, or by treatment with rapamycin, increases the sAHP and recapitulates much of the hippocampal Ca2+ dysregulation phenotype. Moreover, in microarray studies, we found FKBP1b gene expression was downregulated in hippocampus of aging rats and early-stage Alzheimer’s disease subjects. These results suggest the novel hypothesis that declining FKBP function is a key factor in aging-related Ca2+ dysregulation in the brain and point to potential new therapeutic targets for counteracting unhealthy brain aging.
Keywords: Calcium, Ryanodine Receptor, Aging, FKBP1b
1. Introduction
Recent estimates project that 20% of the population of Western countries will be over 65 years of age by 2030, up from just 10% in the year 2000. Further, such changes in the aging population represent a global trend (Lutz et al., 2008). Because Alzheimer’s disease (AD) incidence rises rapidly after age 65, the World Health Organization estimates that 65 million people worldwide will have AD by 2030 and 115 million by 2050. Therefore, it is clear that without therapeutic interventions to slow, reverse or prevent progression of unhealthy brain aging and AD, society will face enormous burdens in caring for the cognitively-dysfunctional elderly. Nevertheless, development of disease modifying interventions will very likely require greater understanding of molecular mechanisms underlying unhealthy brain aging than is yet available. The present article reviews several converging lines of evidence that indicate an important role for neuronal Ca2+ dysregulation in unhealthy hippocampal aging, and highlights recent findings that reveal a potential novel molecular mechanism underlying this Ca2+ dysregulation.
2. Neuronal calcium (Ca2+) dysregulation with aging
2.1. Ca2+ dysregulation in hippocampal neurons and unhealthy brain aging
It has been recognized for some time that the Ca2+-dependent slow afterhyperpolarization (sAHP) that follows a burst of action potentials in hippocampal CA1 pyramidal neurons is larger in aged compared to young-adult animals (Deyo et al., 1989; Disterhoft et al., 1996; Kerr et al., 1989; Landfield and Pitler, 1984; Moyer et al., 1992; Potier et al., 1993). The sAHP is generated by Ca2+ influx via voltage-gated Ca2+ channels (VGCCs) that activates hyperpolarizing K+ currents, which in turn dampen postsynaptic excitability. In addition to the larger sAHP, multiple other Ca2+-related electrophysiological processes have since been shown to be increased in hippocampal pyramidal neurons of aged animals, including Ca2+ action potential duration, L-type VGCC (L-VGCC) activity, voltage-activated Ca2+ transients, long-term depression and action potential accommodation (Brewer et al., 2009; Disterhoft et al., 1996; Foster and Norris, 1997; Moyer et al., 1992; Pitler and Landfield, 1990; Potier et al., 1993; Thibault et al., 2001; Thibault and Landfield, 1996). Importantly, several of these enhanced Ca2+-related functions are correlated with impairment of learning or synaptic plasticity (Disterhoft and Oh, 2007; Disterhoft et al., 1996; Kumar and Foster, 2004; Thibault et al., 2001; Thibault and Landfield, 1996; Tombaugh et al., 2005). Together, these Ca2+-related biomarkers of aging appear to comprise a consistent Ca2+ dysregulation phenotype in hippocampal CA1 pyramidal neurons of aging animals. Interestingly, CA1 pyramidal neurons are also among the brain neurons most vulnerable to degeneration induced by AD, ischemia and other pathological conditions (Morrison and Hof, 2002; Wang et al., 2010).
2.2 Ca2+-induced Ca2+ release (CICR) from ryanodine receptors (RyRs) on the endoplasmic reticulum (ER)
While Ca2+ influx from increased L-VGCC activity apparently is an important source of Ca2+ driving hippocampal Ca2+ dysregulation, another major source of elevated Ca2+ appears to be enhanced Ca2+-induced Ca2+ release (CICR) from intracellular Ca2+ stores. CICR is mediated by Ca2+ sensing ryanodine receptors (RyRs) on the ER that release additional Ca2+ in response to Ca2+ influx via plasmalemmal channels. It is well-established that RyRs operate downstream of and in series with L-VGCCs in mediating CICR. L-VGCCs and RyRs are closely juxtaposed in cardiomyocytes (Protasi, 2002; Sedarat et al., 2000) and neurons (Chavis et al., 1996), and interact physically in hippocampus (Berrout and Isokawa, 2009; Kim et al., 2007). To ensure preferential activation by L-VGCCs, RyRs are bound in close apposition to L-VGCCs by junctophilins in a heteromeric junctional membrane complex (Moriguchi et al., 2006; Takeshima et al., 2000). This complex also contains proteins that regulate Ca2+ release from RyRs, including FK506-binding proteins 1a and 1b (FKBP1a/1b, also known as FKBP12/12.6), and calmodulin-3 (Brillantes et al., 1994; Jayaraman et al., 1992; Lehnart et al., 2003; Meissner, 2002; Samso et al., 2009; Wagenknecht et al., 1997; Wright et al., 2008; Zalk et al., 2007). CICR functions to rapidly amplify the Ca2+ influx that enters via membrane Ca2+ channels, and thereby to achieve the high Ca2+ concentrations required for activation of some major Ca2+-dependent functions, such as excitation-contraction coupling in muscle or Ca2+ signaling in large networks (Roderick et al., 2003; Verkhratsky, 2005). Thus, CICR is well-positioned to modulate Ca2+ signaling. On the other hand, if CICR is aberrant, it is also effectively positioned to drive widespread Ca2+ dysregulation.
A significant increase in CICR is seen with age in several models of hippocampal Ca2+ dysregulation, from longer-term cultured embryonic hippocampal neurons (e.g., Clodfelter et al., 2002) to hippocampal slice neurons of adult rats (Gant et al., 2006; Kumar and Foster, 2005). In a large electrophysiological and Ca2+-imaging study of CA1 neurons across the adult lifespan of male F344 rats, we found that several established biomarkers of Ca2+ dysregulation (larger sAHP, greater spike accommodation and increased Ca2+ transients) emerge roughly together in the midlife age range (Gant et al., 2006). This is also the age range in which increased L-VGCC activity and impaired cognition first emerge (Aitken and Meaney, 1989; Bach et al., 1999; Fischer et al., 1992; Frick et al., 1995; Kadar et al., 1994; Kadish et al., 2009; Knuttinen et al., 2001; Markowska, 1999; Norris et al., 2010; Thibault and Landfield, 1996). Of particular note in the lifespan study (Gant et al., 2006), is that ryanodine, at concentrations that block RyRs and CICR, essentially erases the aging-related increases on all measured biomarkers of Ca2+ dysregulation (Fig. 1), including the increase in Ca2+ transients induced by trains of action potentials at 7 Hz (theta). Therefore, much of the aging-related increase in intracellular [Ca2+] during physiologically-relevant stimulation (7 Hz) apparently originates from CICR and likely underlies the age differences in multiple Ca2+-dependent responses of hippocampal CA1 pyramidal neurons. Based on these findings, our initial ‘Ca2+-current hypothesis’ of Ca2+ dysregulation in hippocampal aging (Landfield, 1987) has been extended to incorporate the growing evidence that augmented CICR further amplifies an already increased L-VGCC source of Ca2+ and is a major contributing factor to Ca2+ dysregulation in brain aging (Gant et al., 2006; Thibault et al., 2007).
Figure 1. The AHP and intracellular Ca2+ are altered by aging.
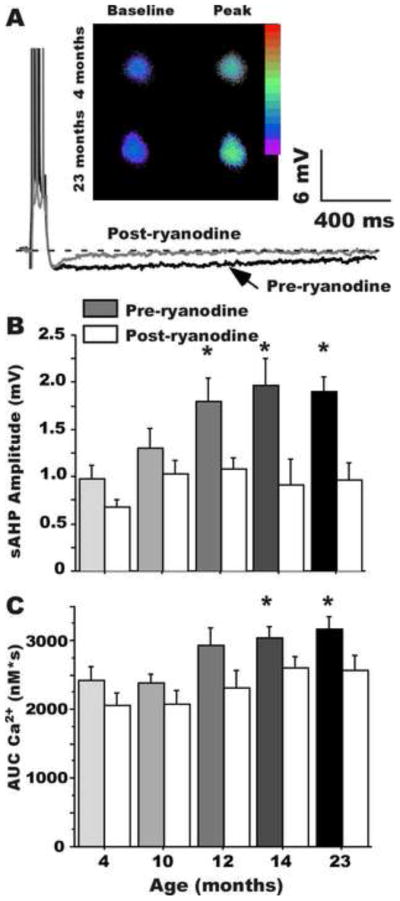
A: Example of the sAHP in hippocampal neurons from a young and an aging rat, showing the increased AHP magnitude with aging (darker trace); also shown are examples of the increase in intracellular Ca2+ transients (inset). Age-related increases in sAHP amplitude (B) and Ca2+ transients (C) begin by 12 months-of-age and are blocked by ryanodine, indicating the increases depend on CICR from RyRs (* p < .05). (From Gant et al., 2006)
2.3 Different patterns of aging-related Ca2+ dysregulation in other neuron types and preparations
From the outset of studies on Ca2+ dysregulation and brain aging, it has been apparent that manifestations of aging-related Ca2+ dyshomeostasis varied widely across neuron types, regions and preparations (Gibson and Peterson, 1987; Khachaturian, 1984; Michaelis et al., 1984; Peterson and Gibson, 1984), some differing substantially from the hippocampal pyramidal cell Ca2+ dysregulation phenotype (Landfield and Pitler, 1984; Thibault et al., 2007). For example, aged rodent dentate gyrus granule neurons are characterized by smaller Ca2+ currents, possibly because of elevated intracellular free Ca2+ and resultant Ca2+-dependent Ca2+ inactivation (Reynolds and Carlen, 89). Rodent cerebellar granule neurons exhibit aging-related increases in resting Ca2+, and lower-amplitude but longer-lasting Ca2+ transients from ER release (Kirischuk et al., 1996), perhaps linked to altered mitochondrial function (Toescu and Verkhratsky, 2004). Furthermore, an aging-related increase in Ca2+ buffering capacity has been found consistently in basal forebrain neurons (see Murchison and Griffith, 2007; Murchison et al., 2009), and may also occur in hippocampal CA1 neurons (Oh et al., 2013). Conversely, aging-related reductions in apparent Ca2+ buffering capacity (e.g., calbindin, mitochondria) have been found, in the rat perirhinal cortex (Moyer et al., 2011) and some other regions (Brown et al., 2004). Recent work on mouse hippocampal CA3 interneurons (Lu et al., 2011) found no aging effects on sAHPs, intracellular Ca2+ transients and resting free Ca2+ under baseline conditions. However, during kainate activation these processes exhibited aging effects that resembled those noted above in hippocampal pyramidal neurons. Clearly, resolving whether this variation in Ca2+-related manifestations of brain aging reflects modifications induced by different neuronal architectures, preparations or physiological processes acting on a common underlying mechanism of Ca2+ dysregulation, or instead indicates that there are multiple cell-type-specific mechanisms of neuronal Ca2+ dyshomeostasis, will require considerable additional study.
2.4 Altered Ca2+ regulation in mouse models of Alzheimer’s disease
As noted, aging is the major risk factor for idiopathic AD and it therefore seems reasonable that mechanisms of unhealthy brain aging related to conversion of normal aging to AD might well continue to act in AD brain. Accordingly, it is interesting that elevated intracellular Ca2+ release in neurons also has been found in transgenic mouse models of AD (Chakroborty et al., 2009; Leissring et al., 2000; Mattson et al., 1998; Stutzmann et al., 2006; Supnet and Bezprozvanny, 2011; Tu et al., 2006). Such results are generally similar to those described above for studies of normal aging in hippocampal neurons, in that Ca2+ dyshomeostasis was in part manifested by altered CICR. On the other hand, in contrast to normal aging, hippocampal L-VGCC activity was recently found to be reduced in a transgenic AD model compared to wild-type controls, possibly because of Ca2+ inactivation (Thibault et al., 2012).
Substantial evidence from human AD brain samples, notably an increase in calpain activity (Nixon, 2003), also suggests a role for Ca2+ dyshomeostasis in AD or other neurodegenerative conditions (Corona et al., 2011; Fedrizzi and Carafoli, 2011; Goussakov et al., 2010). Furthermore, studies of brain samples from AD and MCI patients (Bruno et al., 2012) found that mRNA for RyR2 was elevated in temporal cortex in the MCI group compared to cognitively-intact controls. Potentially decreased Ca2+ buffering, indicated by reduced calbindin immunoreactivity, has been found in basal forebrain regions of human AD samples (Riascos et al., 2011). In addition, although the results are controversial, several clinical trials have suggested that treatment with Ca2+ channel blockers may delay the onset of dementia (Forette et al., 2002; Goodison et al., 2012; Khachaturian et al., 2006; Watfa et al., 2010).
3. Molecular mechanisms
As discussed, multiple Ca2+-related processes have been implicated in brain aging, among which L-VGCCs and RyRs have been identified as likely primary sources of excess Ca2+ in aged animal hippocampal CA1 neurons. Despite these substantial advances in elucidating aging effects on neuronal physiology, little is yet known about the molecular bases of these physiological alterations or specifically, about the molecular mechanisms underlying age-related alterations in L-VGCC or RyR activity. There is some evidence of modest upregulation of the less common isoform (CaV1.3) of the L-VGCC pore-forming subunit (Chen et al., 2000; Herman et al., 1998; Veng et al., 2003), and conditional knock down of CaV1.3 was found to reduce the sAHP of mice (Gamelli et al., 2011). Nevertheless, our gene array analyses (see below) have not confirmed upregulation of expression, at the mRNA level, of any L-VGCC pore-forming subunits (Rowe et al., 2007), and to date have found only modest late-life increases in RyR expression (Kadish et al., 2009). Therefore, at this point changes in brain L-VGCC or RyR expression do not appear to be compelling explanations for their increased activity with aging.
3.1 Whole-genome expression profiles associated with hippocampal aging and cognitive decline
In parallel with the electrophysiological/imaging studies, we have conducted a series of microarray analyses aimed at identifying whole-genome expression profiles associated with aging-related cognitive impairment, including three studies of F344 rat hippocampal expression (Blalock et al., 2003; Kadish et al., 2009; Rowe et al., 2007). These studies differed from one another somewhat in the age of animals used and in time points of gene expression analysis following Morris water maze (MWM) training. However, the Kadish et al. (2009) study examined hippocampal expression changes and cognition across the adult lifespan and therefore closely parallels our age course study of hippocampal electrophysiological and [Ca2+]i changes (Gant et al., 2006). In the lifespan gene array study (Kadish et al., 2009), we examined the age course of changes in hippocampal aging-related genes, and pathways they represented, correlating these with cognitive function at 5 age points of the rat adult lifespan (3-, 6-, 9-, 12- and 23-months-old). Statistically well-powered groups, with one chip per animal were employed, allowing detection of modest expression differences with high reliability. False discovery rates were held down by use of pre-analytic filtering algorithms, well-powered tests and pathway analyses, allowing the studies to take advantage of the discovery power of microarrays while mitigating both Type I and Type II error (Blalock et al., 2003; Blalock et al., 2005; Peng et al., 2003).
The lifespan analysis identified multiple brain processes that begin to change early in aging, and consequently might be strong candidates for initiators of unhealthy brain aging cascades that induce onset of cognitive decline. Of particular interest in the present context, however, data mining subsequent to publication revealed aging-related changes in a group of genes encoding immunophilins and other proteins relevant to regulation of CICR, as described below.
3.2 The FKBP-Ca2+ regulating pathway: a possible molecular mechanism underlying Ca2+ dyshomeostasis
FK506-binding proteins (FKBP) 1a and FKBP1b (also known as FKBP12 and 12.6, respectively) are low molecular weight members of the FKBP family of immunophilins, proteins that bind the immunosuppressant drugs FK506 and rapamycin. Many immunophilins exhibit peptidyl-prolyl isomerase activity and function as protein chaperones and stabilizers (Eitoku et al., 2008; Jakob et al., 2009; Kang et al., 2008; Marks, 1997). In myocytes, FKBP1a and FKBP1b also play a major role in Ca2+ regulation, binding and stabilizing RyRs in the closed state and inhibiting CICR from sarcoplasmic reticulum stores. In cardiomyocytes, genetic depletion of FKBP1b results in Ca2+ leakage from RyRs and cardiac failure (Lehnart et al., 2003; Zalk et al., 2007). In addition, treatment with rapamycin displaces FKBPs from RyRs and increases Ca2+ release from intracellular stores (Lehnart et al., 2003; Long et al., 2007).
However, there have been few studies on the roles of FKBPs in brain neurons. We were prompted to investigate this pathway in neurons by our findings implicating RYRs in brain Ca2+ dysregulation (Gant et al., 2006 and Fig. 1) and by our unexpected observation that rapamycin increased Ca2+ currents (unlike FK506, which inhibits Ca2+ currents) (Norris et al., 2002; Norris et al., 2010). Moreover, our microarray analyses showed that hippocampal Fkbp1b gene expression is downregulated with aging; Fkbp1b expression begins to decline early in the lifespan and continues to drop through mid- and late-life, when cognitive deficits typically emerge. Fkbp1a also declines with aging although its decrease is somewhat more variable (Fig. 2). Together, these lines of evidence raised the possibility that FKBP pathways might also regulate CICR in brain neurons and that their decline might play a critical role in Ca2+ dysregulation in aging.
Figure 2. Age-dependent changes in expression of Fkbp1b, Fkbp1a, Ryr2, and Frap1/mTOR mRNA.
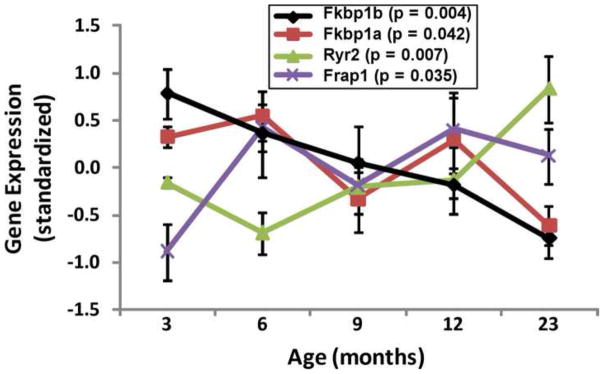
(From data in Kadish et al., 2009)
3.3 Testing predictions of a novel hypothesis implicating FKBP-Ca2+ release pathways in aging-related brain Ca2+ dysregulation
Accordingly, we undertook studies to test specific predictions of the hypothesis that the FKBP-RyR pathway is a major player in aging-related hippocampal Ca2+ dysregulation. To test the possibility that a decline of FKBP function in hippocampal neurons might play a role in aging-related Ca2+ dysregulation, we used two distinct but complementary approaches to disrupt FKBP1b function. In one, hippocampal embryonic cell cultures or hippocampal slices from young-adult rats were exposed to rapamycin, which displaces FKBPs from RyRs. In the second approach, siRNA (siFkbp1b) was used to selectively knock down Fkbp1b expression in vitro; for in vivo studies, adeno-associated viral (AAV) vectors bearing shRNA constructs were microinjected into the hippocampus to knock down Fkbp1b expression (Gant et al., 2011).
3.4 In vitro rapamycin treatment and siRNA knockdown of FKBP1b
In the in vitro studies, incubation of hippocampal cell cultures with rapamycin for 1 hour had no effect on VGCC currents, but larger currents were observed following 24 or 96 hours of incubation (Fig. 3). The L-VGCC blocker, nimodipine reversed the effect of rapamycin on VGCC current, indicating that the rapamycin-induced increase in current was primarily mediated via L-type VGCCs. Moreover, following 3–4 days of in vitro knockdown of FKBP1b by targeted siRNA, VGCC activity was enhanced, much as it was by rapamycin (Fig. 3). Treatments with scrambled siRNA or siRNA vehicle had no effect on Ca2+ currents. Confirming efficacy of knockdown, qPCR analyses showed that FKBP1b mRNA was significantly reduced following incubation with siFkbp1b (Gant et al., 2011).
Figure 3. Knockdown of FKBP1b or treatment with rapamycin (Rap) increased Ca2+ channel current in cultured hippocampal neurons.
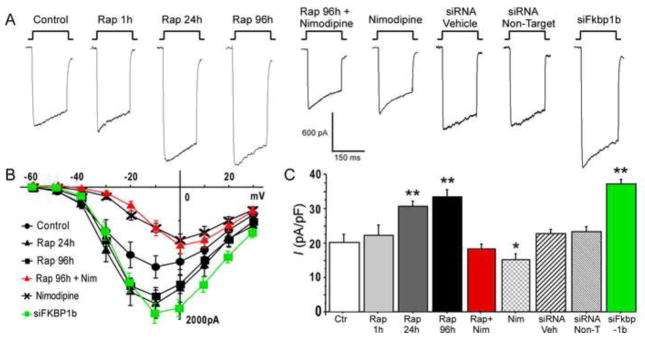
A, Representative whole-cell patch-clamp current traces from each experimental condition; B, Mean current/voltage (I/V) relationships for 6 of the 9 experimental conditions. The I–V curves for Rap 1h, siRNA Veh and siRNA Non-Target were not different from the control condition and are omitted for illustrative clarity. Exposure to siFkbp1b for 96 h or to rapamycin for 24/96 h induced enhancement of VGCC current. Treatment with siFkbp1b or with rapamycin altered the amplitude but not the voltage dependence of Ca2+ current, whereas nimodipine (Nim) shifted peak current to more positive voltage; C, Means +/− S.E.M. of peak Ca2+ current density for the 9 conditions. Asterisks indicate significant differences from the control condition (* p < 0.05 and **p < 0.0001). n = control (18), Rap 1h (4), Rap 24h (20), Rap 96h (20), Rap + Nim (10), Nim (12), siRNA Veh (23), siRNA Non-Target (19), siFkbp1b (23). (From Gant et al., 2011)
3.5 In situ rapamycin treatment of hippocampal slices and in vivo shRNA knockdown of FKBP1b
To determine the effect of rapamycin on Ca2+ responses in adult neurons, we incubated young-adult rat hippocampal slices in rapamycin for electrophysiological recording and Ca2+ imaging. These studies revealed that rapamycin substantially enhanced both the sAHP and neuronal Ca2+ transients, especially the CICR component of the Ca2+ transient. Perfusion of the recording chamber with high concentrations of ryanodine, sufficient to block ryanodine-dependent CICR, reversed the effects of rapamycin on the sAHP and Ca2+ transients.
In order to selectively manipulate gene expression in defined hippocampal regions of adult rats, we conducted a series of comparative studies to determine the ability of various microinjected adeno-associated virus (AAV) serotypes expressing green fluorescent protein (GFP) to transduce pyramidal neurons in the hippocampus. GFP expression showed that all serotypes successfully transduced hippocampal pyramidal neurons and that expression of GFP was limited to the hippocampus proper. Serotype 8 exhibited the least amount of longitudinal spread, while serotypes 1 and 9 exhibited longitudinal spread sufficient to transduce neurons in the hippocampus from the extreme rostral to caudal portions. Serotype 1 was chosen as the preferred serotype for further studies. Our target injection site was Stratum Oriens at the peak of the CA1 pyramidal cell layer. Figure 4 shows sections confirming viral delivery of the GFP transgene to hippocampal CA1 neurons and illustrates that the vector did not substantially cross the hippocampal fissure or the midline following unilateral injection of AAV.
Figure 4. Green Flourescent Protein (GFP) expression in the hippocampus.

A: GFP expression in the hippocampus following infusion with serotype 2.1 AAV expressing GFP. Note the high level of expression in the pyramidal cells corresponding to the CA regions. Solid line box inset is shows close-up of pyramidal cells expressing GFP from the CA1 region (Dotted box). B: Note that GFP expression is only found in the hippocampus that was injected during a unilateral injection. The contralateral side within the same animal contained no cells expressing GFP. Midline of the hemispheres is denoted with a dotted line.
We then conducted a major study testing predictions of the FKBP-Ca2+ dysregulation hypothesis, employing unilateral hippocampal knockdown of FKBP1b in vivo. In this study, one hippocampus (ipsilateral) of young-adult rats was microinjected with AAV expressing shFkbp1b, while the other hippocampus (contralateral) received either empty-vector control AAV(AAV-0) or was not treated (these two control conditions did not differ on any variable and were combined statistically). Approximately, four-to-five weeks after shFkbp1b injection, intracellular electrophysiological recordings of the sAHP were obtained from CA1 pyramidal neurons in acute hippocampal slices from both transduced (ipsilateral) and control (contralateral) sides of 9 animals.
As shown in Figure 5, the Results of the electrophysiological studies were dramatic. sAHP measures were obtained from a total of 17 neurons in slices from the ipsilateral and 17 from the contralateral hippocampi of the 9 injected young rats. Statistical analysis revealed that sAHP amplitude and duration were increased by approximately 100% in the transduced hippocampus. Moreover, the sAHPs of control neurons were similar to those typically observed in young rats, whereas values from knockdown side neurons were similar to those observed in aged rats in several prior studies (e.g., Fig. 1). Our qPCR and immunohistochemical data from this knockdown study clearly showed that FKBP1b knockdown was achieved in hippocampus at both the gene (25–35% by qPCR) and protein (Fig. 6) levels.
Figure 5. Knockdown of FKBP1b in vivo enhanced the slow AHP.

Top, Representative sharp electrode intracellular recordings showing four triggered action potentials, followed by an AHP (dashed line indicates baseline) in CA1 neurons from slices of a control (left) and an AAV–shFkbp1b-injected (right) hippocampus. Middle, Fluorescent imaging in an animal receiving a similar unilateral AAV injection expressing GFP only, showing strong expression in the injected, but not the contralateral hippocampus, and also illustrating general placement of recording pipettes in the pyramidal neuron somal layer (stratum pyramidale) of field CA1. Bottom, sAHP amplitude (left) and duration (right) were significantly increased by FKBP1b knockdown (shFkbp1b injection) (*p < 0.0025 for either variable, t test; n = 17 neurons per group). (From Gant et al., 2011)
Figure 6. shFkbp1b mediated knockdown of Fkbp1b validated at the protein level.
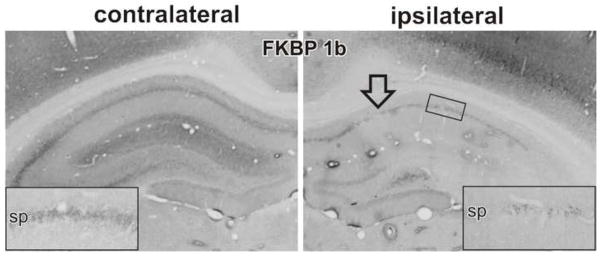
In vivo direct hippocampal shFkbp1b injection into one hemisphere (ipsilateral) is compared to the non-injected contralateral hemisphere. To validate knockdown at the protein level, immunohistochemical staining of FKBP11b/12.6 was performed. Arrow indicates injection site. Inset shows magnified view of CA1 region. (Modified from Gant et al., 2011)
In sum, rapamycin or FKBP1b knockdown in young rat neurons closely recapitulated patterns of Ca2+ dysregulation seen with aging, findings that are clearly consistent with a key prediction of the hypothesis that FKBP1b disruption is an important molecular mechanism underlying aging-related Ca2+-dysregulation in the hippocampus (Gant et al., 2011). It should also be noted that extensive studies are presently underway testing the converse prediction, namely, that in vivo overexpression of Fkbp1b in hippocampus of aging rats can reverse the aging-related pattern of Ca2+ dysregulation. Initial results are highly consistent with this prediction.
4. Interactions of FKBP1b/1a with multiple targets
4.1 Do FKBPs regulate L-VGCCs as well as RyRs in the brain?
As described above, it has been well established that FKBPs inhibit Ca2+ release from RyRs in muscle cells (Zalk et al., 2007). However, the studies of the effects of disrupting FKBP1b on the ryanodine-sensitive sAHP and Ca2+ transient in hippocampus (Fig. 5) provide essentially the first evidence that FKBPs inhibit RyR-mediated CICR in brain neurons as well as in myocytes. Moreover, the findings that FKBP1b apparently inhibits VGCC current (Fig. 3) provides the first evidence in any cell type that FKBP1b inhibits Ca2+ entry via VGCCs as well as intracellular release from RyRs (Gant et al., 2011). It is not clear what pathway might mediate FKBP regulation of VGCCs. The L-VGCC lies upstream of the RyR that is directly bound by FKBPs in the VGCC-RyR circuit that amplifies Ca2+ through CICR (see Berridge, 1997; Verkhratsky, 2004). Therefore, it is feasible that FKBPs regulate Ca2+ currents through their actions on RyRs and unrecognized secondary retrograde effects from RyRs to VGCCs. Nevertheless, a more parsimonious explanation seems to be that FKBPs regulate VGCC activity directly, through an as yet undetermined interaction.
4.2 Inhibition of the mTOR pathway by FKBPs
FKBP1a and FKBP1b interact with cellular pathways in addition to the RyR complex. In particular, FKBPs also tonically inhibit the mTOR pathway that plays an important role in cell growth and plasticity in the brain (Hoeffer et al., 2008; Jacinto and Hall, 2003). Thus, a decline in FKBP1b function with aging, as in our proposed model (Fig. 7) should result in reduced inhibition and increased activity of both the RyR and mTOR pathways. Rapamycin, on the other hand, has opposite effects on the FKBP-RyR and FKBP-mTOR interactions, disrupting FKBP inhibition of RyRs, but strengthening FKBP-mediated inhibition of mTOR. Conceivably, this rapamycin-enhanced mTOR inhibition may account for why rapamycin has been reported to exert some anti-aging-like effects, including extension of lifespan (Caccamo et al.; Harrison et al., 2009; Wilkinson et al., 2012). That is, although BDNF, insulin and other growth factors that mediate neurite extension, plasticity and neurogenesis signal through mTOR (Lynch et al., 2008; Scharfman and MacLusky, 2006), beneficial effects of inhibiting mTOR may result because of excessive mTOR activation with aging. Nevertheless, rapamycin’s anti-aging-like strengthening of FKBP-dependent inhibitory actions on mTOR appear unrelated to rapamycin’s aging-like weakening of FKBP-dependent inhibition of RyR-mediated Ca2+ release, and likely do not obviate the interpretation that rapamycin disrupts the FKBP-RyR interaction (see model-Fig. 7).
Figure 7. Working model of FKBP1b’s role in aging-related Ca2+ dyshomeostasis.

A: In young subjects, neuronal FKBP1b exerts strong tonic inhibitory effects on two pathways, the Ca2+ regulatory pathway and the mTOR signaling pathway. In the Ca2+ regulatory pathway, FKBP1b inhibits cytosolic Ca2+ rises generated by extracellular influx via L-VGCCs (1) and intracellular release from RYRs (2), by inhibiting both channel types directly (3 & 4). In the mTOR pathway, FKBP inhibition of mTOR (5) helps maintain the balance between growth-stimulating and autophagy-suppressing effects of mTOR. B: With aging (mimicked by FKBP1b knockdown), a decline of FKBP1b expression/function leads to weakened inhibition of both pathways, and concomitant increases in both Ca2+ and mTOR signaling. Increased Ca2+ signaling results in dampened neuronal excitability and function, whereas mTOR disinhibition leads to aberrant increases in growth signaling as well as decreased autophagy. C: Rapamycin in young subjects exerts opposite effects on FKBP inhibition in the two pathways, mimicking aging’s effect of weakened FKBP inhibition in the Ca2+ pathway (B), but paradoxically augmenting FKBP inhibition of the mTOR pathway (C). (From Gant et al. 2011)
5. A molecular model for aging-related Ca2+ dysregulation in hippocampus
Together, the data provide increasingly compelling support for the hypothesis that an aging-related decline in function/expression of hippocampal FKBP1b (and possibly, FKBP1a) is a key factor in development of the hippocampal Ca2+ dysregulation phenotype (Gant et al., 2011). This decline may result from decreased expression and/or posttranslational modification. Figure 7 schematically illustrates the model, incorporating FKBP1b regulation of VGCCs as well as RyRs, and shows putative aging effects on multiple target pathways of FKBPs. Also illustrated is the opposite regulation of the FKBP-RyR and FKBP-mTOR pathways by rapamycin.
5.1 Possible causes of age-related decline in FKBP1b function
The finding that disrupting hippocampal FKBP function can induce the Ca2+ dysregulation phenotype is consistent with a model in which similar disruption occurs with aging and raises the question of what factors might trigger an initial aging-related decline in FKBP function. One possibility is that a decrease in the biosynthesis of energy-expensive proteins, notably including FKBPs, might result from a metabolic shift that appears to develop in neurons and glia of the hippocampus during aging of females (Brinton, 2008; Yau and Seckl, 2012) and males (Kadish et al., 2009; Rowe et al., 2007). These metabolic alterations in turn may depend on age-related variations in endocrine status, as some hormones (e.g., estrogens and progestins) appear to interact strongly with brain aging and brain metabolism (Brewer et al., 2009; Foster, 2012; Simpkins and Singh, 2008; Sohrabji and Bake, 2006; Xin et al., 2002). Additionally, adrenal stress hormones (glucocorticoids) exert major effects on metabolism and have long been linked to brain aging (Porter and Landfield, 1998). The impact of glucocorticoids on the brain is altered with aging (Blalock et al., 2010; Chen et al., 2013; Holmes and Seckl, 2006; Landfield et al., 2007). Glucocorticoids also enhance the Ca2+-dependent sAHP in the hippocampus (Joels and de Kloet, 1989; Kerr et al., 1989), consistent with an effect on FKBPs. Similarly, mineralocorticoids have been shown to decrease cardiac FKBP1b expression and elevate RyR-mediated Ca2+ release (Gomez et al., 2009). Thus, it appears plausible that hormonal and/or metabolic changes may contribute to aging-related declines in FKBP expression/function. Regardless of the causal factors, however, declining FKBP function during brain aging appears to provide important new clues in the search for novel targets that can alter the progression of unhealthy brain aging and/or Alzheimer’s disease.
Acknowledgments
Supported by NIH AG004542
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References Cited
- Aitken DH, Meaney MJ. Temporally graded, age-related impairments in spatial memory in the rat. Neurobiol Aging. 1989;10:273–276. doi: 10.1016/0197-4580(89)90062-6. [DOI] [PubMed] [Google Scholar]
- Bach ME, Barad M, Son H, Zhuo M, Lu YF, Shih R, Mansuy I, Hawkins RD, Kandel ER. Age-related defects in spatial memory are correlated with defects in the late phase of hippocampal long-term potentiation in vitro and are attenuated by drugs that enhance the cAMP signaling pathway. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:5280–5285. doi: 10.1073/pnas.96.9.5280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge MJ. Elementary and global aspects of calcium signalling. J Physiol. 1997;499 (Pt 2):291–306. doi: 10.1113/jphysiol.1997.sp021927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berrout J, Isokawa M. Homeostatic and stimulus-induced coupling of the L-type Ca2+ channel to the ryanodine receptor in the hippocampal neuron in slices. Cell Calcium. 2009;46:30–38. doi: 10.1016/j.ceca.2009.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blalock EM, Chen KC, Sharrow K, Herman JP, Porter NM, Foster TC, Landfield PW. Gene microarrays in hippocampal aging: statistical profiling identifies novel processes correlated with cognitive impairment. J Neurosci. 2003;23:3807–3819. doi: 10.1523/JNEUROSCI.23-09-03807.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blalock EM, Chen KC, Stromberg AJ, Norris CM, Kadish I, Kraner SD, Porter NM, Landfield PW. Harnessing the power of gene microarrays for the study of brain aging and Alzheimer’s disease: statistical reliability and functional correlation. Ageing research reviews. 2005;4:481–512. doi: 10.1016/j.arr.2005.06.006. [DOI] [PubMed] [Google Scholar]
- Blalock EM, Grondin R, Chen KC, Thibault O, Thibault V, Pandya JD, Dowling A, Zhang Z, Sullivan P, Porter NM, Landfield PW. Aging-related gene expression in hippocampus proper compared with dentate gyrus is selectively associated with metabolic syndrome variables in rhesus monkeys. J Neurosci. 2010;30:6058–6071. doi: 10.1523/JNEUROSCI.3956-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer LD, Dowling AL, Curran-Rauhut MA, Landfield PW, Porter NM, Blalock EM. Estradiol reverses a calcium-related biomarker of brain aging in female rats. J Neurosci. 2009;29:6058–6067. doi: 10.1523/JNEUROSCI.5253-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brillantes AB, Ondrias K, Scott A, Kobrinsky E, Ondriasova E, Moschella MC, Jayaraman T, Landers M, Ehrlich BE, Marks AR. Stabilization of calcium release channel (ryanodine receptor) function by FK506-binding protein. Cell. 1994;77:513–523. doi: 10.1016/0092-8674(94)90214-3. [DOI] [PubMed] [Google Scholar]
- Brinton RD. Estrogen regulation of glucose metabolism and mitochondrial function: therapeutic implications for prevention of Alzheimer’s disease. Advanced drug delivery reviews. 2008;60:1504–1511. doi: 10.1016/j.addr.2008.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MR, Geddes JW, Sullivan PG. Brain region-specific, age-related, alterations in mitochondrial responses to elevated calcium. J Bioenerg Biomembr. 2004;36:401–406. doi: 10.1023/B:JOBB.0000041775.10388.23. [DOI] [PubMed] [Google Scholar]
- Bruno AM, Huang JY, Bennett DA, Marr RA, Hastings ML, Stutzmann GE. Altered ryanodine receptor expression in mild cognitive impairment and Alzheimer’s disease. Neurobiology of aging. 2012;33:1001, e1001–1006. doi: 10.1016/j.neurobiolaging.2011.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caccamo A, Majumder S, Richardson A, Strong R, Oddo S. Molecular interplay between mTOR, A{beta} and tau: Effects on cognitive impairments. J Biol Chem. doi: 10.1074/jbc.M110.100420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakroborty S, Goussakov I, Miller MB, Stutzmann GE. Deviant ryanodine receptor-mediated calcium release resets synaptic homeostasis in presymptomatic 3xTg-AD mice. J Neurosci. 2009;29:9458–9470. doi: 10.1523/JNEUROSCI.2047-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavis P, Fagni L, Lansman JB, Bockaert J. Functional coupling between ryanodine receptors and L-type calcium channels in neurons. Nature. 1996;382:719–722. doi: 10.1038/382719a0. [DOI] [PubMed] [Google Scholar]
- Chen KC, Blalock EM, Curran-Rauhut MA, Kadish I, Blalock SJ, Brewer L, Porter NM, Landfield PW. Glucocorticoid-Dependent Hippocampal Transcriptome in Male Rats: Pathway-Specific Alterations with Aging. Endocrinology. 2013 doi: 10.1210/en.2013-1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen KC, Blalock EM, Thibault O, Kaminker P, Landfield PW. Expression of alpha 1D subunit mRNA is correlated with L-type Ca2+ channel activity in single neurons of hippocampal “zipper” slices. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:4357–4362. doi: 10.1073/pnas.070056097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clodfelter GV, Porter NM, Landfield PW, Thibault O. Sustained Ca2+-induced Ca2+-release underlies the post-glutamate lethal Ca2+ plateau in older cultured hippocampal neurons. Eur J Pharmacol. 2002;447:189–200. doi: 10.1016/s0014-2999(02)01843-5. [DOI] [PubMed] [Google Scholar]
- Corona C, Pensalfini A, Frazzini V, Sensi SL. New therapeutic targets in Alzheimer’s disease: brain deregulation of calcium and zinc. Cell Death Dis. 2011;2:e176. doi: 10.1038/cddis.2011.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deyo RA, Straube KT, Disterhoft JF. Nimodipine facilitates associative learning in aging rabbits. Science. 1989;243:809–811. doi: 10.1126/science.2916127. [DOI] [PubMed] [Google Scholar]
- Disterhoft JF, Oh MM. Alterations in intrinsic neuronal excitability during normal aging. Aging cell. 2007;6:327–336. doi: 10.1111/j.1474-9726.2007.00297.x. [DOI] [PubMed] [Google Scholar]
- Disterhoft JF, Thompson LT, Moyer JR, Jr, Mogul DJ. Calcium-dependent afterhyperpolarization and learning in young and aging hippocampus. Life sciences. 1996;59:413–420. doi: 10.1016/0024-3205(96)00320-7. [DOI] [PubMed] [Google Scholar]
- Eitoku M, Sato L, Senda T, Horikoshi M. Histone chaperones: 30 years from isolation to elucidation of the mechanisms of nucleosome assembly and disassembly. Cell Mol Life Sci. 2008;65:414–444. doi: 10.1007/s00018-007-7305-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedrizzi L, Carafoli E. Ca2+ dysfunction in neurodegenerative disorders: Alzheimer’s disease. Biofactors. 2011;37:189–196. doi: 10.1002/biof.157. [DOI] [PubMed] [Google Scholar]
- Fischer W, Chen KS, Gage FH, Bjorklund A. Progressive decline in spatial learning and integrity of forebrain cholinergic neurons in rats during aging. Neurobiol Aging. 1992;13:9–23. doi: 10.1016/0197-4580(92)90003-g. [DOI] [PubMed] [Google Scholar]
- Forette F, Seux ML, Staessen JA, Thijs L, Babarskiene MR, Babeanu S, Bossini A, Fagard R, Gil-Extremera B, Laks T, Kobalava Z, Sarti C, Tuomilehto J, Vanhanen H, Webster J, Yodfat Y, Birkenhager WH. The prevention of dementia with antihypertensive treatment: new evidence from the Systolic Hypertension in Europe (Syst-Eur) study. Archives of internal medicine. 2002;162:2046–2052. doi: 10.1001/archinte.162.18.2046. [DOI] [PubMed] [Google Scholar]
- Foster TC. Role of estrogen receptor alpha and beta expression and signaling on cognitive function during aging. Hippocampus. 2012;22:656–669. doi: 10.1002/hipo.20935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster TC, Norris CM. Age-associated changes in Ca(2+)-dependent processes: relation to hippocampal synaptic plasticity. Hippocampus. 1997;7:602–612. doi: 10.1002/(SICI)1098-1063(1997)7:6<602::AID-HIPO3>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Frick KM, Baxter MG, Markowska AL, Olton DS, Price DL. Age-related spatial reference and working memory deficits assessed in the water maze. Neurobiol Aging. 1995;16:149–160. doi: 10.1016/0197-4580(94)00155-3. [DOI] [PubMed] [Google Scholar]
- Gamelli AE, McKinney BC, White JA, Murphy GG. Deletion of the L-type calcium channel Ca(V) 1.3 but not Ca(V) 1.2 results in a diminished sAHP in mouse CA1 pyramidal neurons. Hippocampus. 2011;21:133–141. doi: 10.1002/hipo.20728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gant JC, Chen KC, Norris CM, Kadish I, Thibault O, Blalock EM, Porter NM, Landfield PW. Disrupting function of FK506-binding protein 1b/12.6 induces the Ca(2)+-dysregulation aging phenotype in hippocampal neurons. J Neurosci. 2011;31:1693–1703. doi: 10.1523/JNEUROSCI.4805-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gant JC, Sama MM, Landfield PW, Thibault O. Early and simultaneous emergence of multiple hippocampal biomarkers of aging is mediated by Ca2+-induced Ca2+ release. J Neurosci. 2006;26:3482–3490. doi: 10.1523/JNEUROSCI.4171-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson GE, Peterson C. Calcium and the aging nervous system. Neurobiology of aging. 1987;8:329–343. doi: 10.1016/0197-4580(87)90072-8. [DOI] [PubMed] [Google Scholar]
- Gomez AM, Rueda A, Sainte-Marie Y, Pereira L, Zissimopoulos S, Zhu X, Schaub R, Perrier E, Perrier R, Latouche C, Richard S, Picot MC, Jaisser F, Lai FA, Valdivia HH, Benitah JP. Mineralocorticoid modulation of cardiac ryanodine receptor activity is associated with downregulation of FK506-binding proteins. Circulation. 2009;119:2179–2187. doi: 10.1161/CIRCULATIONAHA.108.805804. [DOI] [PubMed] [Google Scholar]
- Goodison WV, Frisardi V, Kehoe PG. Calcium channel blockers and Alzheimer’s disease: potential relevance in treatment strategies of metabolic syndrome. Journal of Alzheimer’s disease: JAD. 2012;30(Suppl 2):S269–282. doi: 10.3233/JAD-2012-111664. [DOI] [PubMed] [Google Scholar]
- Goussakov I, Miller MB, Stutzmann GE. NMDA-mediated Ca(2+) influx drives aberrant ryanodine receptor activation in dendrites of young Alzheimer’s disease mice. J Neurosci. 2010;30:12128–12137. doi: 10.1523/JNEUROSCI.2474-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, Nadon NL, Wilkinson JE, Frenkel K, Carter CS, Pahor M, Javors MA, Fernandez E, Miller RA. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009;460:392–395. doi: 10.1038/nature08221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman JP, Chen KC, Booze R, Landfield PW. Up-regulation of alpha1D Ca2+ channel subunit mRNA expression in the hippocampus of aged F344 rats. Neurobiology of aging. 1998;19:581–587. doi: 10.1016/s0197-4580(98)00099-2. [DOI] [PubMed] [Google Scholar]
- Hoeffer CA, Tang W, Wong H, Santillan A, Patterson RJ, Martinez LA, Tejada-Simon MV, Paylor R, Hamilton SL, Klann E. Removal of FKBP12 enhances mTOR-Raptor interactions, LTP, memory, and perseverative/repetitive behavior. Neuron. 2008;60:832–845. doi: 10.1016/j.neuron.2008.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes MC, Seckl JR. The role of 11beta-hydroxysteroid dehydrogenases in the brain. Mol Cell Endocrinol. 2006;248:9–14. doi: 10.1016/j.mce.2005.12.002. [DOI] [PubMed] [Google Scholar]
- Jacinto E, Hall MN. Tor signalling in bugs, brain and brawn. Nat Rev Mol Cell Biol. 2003;4:117–126. doi: 10.1038/nrm1018. [DOI] [PubMed] [Google Scholar]
- Jakob RP, Zoldak G, Aumuller T, Schmid FX. Chaperone domains convert prolyl isomerases into generic catalysts of protein folding. Proc Natl Acad Sci U S A. 2009;106:20282–20287. doi: 10.1073/pnas.0909544106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayaraman T, Brillantes AM, Timerman AP, Fleischer S, Erdjument-Bromage H, Tempst P, Marks AR. FK506 binding protein associated with the calcium release channel (ryanodine receptor) J Biol Chem. 1992;267:9474–9477. [PubMed] [Google Scholar]
- Joels M, de Kloet ER. Effects of glucocorticoids and norepinephrine on the excitability in the hippocampus. Science (New York, N Y. 1989;245:1502–1505. doi: 10.1126/science.2781292. [DOI] [PubMed] [Google Scholar]
- Kadar T, Arbel I, Silbermann M, Levy A. Morphological hippocampal changes during normal aging and their relation to cognitive deterioration. J Neural Transm Suppl. 1994;44:133–143. doi: 10.1007/978-3-7091-9350-1_10. [DOI] [PubMed] [Google Scholar]
- Kadish I, Thibault O, Blalock EM, Chen KC, Gant JC, Porter NM, Landfield PW. Hippocampal and cognitive aging across the lifespan: a bioenergetic shift precedes and increased cholesterol trafficking parallels memory impairment. J Neurosci. 2009;29:1805–1816. doi: 10.1523/JNEUROSCI.4599-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang CB, Hong Y, Dhe-Paganon S, Yoon HS. FKBP family proteins: immunophilins with versatile biological functions. Neurosignals. 2008;16:318–325. doi: 10.1159/000123041. [DOI] [PubMed] [Google Scholar]
- Kerr DS, Campbell LW, Hao SY, Landfield PW. Corticosteroid modulation of hippocampal potentials: increased effect with aging. Science (New York, N Y. 1989;245:1505–1509. doi: 10.1126/science.2781293. [DOI] [PubMed] [Google Scholar]
- Khachaturian AS, Zandi PP, Lyketsos CG, Hayden KM, Skoog I, Norton MC, Tschanz JT, Mayer LS, Welsh-Bohmer KA, Breitner JC. Antihypertensive medication use and incident Alzheimer disease: the Cache County Study. Archives of neurology. 2006;63:686–692. doi: 10.1001/archneur.63.5.noc60013. [DOI] [PubMed] [Google Scholar]
- Khachaturian ZS. Towards theories of brain aging. In: Kay DS, Burrows GW, editors. Handbook of studies on Psychiatry and Old Age. Elsevier; Amsterdam: 1984. pp. 7–30. [Google Scholar]
- Kim S, Yun HM, Baik JH, Chung KC, Nah SY, Rhim H. Functional interaction of neuronal Cav1.3 L-type calcium channel with ryanodine receptor type 2 in the rat hippocampus. J Biol Chem. 2007;282:32877–32889. doi: 10.1074/jbc.M701418200. [DOI] [PubMed] [Google Scholar]
- Kirischuk S, Voitenko N, Kostyuk P, Verkhratsky A. Age-associated changes of cytoplasmic calcium homeostasis in cerebellar granule neurons in situ: investigation on thin cerebellar slices. Experimental gerontology. 1996;31:475–487. doi: 10.1016/0531-5565(95)02070-5. [DOI] [PubMed] [Google Scholar]
- Knuttinen MG, Gamelli AE, Weiss C, Power JM, Disterhoft JF. Age-related effects on eyeblink conditioning in the F344 x BN F1 hybrid rat. Neurobiology of aging. 2001;22:1–8. doi: 10.1016/s0197-4580(00)00194-9. [DOI] [PubMed] [Google Scholar]
- Kumar A, Foster TC. Enhanced long-term potentiation during aging is masked by processes involving intracellular calcium stores. Journal of neurophysiology. 2004;91:2437–2444. doi: 10.1152/jn.01148.2003. [DOI] [PubMed] [Google Scholar]
- Kumar A, Foster TC. Intracellular calcium stores contribute to increased susceptibility to LTD induction during aging. Brain research. 2005;1031:125–128. doi: 10.1016/j.brainres.2004.10.023. [DOI] [PubMed] [Google Scholar]
- Landfield PW. ‘Increased calcium-current’ hypothesis of brain aging. Neurobiol Aging. 1987;8:346–347. doi: 10.1016/0197-4580(87)90074-1. [DOI] [PubMed] [Google Scholar]
- Landfield PW, Blalock EM, Chen KC, Porter NM. A new glucocorticoid hypothesis of brain aging: implications for Alzheimer’s disease. Curr Alzheimer Res. 2007;4:205–212. doi: 10.2174/156720507780362083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landfield PW, Pitler TA. Prolonged Ca2+-dependent afterhyperpolarizations in hippocampal neurons of aged rats. Science (New York, N Y. 1984;226:1089–1092. doi: 10.1126/science.6494926. [DOI] [PubMed] [Google Scholar]
- Lehnart SE, Huang F, Marx SO, Marks AR. Immunophilins and coupled gating of ryanodine receptors. Curr Top Med Chem. 2003;3:1383–1391. doi: 10.2174/1568026033451907. [DOI] [PubMed] [Google Scholar]
- Leissring MA, Akbari Y, Fanger CM, Cahalan MD, Mattson MP, LaFerla FM. Capacitative calcium entry deficits and elevated luminal calcium content in mutant presenilin-1 knockin mice. J Cell Biol. 2000;149:793–798. doi: 10.1083/jcb.149.4.793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long C, Cook LG, Wu GY, Mitchell BM. Removal of FKBP12/12.6 from endothelial ryanodine receptors leads to an intracellular calcium leak and endothelial dysfunction. Arterioscler Thromb Vasc Biol. 2007;27:1580–1586. doi: 10.1161/ATVBAHA.107.144808. [DOI] [PubMed] [Google Scholar]
- Lu CB, Hamilton JB, Powell AD, Toescu EC, Vreugdenhil M. Effect of ageing on CA3 interneuron sAHP and gamma oscillations is activity-dependent. Neurobiology of aging. 2011;32:956–965. doi: 10.1016/j.neurobiolaging.2009.05.006. [DOI] [PubMed] [Google Scholar]
- Lutz W, Sanderson W, Scherbov S. The coming acceleration of global population ageing. Nature. 2008;451:716–719. doi: 10.1038/nature06516. [DOI] [PubMed] [Google Scholar]
- Lynch G, Rex CS, Chen LY, Gall CM. The substrates of memory: defects, treatments, and enhancement. Eur J Pharmacol. 2008;585:2–13. doi: 10.1016/j.ejphar.2007.11.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markowska AL. Sex dimorphisms in the rate of age-related decline in spatial memory: relevance to alterations in the estrous cycle. J Neurosci. 1999;19:8122–8133. doi: 10.1523/JNEUROSCI.19-18-08122.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks AR. Intracellular calcium-release channels: regulators of cell life and death. Am J Physiol. 1997;272:H597–605. doi: 10.1152/ajpheart.1997.272.2.H597. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Guo Q, Furukawa K, Pedersen WA. Presenilins, the endoplasmic reticulum, and neuronal apoptosis in Alzheimer’s disease. J Neurochem. 1998;70:1–14. doi: 10.1046/j.1471-4159.1998.70010001.x. [DOI] [PubMed] [Google Scholar]
- Meissner G. Regulation of mammalian ryanodine receptors. Front Biosci. 2002;7:d2072–2080. doi: 10.2741/A899. [DOI] [PubMed] [Google Scholar]
- Michaelis ML, Johe K, Kitos TE. Age-dependent alterations in synaptic membrane systems for Ca2+ regulation. Mechanisms of ageing and development. 1984;25:215–225. doi: 10.1016/0047-6374(84)90142-8. [DOI] [PubMed] [Google Scholar]
- Moriguchi S, Nishi M, Komazaki S, Sakagami H, Miyazaki T, Masumiya H, Saito SY, Watanabe M, Kondo H, Yawo H, Fukunaga K, Takeshima H. Functional uncoupling between Ca2+ release and afterhyperpolarization in mutant hippocampal neurons lacking junctophilins. Proc Natl Acad Sci U S A. 2006;103:10811–10816. doi: 10.1073/pnas.0509863103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison JH, Hof PR. Selective vulnerability of corticocortical and hippocampal circuits in aging and Alzheimer’s disease. Progress in brain research. 2002;136:467–486. doi: 10.1016/s0079-6123(02)36039-4. [DOI] [PubMed] [Google Scholar]
- Moyer JR, Jr, Furtak SC, McGann JP, Brown TH. Aging-related changes in calcium-binding proteins in rat perirhinal cortex. Neurobiology of aging. 2011;32:1693–1706. doi: 10.1016/j.neurobiolaging.2009.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyer JR, Jr, Thompson LT, Black JP, Disterhoft JF. Nimodipine increases excitability of rabbit CA1 pyramidal neurons in an age- and concentration-dependent manner. Journal of neurophysiology. 1992;68:2100–2109. doi: 10.1152/jn.1992.68.6.2100. [DOI] [PubMed] [Google Scholar]
- Murchison D, Griffith WH. Calcium buffering systems and calcium signaling in aged rat basal forebrain neurons. Aging cell. 2007;6:297–305. doi: 10.1111/j.1474-9726.2007.00293.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murchison D, McDermott AN, Lasarge CL, Peebles KA, Bizon JL, Griffith WH. Enhanced calcium buffering in F344 rat cholinergic basal forebrain neurons is associated with age-related cognitive impairment. Journal of neurophysiology. 2009;102:2194–2207. doi: 10.1152/jn.00301.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nixon RA. The calpains in aging and aging-related diseases. Ageing research reviews. 2003;2:407–418. doi: 10.1016/s1568-1637(03)00029-1. [DOI] [PubMed] [Google Scholar]
- Norris CM, Blalock EM, Chen KC, Porter NM, Landfield PW. Calcineurin enhances L-type Ca(2+) channel activity in hippocampal neurons: increased effect with age in culture. Neuroscience. 2002;110:213–225. doi: 10.1016/s0306-4522(01)00574-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris CM, Blalock EM, Chen KC, Porter NM, Thibault O, Kraner SD, Landfield PW. Hippocampal ‘zipper’ slice studies reveal a necessary role for calcineurin in the increased activity of L-type Ca(2+) channels with aging. Neurobiology of aging. 2010;31:328–338. doi: 10.1016/j.neurobiolaging.2008.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh MM, Oliveira FA, Waters J, Disterhoft JF. Altered calcium metabolism in aging CA1 hippocampal pyramidal neurons. J Neurosci. 2013;33:7905–7911. doi: 10.1523/JNEUROSCI.5457-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng X, Wood CL, Blalock EM, Chen KC, Landfield PW, Stromberg AJ. Statistical implications of pooling RNA samples for microarray experiments. BMC Bioinformatics. 2003;4:26. doi: 10.1186/1471-2105-4-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson C, Gibson GE. Synaptosomal calcium metabolism during hypoxia and 3,4-diaminopyridine treatment. Journal of neurochemistry. 1984;42:248–253. doi: 10.1111/j.1471-4159.1984.tb09725.x. [DOI] [PubMed] [Google Scholar]
- Pitler TA, Landfield PW. Aging-related prolongation of calcium spike duration in rat hippocampal slice neurons. Brain research. 1990;508:1–6. doi: 10.1016/0006-8993(90)91109-t. [DOI] [PubMed] [Google Scholar]
- Porter NM, Landfield PW. Stress hormones and brain aging: adding injury to insult? Nat Neurosci. 1998;1:3–4. doi: 10.1038/196. [DOI] [PubMed] [Google Scholar]
- Potier B, Lamour Y, Dutar P. Age-related alterations in the properties of hippocampal pyramidal neurons among rat strains. Neurobiology of aging. 1993;14:17–25. doi: 10.1016/0197-4580(93)90016-5. [DOI] [PubMed] [Google Scholar]
- Protasi F. Structural interaction between RYRs and DHPRs in calcium release units of cardiac and skeletal muscle cells. Front Biosci. 2002;7:d650–658. doi: 10.2741/A801. [DOI] [PubMed] [Google Scholar]
- Riascos D, de Leon D, Baker-Nigh A, Nicholas A, Yukhananov R, Bu J, Wu CK, Geula C. Age-related loss of calcium buffering and selective neuronal vulnerability in Alzheimer’s disease. Acta Neuropathol. 2011;122:565–576. doi: 10.1007/s00401-011-0865-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roderick HL, Berridge MJ, Bootman MD. Calcium-induced calcium release. Curr Biol. 2003;13:R425. doi: 10.1016/s0960-9822(03)00358-0. [DOI] [PubMed] [Google Scholar]
- Rowe WB, Blalock EM, Chen KC, Kadish I, Wang D, Barrett JE, Thibault O, Porter NM, Rose GM, Landfield PW. Hippocampal expression analyses reveal selective association of immediate-early, neuroenergetic, and myelinogenic pathways with cognitive impairment in aged rats. J Neurosci. 2007;27:3098–3110. doi: 10.1523/JNEUROSCI.4163-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samso M, Feng W, Pessah IN, Allen PD. Coordinated movement of cytoplasmic and transmembrane domains of RyR1 upon gating. PLoS biology. 2009;7:e85. doi: 10.1371/journal.pbio.1000085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharfman HE, MacLusky NJ. Estrogen and brain-derived neurotrophic factor (BDNF) in hippocampus: complexity of steroid hormone-growth factor interactions in the adult CNS. Frontiers in neuroendocrinology. 2006;27:415–435. doi: 10.1016/j.yfrne.2006.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedarat F, Xu L, Moore ED, Tibbits GF. Colocalization of dihydropyridine and ryanodine receptors in neonate rabbit heart using confocal microscopy. Am J Physiol Heart Circ Physiol. 2000;279:H202–209. doi: 10.1152/ajpheart.2000.279.1.H202. [DOI] [PubMed] [Google Scholar]
- Simpkins JW, Singh M. More than a decade of estrogen neuroprotection. Alzheimer’s & dementia: the journal of the Alzheimer’s Association. 2008;4:S131–136. doi: 10.1016/j.jalz.2007.10.009. [DOI] [PubMed] [Google Scholar]
- Sohrabji F, Bake S. Age-related changes in neuroprotection: is estrogen pro-inflammatory for the reproductive senescent brain? Endocrine. 2006;29:191–197. doi: 10.1385/ENDO:29:2:191. [DOI] [PubMed] [Google Scholar]
- Stutzmann GE, Smith I, Caccamo A, Oddo S, Laferla FM, Parker I. Enhanced ryanodine receptor recruitment contributes to Ca2+ disruptions in young, adult, and aged Alzheimer’s disease mice. J Neurosci. 2006;26:5180–5189. doi: 10.1523/JNEUROSCI.0739-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supnet C, Bezprozvanny I. Presenilins function in ER calcium leak and Alzheimer’s disease pathogenesis. Cell Calcium. 2011;50:303–309. doi: 10.1016/j.ceca.2011.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeshima H, Komazaki S, Nishi M, Iino M, Kangawa K. Junctophilins: a novel family of junctional membrane complex proteins. Molecular cell. 2000;6:11–22. doi: 10.1016/s1097-2765(00)00003-4. [DOI] [PubMed] [Google Scholar]
- Thibault O, Gant JC, Landfield PW. Expansion of the calcium hypothesis of brain aging and Alzheimer’s disease: minding the store. Aging cell. 2007;6:307–317. doi: 10.1111/j.1474-9726.2007.00295.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thibault O, Hadley R, Landfield PW. Elevated postsynaptic [Ca2+]i and L-type calcium channel activity in aged hippocampal neurons: relationship to impaired synaptic plasticity. J Neurosci. 2001;21:9744–9756. doi: 10.1523/JNEUROSCI.21-24-09744.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thibault O, Landfield PW. Increase in single L-type calcium channels in hippocampal neurons during aging. Science (New York, N Y. 1996;272:1017–1020. doi: 10.1126/science.272.5264.1017. [DOI] [PubMed] [Google Scholar]
- Thibault O, Pancani T, Landfield PW, Norris CM. Reduction in neuronal L-type calcium channel activity in a double knock-in mouse model of Alzheimer’s disease. Biochimica et biophysica acta. 2012;1822:546–549. doi: 10.1016/j.bbadis.2012.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toescu EC, Verkhratsky A. Ca2+ and mitochondria as substrates for deficits in synaptic plasticity in normal brain ageing. Journal of cellular and molecular medicine. 2004;8:181–190. doi: 10.1111/j.1582-4934.2004.tb00273.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tombaugh GC, Rowe WB, Rose GM. The slow afterhyperpolarization in hippocampal CA1 neurons covaries with spatial learning ability in aged Fisher 344 rats. J Neurosci. 2005;25:2609–2616. doi: 10.1523/JNEUROSCI.5023-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu H, Nelson O, Bezprozvanny A, Wang Z, Lee SF, Hao YH, Serneels L, De Strooper B, Yu G, Bezprozvanny I. Presenilins form ER Ca2+ leak channels, a function disrupted by familial Alzheimer’s disease-linked mutations. Cell. 2006;126:981–993. doi: 10.1016/j.cell.2006.06.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veng LM, Mesches MH, Browning MD. Age-related working memory impairment is correlated with increases in the L-type calcium channel protein alpha1D (Cav1.3) in area CA1 of the hippocampus and both are ameliorated by chronic nimodipine treatment. Brain Res Mol Brain Res. 2003;110:193–202. doi: 10.1016/s0169-328x(02)00643-5. [DOI] [PubMed] [Google Scholar]
- Verkhratsky A. Endoplasmic reticulum calcium signaling in nerve cells. Biol Res. 2004;37:693–699. doi: 10.4067/s0716-97602004000400027. [DOI] [PubMed] [Google Scholar]
- Verkhratsky A. Physiology and pathophysiology of the calcium store in the endoplasmic reticulum of neurons. Physiol Rev. 2005;85:201–279. doi: 10.1152/physrev.00004.2004. [DOI] [PubMed] [Google Scholar]
- Wagenknecht T, Radermacher M, Grassucci R, Berkowitz J, Xin HB, Fleischer S. Locations of calmodulin and FK506-binding protein on the three-dimensional architecture of the skeletal muscle ryanodine receptor. J Biol Chem. 1997;272:32463–32471. doi: 10.1074/jbc.272.51.32463. [DOI] [PubMed] [Google Scholar]
- Wang X, Michaelis ML, Michaelis EK. Functional genomics of brain aging and Alzheimer’s disease: focus on selective neuronal vulnerability. Current genomics. 2010;11:618–633. doi: 10.2174/138920210793360943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watfa G, Rossignol P, Kearney-Schwartz A, Fay R, Bracard S, Felblinger J, Boivin JM, Lacolley P, Zannad F, Benetos A. Use of calcium channel blockers is associated with better cognitive performance in older hypertensive patients with subjective memory complaints. J Hypertens. 2010;28:2485–2493. doi: 10.1097/HJH.0b013e32833e4108. [DOI] [PubMed] [Google Scholar]
- Wilkinson JE, Burmeister L, Brooks SV, Chan CC, Friedline S, Harrison DE, Hejtmancik JF, Nadon N, Strong R, Wood LK, Woodward MA, Miller RA. Rapamycin slows aging in mice. Aging cell. 2012;11:675–682. doi: 10.1111/j.1474-9726.2012.00832.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright NT, Prosser BL, Varney KM, Zimmer DB, Schneider MF, Weber DJ. S100A1 and calmodulin compete for the same binding site on ryanodine receptor. J Biol Chem. 2008;283:26676–26683. doi: 10.1074/jbc.M804432200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin HB, Senbonmatsu T, Cheng DS, Wang YX, Copello JA, Ji GJ, Collier ML, Deng KY, Jeyakumar LH, Magnuson MA, Inagami T, Kotlikoff MI, Fleischer S. Oestrogen protects FKBP12.6 null mice from cardiac hypertrophy. Nature. 2002;416:334–338. doi: 10.1038/416334a. [DOI] [PubMed] [Google Scholar]
- Yau JL, Seckl JR. Local amplification of glucocorticoids in the aging brain and impaired spatial memory. Frontiers in aging neuroscience. 2012;4:24. doi: 10.3389/fnagi.2012.00024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zalk R, Lehnart SE, Marks AR. Modulation of the ryanodine receptor and intracellular calcium. Annu Rev Biochem. 2007;76:367–385. doi: 10.1146/annurev.biochem.76.053105.094237. [DOI] [PubMed] [Google Scholar]