

Abstract
The objectives of the present work were to use blends of Eudragit L and hydroxypropyl methylcellulose acetate succinate (HPMCAS) as enteric film coatings for lansoprazole (LSP) pellets. The enteric-coated pellets were prepared with a fluid-bed coater. The influence of the blend ratio, type of plasticizer, plasticizer level, coating level, and curing conditions on gastric stability in vitro drug release and drug stability was evaluated. Furthermore, the bioavailability of the blend-coated pellets in beagle dogs was also performed. The blend-coated pellets exhibited significant improvement of gastric stability and drug stability compared to the pure polymer-coated pellets. Moreover, the AUC values of blend-coated pellets were greater than that of the pure polymer-coated pellets. It was concluded that the using blends of Eudragit L and HPMCAS as enteric film coatings for LSP pellets improved the drug stability and oral bioavailability.
Electronic supplementary material
The online version of this article (doi:10.1208/s12249-013-0035-1) contains supplementary material, which is available to authorized users.
KEY WORDS: bioavailability, drug stability, enteric coating, lansoprazole, pellets
INTRODUCTION
Proton pump inhibitors (PPIs) are a commonly prescribed class of medications whose main action is to create a pronounced and long-lasting reduction of stomach acid production. They are commonly used to treat gastroesophageal reflux disease, frequent heartburn, and acid regurgitation, esophagitis, duodenal and gastric ulcers, H. pylori eradication, gastrointestinal lesions caused by nonsteroidal anti-inflammatory drugs, and Zollinger–Ellison syndrome (1–4).
Lansoprazole (LSP) is one of the PPIs, consisting of a substituted benzimidazole ring and a pyridine ring connected by a sulfoxide-containing chain. It is a lipophilic and weak base with pKa values of 4.15 and 1.33, while the N–H proton in the benzimidazole ring is responsible for the acidity of the molecule (pKa 8.84) (5–7). Compared to other PPIs, it is more instable to heat, light, and acidic medium. Especially, the drug degrades rapidly in acid medium (8). Thus, an enteric coating must be applied to the solid dosage form to prevent the drug from degradation in stomach and allow drug release in small intestine.
The enteric film coating polymers includes cellulose acetate phthalate, cellulose acetate trimellitate, hydroxypropyl methylcellulose acetate succinate (HPMCAS), hydroxypropyl methylcellulose phthalate, polyvinyl acetate phthalate, and methacrylic acid/methyl methacrylate copolymers (Eudragit series). The polymers contain carboxylic groups and thus reveal pH-dependent solubility; at higher pH, the carboxylic groups became ionized and make the polymers dissolve; at lower pH, the carboxylic groups are not ionized and render them insoluble (9,10). Among the polymers, Eudragit L, containing an anionic copolymer based on methacrylic acid/ethyl acrylate (1:1), is the most widely used in the pharmaceutical industry. It is produced by spray-drying of Eudragit L 30D and easily redispersed into water with the aid of small amounts of alkali or organic base, resulting in redispersed latex with a final pH 2–3. The polymer becomes ionized at pH values of about 5 or higher. HPMCAS is another enteric coating agent, whose pKa is 5. It is a cellulose ester and contains methyl, hydroxypropyl, acetyl, and succinoyl groups on a cellulose backbone. Less than 10% and more than 50% are ionized at pH values below 4 and above 5, respectively. Because of the presence of hydrophobic methoxy and acetate substituents, it is water insoluble when unionized and remains predominantly colloidal at intestinal pH (11).
However, enteric polymers influence the drug stability and bioavailability greatly, since an interaction between the free carboxyl groups contained in enteric polymer and the drug would occur (10,12,13). A conventional enteric coating applied to LSP formulations contains one enteric polymer. By contrast, the use of polymer blends as coating materials offer many advantages as follows (14–19): (1) facilitated adjustment of desired drug release patterns, mechanical properties, and drug release mechanisms, (2) improved film formation and storage stability, and (3) the possibility to develop novel strategies for site-specific drug delivery within the gastrointestinal tract. Considering the excellent enteric-coating protection of Eudragit L and lower influence of HPMCAS on PPIs stability (13,20–22), we used blends of Eudragit L and HPMCAS as enteric film coatings for LSP pellets. To date, few reports have indicated that polymer blends are utilized as enteric coating materials for PPIs formulation.
Thus, the aim of the present work was to (a) use blends of Eudragit L and HPMCAS as enteric film coating for LSP pellets; (b) evaluate the blend ratio, type of plasticizer, plasticizer level, coating level, and curing conditions on gastric stability, in vitro drug release, and drug stability; and (c) study the bioavailability of the blend-coated pellets in beagle dogs.
MATERIALS AND METHODS
Materials
LSP was purchased from ZhuhaiRuntong Pharma Ltd. (Zhuhai, China). Non-pareil pellets (sugar spheres 0.5–0.7 mm in diameter) were provided by Gaocheng Biotech & Health Co., Ltd. (Hangzhou, China). Hydroxypropyl methylcellulose (HPMC, 60RT5) was purchased from Feichengruitai Ltd. (Shandong, China). Eudragit L was a gift from Evonik Degussa Co., Ltd. (Darmstadt, Germany). Sodium carbonate was purchased from Er-Kang Pharma Ltd. (Hunan, China). HPMCAS-HF (AQOAT) was purchased from Shin-Etsu Chemical Co., Ltd. (Tokyo, Japan). Triethyl citrate (TEC) was obtained from Alladin Reagent (Shanghai, China). Polyethylene glycol 6000 (PEG 6000), dibutyl sebacate (DBS), and diisobutyl phthalate (DBP) were from SCR Co., Ltd. (Beijing, China). Other reagents were of analytical grade.
Preparation of Film-Coated Pellets of LSP
The drug-layered pellets were prepared by layering the drug suspensions on nonpareil pellets, achieving 20% drug content (22,23). Before an enteric film coating, a HPMC sub-coating with 40% weight gain was layered onto the pellets. The subcoated pellets were then enteric coated by HPMCAS, Eudragit L dispersion, or their blends. The aqueous polymer dispersion were separately plasticized overnight with 5–30% plasticizer (w/w, based on the total polymer mass) and adjusted to 10% (w/w) polymer content with purified water. The blends were obtained by adding the HPMCAS dispersion into the Eudragit L dispersion under the condition of electromagnetic stirring. After talc (50% w/w, based on the total polymer mass) was added to the coating formulations, the coating dispersions were sprayed onto the subcoated pellets until a coating level of 10–50% (w/w) was achieved. The polymer content prior to coating was adjusted to 10%. The following Eudragit L/HPMCAS blend ratios were studied: 1:0, 1:1, 1:2, 1:3, 1:4, and 0:1 (w/w).
The process parameters were atomizing pressure = 1.5–2.0 bars, inlet air temperature = 45–50°C, inlet air = 45–50 m3/h, exhaust air temperature = 30–35°C, pellet bed temperature = 40–45°C, and spray rate = 1.5–2.0 mL/min. After the coating process, a pellet curing was performed. The pellets were further fluidized in the coater for 15 min at 40°C and subsequently cured for 2–24 h at 40 or 60°C in an oven.
In Vitro Drug Release
After immersed in acidic medium (0.1 M HCl) for 1 h, the drug release from the pellets was measured in a paddle USP apparatus (75 rpm, 37°C, 900 mL (pH 6.8)). At specific time intervals, samples were withdrawn and analyzed using an HPLC assay described below.
Scanning Electron Microscopy
The micrographs of the coated pellets were taken with a scanning electron microscopy (SEM) (S-3500N, SEM, Hitachi, Tokyo, Japan) to examine the surfaces and morphology of the pellets. The pellets were mechanically cleaved transversely and sputtered with gold for 5 min by a sputter.
Stability Studies
Gastric Stability
The gastric-stability study was performed by exposing the pellets in acid medium (500 mL, 0.1 M HCl) for 1 h. The amount of drug degradation was determined by a UV method described below.
Accelerated Stability
The coated pellets that were packaged with aluminium foil were stored under accelerated conditions of 40°C/75% RH. The gastric stability and drug content of the pellets was measured at initial, 1, 3, and 6 months.
Drug Content Estimation of Pellets
The acidic degradation of LSP was composed of several compounds making it difficult to determine by HPLC. Thus, according to USP method, the amount of drug degradation (drug release) in acidic condition was determined with a UV spectrophotometer (UV2200, Shimazu, Japan) at a wavelength of 245 nm. The drug content and drug release in PBS buffer was evaluated by HPLC assay (Waters 515 pump/2487 UV detector, Waters, Milford, MA, USA). The separations were performed at 30°C using a 250 mm × 4.6 mm column (DiamonsilTM C18). The mobile phase was consisted of water/acetonitrile/triethylamine (60/40/1, pH 7.0) and was pumped at a flow rate of 1.0 mL/min. The eluent was detected by UV detector at 285 nm.
Bioavailability in Dogs
The bioavailability of HPMCAS, Eudragit L dispersion, and Eudragit L/HPMCAS blend (1:3)-coated pellets (coating level 30%) of LSP were assessed and compared in dogs in a randomized cross-over study. The wash out period was 1 week. Six male beagle dogs (8–10 kg) used in the experiments received care in compliance with the Principles of Laboratory Animal Care and the Guide for the Care and Use of Laboratory Animals. Experiments followed protocol approved by the Hebei Medical University Institutional Animal Care and Use Committee.
The dogs were fasted 12 h before administration. The hard gelatin capsules filled with pellets was orally administered to the dogs at a dosage of 5 mg/kg. All of the formulations were administered with water of 20 mL. Blood samples (2 mL) were collected from saphenous vein into heparinized tubes at the following time points: 0.25, 0.5, 0.75, 1, 1.5, 2, 2.5, 3, 4, 5, 6, 8, and 12 h. The heparinized blood samples were immediately centrifuged at 4,000×g for 10 min in a desktop centrifuge (Anke TGL-16G, China), and the plasma was separated and transferred to microcentrifuge tubes. The plasma samples were frozen at −18°C until analysis. Frozen plasma samples were prepared by a procedure reported by Ito et al. (24), which the drug samples was measured by a validated HPLC method described below.
HPLC Analysis
The LSP concentrations in plasma were determined using an HPLC assay reported by Dugger and coworkers (25). Briefly, a 1-mL aliquot of plasma was extracted with 6 mL of extraction solution containing internal standard (megestrol acetate, 100 μg/mL in methyl-t-butyl ether). After mixing and centrifugation, the organic phase was removed and evaporated to dryness under nitrogen stream. The residue was reconstituted in 100 mL of methanol and centrifuged at 10,000×g for 5 min, and then 50 μL of the supernatant liquid was injected onto the HPLC system.
The HPLC system consisted of a Waters 2487 detector (UV) and an Empower workstation. The separations were performed at 25°C using a 250 mm × 4.6 mm column (DiamonsilTM C18). The mobile phase was consisted of 46% acetonitrile and 54% KH2PO4 buffer (pH 4.5) and was pumped at a flow rate of 1.5 mL/min. The eluent was detected by UV detector at 266 nm and corresponding peak areas were recorded.
Data Analysis
Pharmacokinetic parameters were calculated by non-compartmental analysis based on statistical moment theory using Microsoft Excel 2003. The pharmacokinetic parameters, such as maximum plasma concentration (Cmax) and time of maximum concentration (Tmax), were obtained directly from the plasma concentration-time plots. The area under the plasma concentration-time curve up to the last time (t) (AUC0–t) was calculated using the linear trapezoidal rule.
The results were expressed as mean ± standard deviation. One-way analysis of variance was performed to assess the statistical significance of differences among samples. Results with P < 0.05 were considered statistically significant.
RESULTS AND DISCUSSION
Preparation and Characterization of Pellets
The enteric-coated pellets of LSP could be successfully prepared by the blends of Eudragit L and HPMCAS. As shown in Fig. 1, the surface of the Eudragit L-coated pellets was smooth, while the HPMCAS-coated pellets possessed rough surface. It was ascribed to the difference in the particle size of the two dispersions (16), which the particle size of HPMCAS and Eudragit L dispersion was about 5 μm and 300 nm, respectively. Increasing the particle size decreased the ability of polymer particles to fuse into a homogeneous film, forming a rough film (26,27). Thus, the blend (1:3 ratio)-coated pellets also possessed rough surface.
Fig. 1.

Scanning electron pictures of surface of Eudragit L a, HPMCAS b, and Eudragit L/HPMCAS blend (c, 1:3 ratio) coated pellets (coating level 30%). The curing condition for the coated pellets was 12 h at 40°C
The coating process went smoothly when the blends were used as film coatings. During the experiment, we found that the coating process with pure HPMCAS dispersion was poor, which the spray-nozzle blocking and precipitation were observed. Interestingly, the addition of Eudragit L dispersion not only did not cause any flocculation of aqueous HPMCAS dispersion, but also improved the coating process significantly. The absolute value of zeta-potential of Eudragit L dispersion was about 55 mV, while the value of HPMCAS dispersion was only 10 mV. Thus, the force of electrostatic repulsion between the particles in dispersion was enhanced, inhibiting the precipitation and aggregation.
It is well accepted that the drug release of the formulations has significant effect on the bioavailability. Thus, we investigated the effects of various processing parameters including blend ratio, type and amount of plasticizer, coating level, and curing conditions on drug degradation in acid medium and drug release in PBS buffer.
Effect of Blend Ratio on Drug Release
As shown in Fig. 2a, the drug release of Eudragit L/HPMCAS blend-coated pellets was slower than that of pure Eudragit L- or HPMCAS-coated pellets. At the first 30 min, the drug release decreased with an increase in the mounts of HPMCAS. It was attributed to the formation of tighter films when the blends were utilized as film-coatings for the pellets. After immersed in acid medium (0.1 M HCl), the water uptake of blend-coated pellets was significantly less than that of the pure polymer-coated pellets (S1). Thus, the swelling of the coated pellets was suppressed and then affected the rupture of coated pellets, retarding the drug release. Moreover, less water uptake was not beneficial to the production of microenvironments inside the pellets, delaying the dissolution of the enteric polymer coatings (28,29).
Fig. 2.
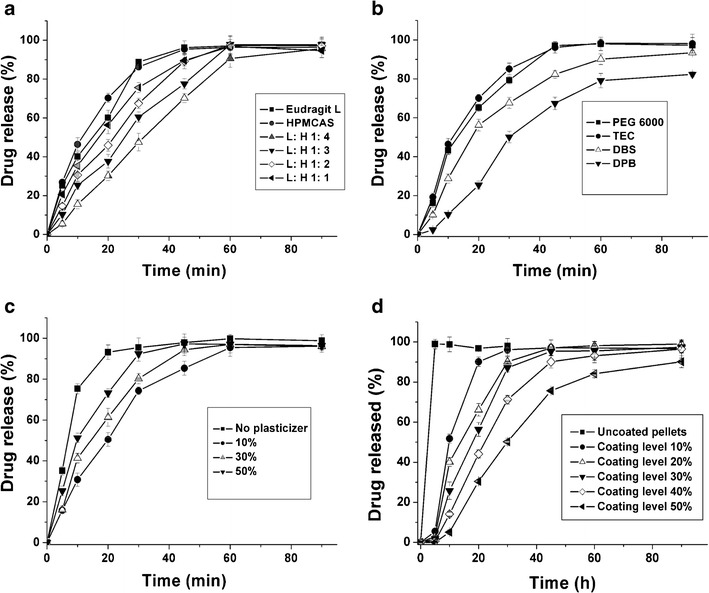
Effect of Eudragit L/HPMCAS (L: H) blend ratio (a, plasticized by TEC, coating level 30%), type of plasticizer (b, 1:3 ratio, coating level 30%), plasticizer level (c, Eudragit L/HPMCAS 3:1 ratio, plasticized by 30% TEC), and coating level (d, 1:3 ratio, plasticized by 30% TEC) on the drug release from the coated pellets. The curing condition for the coated pellets was 12 h at 40°C
Effect of Type of Plasticizer on Drug Release
Clearly, the type of plasticizer strongly affected the drug release (Fig. 2b). The addition of plasticizer into the films induced greater mobility of the polymer chains by replacing polymer–polymer interactions by polymer–plasticizer interactions (30). Thus, lowering the glass transition temperature of the films and enhanced polymer particle coalescence are achieved. The drug release from the pellets coated by the dispersion plasticized by TEC and PEG 6000 was faster when compared to that of DBS and DBP. Due to higher water solubility of TEC and PEG 6000, their films took up water more rapidly, rendering an increase in the permeability of films (17).
Importantly, the type of plasticizer also affected the gastric stability. When the coated pellets were immersed in acid medium for 1 h, the drug degradation from the pellets coated by blend dispersion plasticized by PEG 6000, TEC, DBS, and DBP, was 7.8, 1.1, 3.0, and 3.6%, respectively. The drug release from coated pellets in acid medium relied on the diffusion through the pores produced by leaching out the plasticizer from the films. TEC was a better plasticizer for Eudragit dispersion and it was the only compatible plasticizer for HPMCAS dispersion. Thus, the addition of TEC rendered a formation of more uniform and continuous film, hindering the leaching out of plasticizer. Therefore, it was observed that the pellets coated by blend dispersion plasticized by TEC was more beneficial to the gastric stability of LSP.
Effect of Level of Plasticizer on Drug Release
As shown in Fig. 2c, the level of TEC in the films influenced the drug release. The drug release from the pellets coated by the dispersion without TEC was faster than that of the pellets coated by dispersion plasticized by TEC. It was explained that polymer particle could not coalesce into a uniform film without addition of TEC. The drug release was slightly increased when the plasticizer level was varied from 10 to 50%. It was attributed to the formation of more permeable film coatings due to the presence of higher hydrophilicity of TEC. It was reported that the drug release could be accelerated when water-soluble materials such as HPMC and poly(vinyl alcohol)–poly(ethylene glycol) graft copolymer were added into the film coatings (31,32).
The level of TEC in the films also produced significant effect on the gastric stability. The amount of drug degradation for level of 0, 10, 30, and 50% was 32.56, 4.12, 1.54, and 2.15%, respectively. The presence of TEC in the films decreased the drug degradation significantly, and the amount of degradation from the level of 30% was less than other levels. It was explained that the addition of plasticizer could improve the coalescence between the polymer particles and decreased the drug release.
On the other hand, the plasticizer, TEC, could also behave as a pore-forming agent. When a threshold value of TEC level is reached, a further increase of its level would result in an increase in the permeability of the films and drug release from the coated pellets. Thus, it was observed that the drug degradation level of 50% was slightly greater than that of 30%.
Effect of Coating Level on Drug Release
The coating level produced significant effect on the drug release (Fig. 2d). At the first 30 min, the drug release was gradually decreased when the coating level was increased from 0 to 50%. The gastric stability of uncoated pellets was very poor, indicating that all of the drug was degraded when the pellets were immersed in acid medium for 10 min. Thus, the testing for the gastric stability and drug release of uncoated pellets was not performed. It was observed that the films of coated pellets were excellent without damage in the first 20 min. Thus, it was explained that the increased coating level led to an increase in length of diffusion pathways and increasing the time required for the drug to diffuse through the coating membrane. Importantly, even at 50% coating level, not less than 80% of the drug was released, indicating that a rapid drug release was achieved.
Effect of Curing Condition on Drug Release and Gastric Stability
The drug release and gastric stability was influenced by the curing conditions. Figure 3a, b shows the drug release obtained from the coated pellets collected at specific time intervals during curing procedure. Compared to uncured pellets, the cured pellets, irrespective of the curing time and temperature, exhibited a slower drug release. The drug degradation was 30.5% when the uncured pellets were immersed in acid medium for 1 h, while the drug degradation was less than 9.0% (Fig. 3c). It indicated that film formation was incomplete after coating procedure, thus a curing procedure must impose on the initial coated pellets. After curing at 40°C for 12 h or 60°C for 4 h, the drug release and drug degradation were not altered, suggesting that a stable film coating system was achieved. An increase in temperature resulted in increasing macromolecular mobility and then decreasing the curing time (33,34).
Fig. 3.
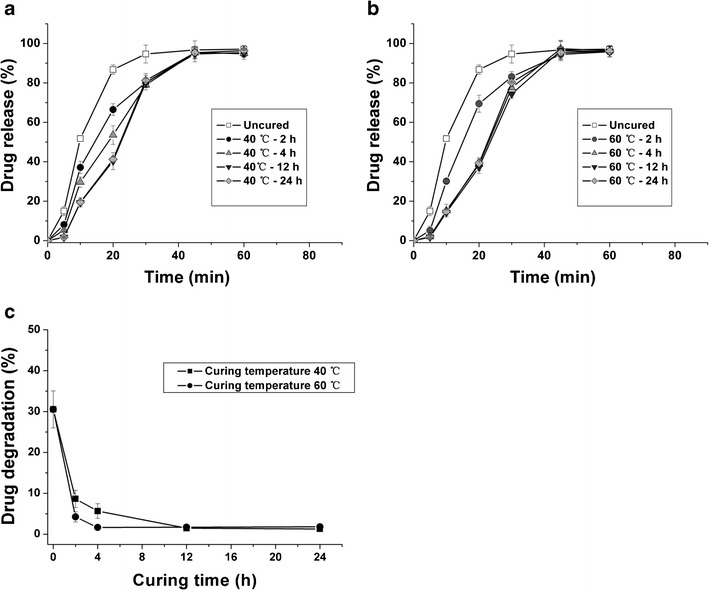
Effect of curing conditions on drug release (a, b) and drug degradation c from pellets coated by Eudragit L/HPMCAS blend (1:3 ratio). The dispersion was plasticized by 30% TEC and the coating level was 30%
Stability Studies
Gastric Stability
As shown in Fig. 4, the drug degradation was about 2 and 5% for HPMCAS- and Eudragit L-coated pellets, whereas the degradation was less than 0.5% for the blend-coated pellets. After storage for 6 months at 40°C/75% RH, the drug degradation obtained from the blend-coated pellets was less than 5%, while the degradation was 15.9 and 10% for Eudragit L- or HPMCAS-coated pellets. Therefore, the blend-coated pellets significantly improved the gastric stability of LSP. During the acid phase, the swelling of the film coating, water penetration into the core, drug dissolution, and subsequent diffusion through the hydrated polymeric film were contributed to the drug release (35,36). HPMCAS is poorly water soluble due to the presence of lots of hydrophobic substituents (37). Thus, the addition of HPMCAS made the coating films more hydrophobic, reducing the water permeation and swelling degree of the film coating. Thus, the blend-coated pellets were beneficial to reducing the drug release in acid medium and improving the gastric stability of LSP. Recently, it was also reported that the blends of two polymers for pellet coating could improve film formation and storage stability, not altering the drug release (19,32).
Fig. 4.

Effect of Eudragit L/HPMCAS (L: H) blend ratio on the drug degradation from coated pellets before and after being stored at 40°C/75% RH for 0, 1, 3, and 6 months. The dispersion was plasticized by 30% TEC and the coating level was 30%. The curing condition for the coated pellets was 12 h at 40°C
Accelerated Stability
After storage for 6 months at 40°C/75% RH, the remaining drug of blend-coated pellets was 86, 82, 76, and 70% for 1:4, 1:3, 1:2, and 1:1 ratio, whereas the remaining drug from Eudragit L- and HPMCAS-coated pellets was 50 and 61% (Fig. 5). The blend-coated pellets, regardless of the blend ratio, significantly improved the drug stability. It was ascribed to the decreased moisture absorption that played a key role in the stability of LSP in formulations (13,23). The enteric polymers, due to presence of an ester structure, were susceptible to hydrolysis in the conditions of humidity, affecting the enteric protection in acid medium. Moreover, the absorbed moisture would make the drug migrate into the enteric film and interact with its acidic carboxyl groups of the polymer. HPMCAS, due to the presence of hydrophobic substituents, was water insoluble, thus its incorporation could reduce the hydrophilic nature of films and then decreased moisture absorption (11).
Fig. 5.

Effect of Eudragit L/HPMCAS (L: H) blend ratio on the drug remaining from coated pellets after being stored at 40°C/75% RH for 1, 3, and 6 months. The dispersion was plasticized by 30% TEC and the coating level was 30%. The curing condition for the coated pellets was 12 h at 40°C
It was observed that the remaining drug was direct ratio to the amount of Eudragit L in the films. It was explained that the amount of free carboxyl groups contained in the enteric polymers was direct ratio to the drug degradation. Compared to Eudragit L, the degradation ability of HPMCAS was weaker since HPMCAS contained less free carboxyl groups in its structure (13). LSP is a member of PPIs, thus a similar effect might be produced.
Pharmacokinetics in Dogs
Mean plasma LSP concentration versus time profiles following a single oral dose of the five formulations are shown in Fig. 6. Mean values of the pharmacokinetic parameters are summarized in Table I.
Fig. 6.

Plasma lansoprazole concentrations after oral administration of coated pellets at a dose of 5 mg/kg in dogs. (n = 6)
Table I.
Pharmacokinetic Parameters of LSP After Oral Administration of Coated Pellets
| PK parameters | Eudragit L | HPMCAS | 3:1 |
|---|---|---|---|
| C max (μg/mL) | 0.95 ± 0.28 | 1.23 ± 0.45 | 1.31 ± 0.32 |
| T max | 1.50 ± 0.35 | 2.00 ± 0.56 | 2.50 ± 0.69* |
| AUC0-t (μg h/mL) | 2.24 ± 0.78 | 2.63 ± 0.37 | 3.39 ± 0.11* |
* Statistically higher than purely polymer-coated pellets (P < 0.05)
The Tmax/Cmax of LSP from Eudragit L/HPMCAS blend (1:3 ratio)-coated pellets was 2.50 ± 0.69 h/1.31 ± 0.32 μg mL−1. In the case of pure Eudragit L- or HPMCAS-coated pellets, the Tmax/Cmax was 1.50 ± 0.35 h/0.95 ± 0.28 μg mL−1 and 2.00 ± 0.56 h/1.23 ± 0.45 μg mL−1, respectively, which Tmax differed from the values obtained from the blend-coated pellets (P < 0.05). Interestingly, the AUC values of blend-coated pellets were 3.39 ± 0.11 μg h mL−1, which was greater than that of the pure polymer-coated pellets (2.24 ± 0.78 and 2.63 ± 0.37 μg h mL−1) (P < 0.05). The relative bioavailability calculated by the ratio of blend-coated formulation AUC to Eudragit L- or HPMCAS-coated formulation AUC was 157.56 ± 34.45 and 133.11 ± 18.61%, respectively. The enhanced absorption was ascribed to the fact that the ionized HPMCAS could improve the solubility of insoluble drugs (11,37). You and coworkers (38) also reported that HPMCAS could function as surfactants to stabilize the emulsions for water-insoluble drugs and enhance the drug absorption. Additionally, compared to pure Eudragit L or HPMCAS, the Eudragit L/HPMCAS blends had different values of carboxylic group contents, which led to variation in disintegration site of enteric-coated pellets in the small intestine and a time lag and then affected the bioavailability of the drugs (39,40). Fourthly, beagle dogs were widely used to study the bioavailability of oral formulations, since the dimensions of the GI tract are similar enough to permit the administration of dosage forms (41). However, the gastric pH in fasted dogs was not similar to that in humans and the fluctuation of pH must be considered. Due to low basal gastric acid secretion, the gastric pH in fasting dogs fluctuated (2.7–8.3), and it was as high as the pH of its duodenal content, which would produce significant effect on the drug absorption of preparations with pH-dependent release (42,43). If the enteric-coated pellets were administered to the fasted dogs, the enteric films might be damaged and led to premature drug release, drug instability, and decreasing bioavailability. The drug release of conventional enteric-coated dosage forms always occurred in the distal small intestine, resulting in a delayed response to medication and decreased the drug bioavailability (28). The blend-coated pellets might get over the physiological variations and improved the conformity of disintegration in gastrointestinal tract.
CONCLUSIONS
The blend of Eudragit L/HPMCAS as enteric film coatings for LSP pellets improved the gastric stability, storage stability, and oral bioavailability. There was of significant influence of the blend ratio, type and amount of plasticizer, coating level, and curing conditions on the drug degradation and drug release.
Electronic supplementary material
{kind=link}
Effect of Eudragit L/HPMCAS (L:H) blend ratio on the water-uptake from coated pellets after immersed in acid medium for 2 h. The dispersion was plasticized by 30% TEC and the coating level was 30%. The curing condition for the coated pellets was 12 h at 40°C. (JPEG 434 kb)
Acknowledgments
Conflict of interest
The authors declare that they have no conflicts of interest to disclose.
References
- 1.Lai KC, Lam SK, Chu KM, Wong BC, Hui WM, Hu WH, et al. Lansoprazole for the prevention of recurrences of ulcer complications from long-term low-dose aspirin use. N Engl J Med. 2002;346(26):2033–2038. doi: 10.1056/NEJMoa012877. [DOI] [PubMed] [Google Scholar]
- 2.Baldi F. Lansoprazole oro-dispersible tablet—pharmacokinetics and therapeutic use in acid-related disorders. Drugs. 2005;65(10):1419–1426. doi: 10.2165/00003495-200565100-00007. [DOI] [PubMed] [Google Scholar]
- 3.Savarino V, Di Mario F, Scarpignato C. Proton pump inhibitors in GORD An overview of their pharmacology, efficacy, and safety. Pharmacol Res. 2009;59(3):135–153. doi: 10.1016/j.phrs.2008.09.016. [DOI] [PubMed] [Google Scholar]
- 4.Edwards SJ, Borrill J. Proton pump inhibitors for the initial treatment of gastroesophageal reflux disease (GORD) symptoms in patients with reflux esophagitis: A systematic review of randomized controlled trials. Value Health. 2007;10(6):A352-A
- 5.Cirilli R, Ferretti R, Gallinella B, Turchetto L, Zanitti L, La Torre F. Development and validation of an enantioselective and chemoselective HPLC method using a Chiralpak IA column to simultaneously quantify (R)-(+)- and (S)-(−)-lansoprazole enantiomers and related impurities. J Pharm Biomed Anal. 2009;50(1):9–14. doi: 10.1016/j.jpba.2009.03.021. [DOI] [PubMed] [Google Scholar]
- 6.Jain KS, Shah AK, Bariwal J, Shelke SM, Kale AP, Jagtap JR, et al. Recent advances in proton pump inhibitors and management of acid-peptic disorders. Bioorg Med Chem. 2007;15(3):1181–1205. doi: 10.1016/j.bmc.2006.07.068. [DOI] [PubMed] [Google Scholar]
- 7.Hellstrom PM, Vitols S. The choice of proton pump inhibitor: does it matter? Basic Clin Pharmacol Toxicol. 2004;94(3):106–111. doi: 10.1111/j.1742-7843.2004.pto940302.x. [DOI] [PubMed] [Google Scholar]
- 8.Albin K, Franc V. Preformulation investigation of the novel proton pump inhibitor lansoprazole. Drug Dev Ind Pharm. 2000;26:781–783. doi: 10.1081/DDC-100101299. [DOI] [PubMed] [Google Scholar]
- 9.de Oliveira HP, Albuquerque JJF, Jr, Nogueiras C, Rieumont J. Physical chemistry behavior of enteric polymer in drug release systems. Int J Pharm. 2009;366:185–189. doi: 10.1016/j.ijpharm.2008.08.041. [DOI] [PubMed] [Google Scholar]
- 10.Missaghi S, Young C, Fegely K, Rajabi-Siahboomi AR. Delayed release film coating applications on oral solid dosage forms of proton pump inhibitors: case studies. Drug Dev Ind Pharm. 2010;36(2):180–189. doi: 10.3109/03639040903468811. [DOI] [PubMed] [Google Scholar]
- 11.Friesen DT, Shanker R, Crew M, Smithey DT, Curatolo WJ, Nightingale JA. Hydroxypropyl methylcellulose acetate succinate-based spray-dried dispersions: an overview. Mol Pharm. 2008;5(6):1003–1019. doi: 10.1021/mp8000793. [DOI] [PubMed] [Google Scholar]
- 12.Storpirtis S, Rodrigues D. In vitro evaluation of dissolution properties and degradation products of omeprazole in enteric-coated pellets. Drug Dev Ind Pharm. 1998;24(11):1101–1107. doi: 10.3109/03639049809089956. [DOI] [PubMed] [Google Scholar]
- 13.Stroyer A, McGinity JW, Leopold CS. Solid-state interactions between the proton pump inhibitor omeprazole and various enteric coating polymers. J Pharm Sci. 2006;95(6):1342–1353. doi: 10.1002/jps.20450. [DOI] [PubMed] [Google Scholar]
- 14.Lecomte F, Siepmann J, Walther M, MacRae RJ, Bodmeier R. Blends of enteric and GIT-insoluble polymers used for film coating: physicochemical characterization and drug release patterns. J Control Release. 2003;89(3):457–471. doi: 10.1016/S0168-3659(03)00155-X. [DOI] [PubMed] [Google Scholar]
- 15.Siepmann F, Siepmann J, Walther M, MacRae RJ, Bodmeier R. Polymer blends for controlled release coatings. J Control Release. 2008;125(1):1–15. doi: 10.1016/j.jconrel.2007.09.012. [DOI] [PubMed] [Google Scholar]
- 16.Siepmann F, Siepmann J, Walther M, MacRae RJ, Bodmeier R. Blends of aqueous polymer dispersions used for pellet coating: importance of the particle size. J Control Release. 2005;105(3):226–239. doi: 10.1016/j.jconrel.2005.03.028. [DOI] [PubMed] [Google Scholar]
- 17.Lecomte F, Siepmann J, Walther M, MacRae RJ, Bodmeier R. Polymer blends used for the aqueous coating of solid dosage forms: importance of the type of plasticizer. J Control Release. 2004;99(1):1–13. doi: 10.1016/j.jconrel.2004.05.011. [DOI] [PubMed] [Google Scholar]
- 18.El-Malah Y, Nazzal S. Novel use of Eudragit NE 30D/Eudragit L 30D-55 blends as functional coating materials in time-delayed drug release applications. Int J Pharm. 2008;357:219–227. doi: 10.1016/j.ijpharm.2008.02.003. [DOI] [PubMed] [Google Scholar]
- 19.Kranz H, Gutsche S. Evaluation of the drug release patterns and long term stability of aqueous and organic-coated pellets by using blends of enteric and gastrointestinal insoluble polymers. Int J Pharm. 2009;380:112–119. doi: 10.1016/j.ijpharm.2009.07.013. [DOI] [PubMed] [Google Scholar]
- 20.Riedel A, Leopold CS. Degradation of omeprazole induced by enteric polymer solutions and aqueous dispersions: HPLC investigations. Drug Dev Ind Pharm. 2005;31(2):151–160. doi: 10.1081/DDC-200047787. [DOI] [PubMed] [Google Scholar]
- 21.Riedel A, Leopold CS. Quantification of omeprazole degradation by enteric coating polymers: an UV–VIS spectroscopy study. Pharmazie. 2005;60(2):126–130. [PubMed] [Google Scholar]
- 22.He W, Tian Z, Yang M, Fan J, Zhang S, Guan P, et al. A comparative study of the effect of different alkaline stabilizers on physicochemical properties of lansoprazole in formulation. Asian J Pharm Sci. 2011;6(3–4):89–100. [Google Scholar]
- 23.He W, Yang M, Fan JH, Feng CX, Zhang SJ, Wang JX, et al. Influences of sodium carbonate on physicochemical properties of lansoprazole in designed multiple coating pellets. AAPS Pharmscitech. 2010;11(3):1287–1293. doi: 10.1208/s12249-010-9493-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ito Y, Arai H, Uchino K, Iwasaki K, Shibata N, Takada K. Effect of adsorbents on the absorption of lansoprazole with surfactant. Int J Pharm. 2005;289:69–77. doi: 10.1016/j.ijpharm.2004.10.010. [DOI] [PubMed] [Google Scholar]
- 25.Dugger HA, Carlson JD, Henderson W, Erdmann GR, Alam SM, Dham R, et al. Bioequivalence evaluation of lansoprazole 30-mg capsules (Lanfast and Lanzor) in healthy volunteers. Eur J Pharm Biopharm. 2001;51(2):153–157. doi: 10.1016/S0939-6411(00)00152-1. [DOI] [PubMed] [Google Scholar]
- 26.Siepmann F, Siepmann J, Walther M, MacRae R, Bodmeier R. Aqueous HPMCAS coatings: effects of formulation and processing parameters on drug release and mass transport mechanisms. Eur J Pharm Biopharm. 2006;63(3):262–269. doi: 10.1016/j.ejpb.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 27.Steward PA, Hearn J, Wilkinson MC. An overview of polymer latex film formation and properties. Adv Colloid Interface Sci. 2000;86(3):195–267. doi: 10.1016/S0001-8686(99)00037-8. [DOI] [PubMed] [Google Scholar]
- 28.Liu F, Lizio R, Meier C, Petereit H-U, Blakey P, Basit AW. A novel concept in enteric coating: a double-coating system providing rapid drug release in the proximal small intestine. J Control Release. 2009;133(2):119–124. doi: 10.1016/j.jconrel.2008.09.083. [DOI] [PubMed] [Google Scholar]
- 29.Liu F, Moreno P, Basit AW. A novel double-coating approach for improved pH-triggered delivery to the ileo-colonic region of the gastrointestinal tract. Eur J Pharm Biopharm. 2011;74:311–315. doi: 10.1016/j.ejpb.2009.11.008. [DOI] [PubMed] [Google Scholar]
- 30.Laboulfie F, Hmati M, Lamure A, Diguet S. Effect of the plasticizer on permeability, mechanical resistance and thermal behaviour of composite coating films. Powder Technol. 2012;(0). doi:10.1016/j.powtec.2012.07.035
- 31.Frohoff-Hulsmann MA, Lippold BC, McGinity JW. Aqueous ethyl cellulose dispersion containing plasticizers of different water solubility and hydroxypropyl methyl-cellulose as coating material for diffusion pellets II: properties of sprayed films. Eur J Pharm Biopharm. 1999;48(1):67–75. doi: 10.1016/S0939-6411(99)00023-5. [DOI] [PubMed] [Google Scholar]
- 32.Siepmann F, Muschert S, Leclercq B, Carlin B, Siepmann J. How to improve the storage stability of aqueous polymeric film coatings. J Control Release. 2008;126(1):26–33. doi: 10.1016/j.jconrel.2007.10.018. [DOI] [PubMed] [Google Scholar]
- 33.Muschert S, Siepmann F, Leclercq B, Siepmann J. Dynamic and static curing of ethylcellulose: PVA-PEG graft copolymer film coatings. Eur J Pharm Biopharm. 2010;78(3):455–461. doi: 10.1016/j.ejpb.2011.02.010. [DOI] [PubMed] [Google Scholar]
- 34.Cuppok Y, Muschert S, Marucci M, Hjaertstam J, Siepmann F, Axelsson A, et al. Drug release mechanisms from Kollicoat SR: Eudragit NE- coated pellets. Int J Pharm. 2011;409:30–37. doi: 10.1016/j.ijpharm.2011.02.026. [DOI] [PubMed] [Google Scholar]
- 35.Ozturk SS, Palsson BO, Donohoe B, Dressman JB. Kinetics of release from enteric-coated tablets. Pharm Res. 1988;5(9):550–565. doi: 10.1023/A:1015937912504. [DOI] [PubMed] [Google Scholar]
- 36.Sauer D, Zheng W, Coots LB, McGinity JW. Influence of processing parameters and formulation factors on the drug release from tablets powder-coated with Eudragit L 100–55. Eur J Pharm Biopharm. 2007;67(2):464–475. doi: 10.1016/j.ejpb.2007.02.021. [DOI] [PubMed] [Google Scholar]
- 37.Curatolo W, Nightingale JA, Herbig SM. Utility of hydroxypropylmethylcellulose acetate succinate (HPMCAS) for initiation and maintenance of drug supersaturation in the GI milieu. Pharm Res. 2009;26(6):1419–1431. doi: 10.1007/s11095-009-9852-z. [DOI] [PubMed] [Google Scholar]
- 38.You J, Cui F-D, Wang Y-S, Yu Y-w, Li X, Li Q-P. The study of the emulsification efficiency of Aerosil and HPMCAS type and their ratio to stabilize emulsions of zedoary turmeric oil. Colloid Surf A-Physicochem Eng Asp. 2006;280:76–80. doi: 10.1016/j.colsurfa.2006.01.034. [DOI] [Google Scholar]
- 39.Tanno FK, Sakuma S, Masaoka Y, Kataoka M, Kozaki T, Kamaguchi R, et al. Site-specific drug delivery to the middle-to-lower region of the small intestine reduces food-drug interactions that are responsible for low drug absorption in the fed state. J PharmSci. 2008;97(12):5341–5353. doi: 10.1002/jps.21382. [DOI] [PubMed] [Google Scholar]
- 40.Tanno FK, Sakuma S, Masaoka Y, Kataoka M, Kozaki T, Kamaguchi R, et al. Site-specific drug delivery to the middle region of the small intestine by application of enteric coating with hypromellose acetate succinate (HPMCAS) J PharmSci. 2008;97(7):2665–2679. doi: 10.1002/jps.21172. [DOI] [PubMed] [Google Scholar]
- 41.Arndt M, Chokshi H, Tang K, Parrott NJ, Reppas C, Dressman JB. Dissolution media simulating the proximal canine gastrointestinal tract in the fasted state. Eur J Pharm Biopharm. 2013;84(3):633–641. doi: 10.1016/j.ejpb.2013.01.010. [DOI] [PubMed] [Google Scholar]
- 42.Akimoto M, Nagahata N, Furuya A, Fukushima K, Higuchi S, Suwa T. Gastric pH profiles of beagle dogs and their use as an alternative to human testing. Eur J Pharm Biopharm. 2000;49(2):99–102. doi: 10.1016/S0939-6411(99)00070-3. [DOI] [PubMed] [Google Scholar]
- 43.Varum FJ, Hatton GB, Basit AW. Food, physiology and drug delivery. Int J Pharm. doi:10.1016/j.ijpharm.2013.04.034 [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Effect of Eudragit L/HPMCAS (L:H) blend ratio on the water-uptake from coated pellets after immersed in acid medium for 2 h. The dispersion was plasticized by 30% TEC and the coating level was 30%. The curing condition for the coated pellets was 12 h at 40°C. (JPEG 434 kb)