

Abstract
The objective of this study is to investigate the effect of lipolysis on the release of poorly water-soluble drug from SMEDDS in the perspective of drug core/shell location. For this purpose, four SMEDDS formulations with various core/shell properties were developed based on long-chain lipid or medium-chain lipid as well as different surfactant/oil ratios. Poorly water-soluble drugs, hymecromone and resveratrol, were significantly solubilized in all SMEDDS formulations and the diluted microemulsions. Fluorescence spectra analysis indicated that hymecromone was mainly located in the shell of microemulsions, while resveratrol was located in the core. The effect of lipolysis on the release rates of drugs with different core/shell locations were investigated by a modified in vitro drug release model. For the drug located in the shell, hymecromone, the release profiles were not affected during the lipolysis process and no significant differences were observed among four formulations. For the drug located in the core, resveratrol, the release rates were increased to various degrees depending on the extent of digestion. In conclusion, the drug core/shell location plays an important role for determining the effect of lipolysis on drug release from SMEDDS formulation.
KEY WORDS: core/shell location, drug release, fluorescence spectra, lipolysis, self-microemulsifying drug delivery system (SMEDDS)
INTRODUCTION
Oral route is the most convenient and acceptable method of drug delivery. However, oral administration of poorly water-soluble drugs is hampered, owing to their low solubility in aqueous solution. It is estimated that over 40% of new chemical entities identified by high-throughput screening are poorly water-soluble (1). The release of these compounds from the dosage form is the limiting step in the absorption process (2). Hence, formulation strategies for enhancing solubility and dissolution rate of poorly water-soluble drugs are developed to improve the oral bioavailability, e.g., micronization, solid dispersion, and cyclodextrin.
Much attention has been focused on microemulsion and self-microemulsifying drug delivery systems (SMEDDS) in the last two decades due to the successful commercialization of Neoral® (cyclosporine A), Norvir® (ritonavir), Fortovase® (saquinavir), and Agenerase® (amprenavir) (3,4). SMEDDS is a preconcentrate of microemulsion containing drug, oil, surfactant, and co-surfactant. It forms isotropic and thermodynamically stable microemulsions in the gastrointestinal tract under gentle digestive motility (5,6). Poorly water-soluble drugs, especially lipophilic drugs, are significantly solubilized in SMEDDS and the diluted microemulsions. Furthermore, they could be solubilized in the hydrophobic core (the oil phase and the palisade layer of surfactant) or the hydrophilic shell (the hydrophilic head group layer of surfactant) of the formed microemulsions (7–9).
As the first limiting step before absorption, drug release is significantly improved by SMEDDS due to the large interfacial surface area. Many studies have demonstrated that enhanced drug release from SMEDDS could improve the bioavailability (6,10–15). However, SMEDDS is digestible and undergoes complicated structural changes in the gastrointestinal tract. This might lead to a difference between in vitro drug release and in vivo drug release and hence results in the failure of predicting the bioavailability. Generally, pancreatic lipase hydrolyzes triglycerides of SMEDDS into fatty acids and 2-monoglycerides. Then the lipolytic products form into mixed micelles and other intermediate phases, such as vesicle, lamellar liquid crystalline, and hexagonal liquid crystalline (16–19). During the process, drug in SMEDDS is not only dissolved into the aqueous phase, but also incorporated into the intermediate phases and the mixed micelles. Theoretically, the breakdown of SMEDDS and the generation of other phases would inevitably change the drug release profile. Confirming the effect of lipolysis on drug release is essential for understanding the fate of drug in the gastrointestinal tract and predicting the in vitro–in vivo correlation. Unfortunately, information concerning the relationship between the degree of digestion and the drug release from SMEDDS is limited. Some researches demonstrated that the release of poorly water-soluble drugs from lipid-based formulations showed various performances under digestion: the dissolution rate might be improved, decreased, or unchanged compared with the undigested samples (20,21). These conflicting results indicate that the effect of lipolysis on the dissolution rate of lipid-based formulations depends on a certain drug-related parameters. In our opinion, the drug core/shell location might play an important role on the effect of drug release during digestion. For the drug located in the core, it should have to pass through the whole surfactant layer into the aqueous solution, and the consumption of oil phase would lead the drug to incorporate into the intermediate phase. For the drug located in the shell, it just needs to diffuse through the polar group layer, which might not be influenced by the digestion. However, the effect of lipolysis on the release of drug with different core/shell location has not been fully demonstrated.
The purpose of this work is to investigate the influence of lipolysis on the release of poorly water-soluble drugs from SMEDDS considering the effect of drug core/shell location. Firstly, four SMEDDS formulations containing medium-chain lipid or long-chain lipid with different surfactant/oil ratios were developed, and they formed microemulsions with different core/shell properties on dilution. Then, poorly water-soluble drugs, hymecromone and resveratrol, were selected as models of nonlipophilic drugs and lipophilic drugs. Fluorescence spectra analysis demonstrated that hymecromone was located in the hydrophilic shell of microemulsions while resveratrol was located in the hydrophobic core. At last, the effect of lipolysis on the release of drug from SMEDDS formulations was studied by a modified in vitro drug release model.
MATERIALS AND METHODS
Materials
Medium-chain triglyceride (MCT) was provided by Qiandao fine chemical industry Co., Ltd. (Hangzhou, China). Cremophor RH40 was purchased from Puruixing Fine Chemicals Co., Ltd. (Shenyang, China). Ethyl oleate and hymecromone (4-methylumbelliferone, 97%) were obtained from Aladdin Chemistry Co., Ltd. (Shanghai, China). 1, 2-Propanediol and castor oil were purchased from Damao Chemical Reagent Factory (Tianjin, China). Maleic acid and Tween 80 were obtained from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China). Taurocholic acid sodium salt (NaTC) was purchased from Bio Basic Inc. (Markham, Ontario, Canada). Lecithin (PC) was purchased from Sangon Biotech Co., Ltd. (Shanghai, China). Tris-(hydroxymethyl)-aminomethane (Tris) and porcine pancreatin (8 × USP specifications activity) were from Sigma Chemical Co. (St. Louis, MO, USA). 4-Bromophenylboronic acid (4-BPB) was purchased from Alfa Aesar (Tianjin, China). Resveratrol (98%) was obtained from Nuote Biological Technology Co., Ltd. (Shanghai, China). Methanol was HPLC grade and supplied from J&K Chemical Ltd. (Shanghai, China).
Preparation of SMEDDS
Medium-chain lipid and long-chain lipid based SMEDDS formulations were prepared with various ratios of oil, surfactant, and co-surfactant (Table I). Medium-chain lipid (MCT) or long-chain lipid (ethyl oleate and castor oil) was selected as the oil phase. Cremophor RH40 and 1, 2-propanediol were selected as surfactant and co-surfactant with the ratio of 2:1. Briefly, oil, surfactant, and co-surfactant were accurately weighed into glass vials according to their ratios. Poorly water-soluble drug (hymecromone or resveratrol) was added to the mixture. Then, the mixture was stirred overnight. All SMEDDS formulations were freshly prepared for each measurement.
Table I.
Compositions of SMEDDS Formulations (wt%)
| SMEDDS | Oil | Surfactant | Co-surfactant | |||
|---|---|---|---|---|---|---|
| SME-1 | MCT | 60% | Cremophor RH40 | 27% | 1, 2-propanediol | 13% |
| SME-2 | MCT | 40% | Cremophor RH40 | 40% | 1, 2-propanediol | 20% |
| SME-3 | Ethyl oleate | 40% | Cremophor RH40 | 40% | 1, 2-propanediol | 20% |
| SME-4 | Castor oil | 40% | Cremophor RH40 | 40% | 1, 2-propanediol | 20% |
Determination of Particle Size and Zeta Potential
One gram of SMEDDS (containing 5 mg drugs or not) was diluted with 100 mL of deionized water, and then mixed thoroughly by stirring for 30 min on a magnetic stirrer. The particle size and zeta potential of the formed microemulsions were determined by dynamic light scattering (DLS) and laser Doppler velocimetry (LDV) using a Zetasizer Nano ZS 90 (Malvern Instruments Corp, Malvern, UK). All the DLS measurements were performed at 25°C and at a scattering angle 90°. The zeta potential values were calculated using the Smoluchowski equation which was automatic integrated with the instrument. For each group, three parallel samples were measured.
Solubility Study
Excess amounts of hymecromone or resveratrol were added into a test tube containing 1 mL of oil, surfactant, co-surfactant, SMEDDS, or microemulsions under different dilutions. After sealing, the mixtures were shaken in a shaking incubator (HZQ-F, Harbin Donglian Electronic Technology Development Co., LTD., Harbin, China) at 37°C, 150 rpm for 72 h. Then, each test tube was centrifuged at 10,000 rpm for 30 min to remove the excess drug. The concentration of hymecromone or resveratrol in the supernatant was measured by HPLC after appropriate dilution with methanol.
Determination of Drug Core/Shell Location
The drug location in microemulsions was determined by fluorescence spectra analysis. The fluorescence spectra were measured on a PerkinElmer LS 55 luminescence spectrometer (Beaconsfield, UK). Hymecromone or resveratrol was dissolved in phosphate buffer solution (PBS; pH 7.0), MCT, micelles, and microemulsions, respectively, with the final concentration of 5 μM. The micelles were obtained by a 20-fold dilution from the mixture of surfactant/co-surfactant (Smix) and the microemulsions were prepared by a 20-fold dilution from SMEDDS. The excitation and emission spectrum of both compounds were recorded. Their maximum excitation wavelengths (λex) were 320 nm. The emission spectra were collected from 350 to 550 nm. The emission and excitation slits were both set at 2.5 nm for resveratrol and 10 nm for hymecromone with emission filter of 1% T attenuator.
In Vitro Lipolysis Study
In vitro lipolysis experiments were conducted using an in vitro lipid digestion model with a pH-Stat automatic titration unit (848 Titrino plus, Metrohm AG, Herisau, Switzerland) (22,23). The lipolysis of SMEDDS and oil solution (MCT, ethyl oleate, and castor oil) were determined in separate experiments. For each experiment, 1 g of SMEDDS (or 0.4 g of oil solution) was added into a thermostated reaction vessel and dispersed in 18 mL of digestion buffer (50 mM Trizma maleate, 150 mM NaCl, 5 mM CaCl2·2H2O, pH 7.5) containing 5 mM NaTC and 1.25 mM PC. The pH was then adjusted to 7.5 with 0.1 M NaOH. The digestion experiments were initiated by the addition of 1 mL of pancreatin extract and the mixture was kept at 37°C with continuously stirring. The pH-Stat automatic titration unit was used to maintain the pH at 7.50 ± 0.05 by titrating with 0.1 M NaOH. The titrant volumes were recorded at predetermined times. The percentage of lipid digested was calculated from the amount of free fatty acids (FFA), which was equal to the amount of consumed NaOH:
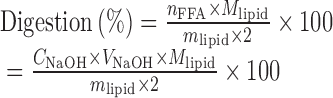 |
1 |
where Digestion (%) is the percentage of lipid digested, nFFA is the number of moles of the generated free fatty acids, Mlipid is the molecular weight of the lipid, mlipid is the total mass of the lipid, CNaOH is the concentration of NaOH used for titration, and VNaOH is the titrant volumes of NaOH.
Pancreatin extract was prepared by adding 1 g of porcine pancreatin (containing pancreatic lipase and co-lipase) to 5 mL of digestion buffer. The mixture was stirred for 15 min followed by centrifugation at 1,600×g and 5°C for 15 min. The supernatant was collected and stored on ice. Pancreatin extracts were freshly prepared for each experiment.
In Vitro Release Study
The in vitro release studies were carried out by a modified dialysis method using a dissolution apparatus (RC806, Tianda Tianfa technology Co., Ltd., Tianjin, China) (6,12,13,24). Briefly, 200 mL of release medium (50 mM PBS containing 0.5% tween 80 (w/v), pH 7.5) was added into the dissolution bath and preheated to 37°C. One gram of SMEDDS (including 5 mg of hymecromone or resveratrol) was placed in a dialysis bag (MWCO 7000, MYM Biological Technology Co., Ltd., Beijing, China) containing 8 mL of PBS. Then 1 mL of pancreatin extract (or 1 mL of PBS as control) was added into the dialysis bag to begin the lipolysis. At last, the dialysis bag was immersed in the release medium and continuously stirred at a paddle speed of 100 rpm. One milliliter of release sample was withdrawn at predetermined time intervals and an aliquot amount of release medium was replaced. The drug concentration of samples was determined by HPLC after appropriate dilution with methanol. Each measurement was taken in triplicate.
HPLC Analysis
The concentration of hymecromone and resveratrol in all samples were determined by HPLC. The HPLC system was equipped with a Waters e2695 separation module, a Waters 2998 photodiode array detector and Empower Pro software (Waters Corporation, Milford, USA). The analysis was carried out on a SinoChrom ODS-BP column (4.6 × 250 mm, 5 μm, Dalian Elite Analytical Instruments, Dalian, China). The mixtures of methanol–water with the ratio of 50:50 and 60:40 were used as mobile phase for hymecromone and resveratrol, respectively. For both compounds, the flow rate was kept at 0.8 mL/min and the injection volume was 20 μL. The UV detector was set at 320 nm for hymecromone and 308 nm for resveratrol.
Statistical Analysis
All values were expressed as the mean ± standard deviation (SD). The statistical differences were determined by Student’s t test and differences were considered as significant at p < 0.05. All statistical analyses were carried out using SigmaStat for Windows Version 3.5 (Systat Software. Inc., Chicago, IL, USA).
RESULTS AND DISCUSSION
Characterization of SMEDDS
The particle size, polydispersity index (PDI), and zeta potential are important characteristics closely related to the core/shell properties of microemulsions. In this study, four SMEDDS formulations that could form microemulsions with different core/shell properties were prepared based on long-chain lipid or medium-chain lipid as well as different surfactant/oil ratios. As shown in Table I, Cremophor RH40 and 1, 2-propanediol were selected as surfactant and co-surfactant with the ratio of 2:1. Medium-chain lipid (MCT) and long-chain lipid (ethyl oleate and castor oil) were selected as the oil phase. The SMEDDS formulations were viscous, homogeneous, and stable, and they were transformed into sky blue opalescent microemulsions on adequate dilution. Table II shows the mean particle size, polydispersity index (PDI), and zeta potential of the formed microemulsions after 100-fold dilution with deionized water. Both SME-1 and SME-2 contained MCT as the oil phase, and they formed blank microemulsions with the average particle sizes of 153.5 ± 4.6 and 66.7 ± 1.2 nm, respectively. The relative smaller particle sizes and polydispersity index of SME-2 microemulsions were mainly caused by higher surfactant/co-surfactant (Smix) content, which could reduce the interfacial tension. SME-3 and SME-4 selected long-chain lipids as the oil phase with the same surfactant/oil ratio of SME-2. The particle size of SME-3 microemulsions was 63.5 ± 0.5 nm, similar to SME-2 microemulsions, while the particle size of SME-4 microemulsions was as large as 205.8 ± 10.8 nm. The larger droplet size and higher PDI of SME-4 formulation might be caused by the fact that castor oil had a larger molecular volume and higher viscosity than MCT and ethyl oleate (25). All formulations have slightly negative zeta potentials (between −5 and −10 mV). Apart from the blank microemulsion, we studied the effect of drug incorporation into microemulsions on the properties of the formulations. The results showed that the particle sizes, PDI, and zeta potentials of drug-loaded microemulsions were much similar to the blank microemulsions.
Table II.
Mean Particle Size, Polydispersity Index, and Zeta Potential of Diluted Microemulsions (Mean ± SD, n = 3)
| Formulation | Mean particle size (nm) | Polydispersity index | Zeta potential (mV) |
|---|---|---|---|
| SME-1 | 153.5 ± 4.6 | 0.513 ± 0.011 | −9.70 ± 0.83 |
| SME-1 with hymecromone | 159.5 ± 3.3 | 0.531 ± 0.022 | −7.50 ± 0.39 |
| SME-1 with resveratrol | 152.7 ± 2.8 | 0.472 ± 0.048 | −6.41 ± 0.73 |
| SME-2 | 66.7 ± 1.2 | 0.213 ± 0.005 | −7.18 ± 0.71 |
| SME-2 with hymecromone | 66.4 ± 1.5 | 0.192 ± 0.014 | −6.09 ± 0.94 |
| SME-2 with resveratrol | 61.2 ± 1.6 | 0.183 ± 0.019 | −7.65 ± 0.43 |
| SME-3 | 63.5 ± 0.5 | 0.183 ± 0.008 | −8.22 ± 1.54 |
| SME-3 with hymecromone | 64.5 ± 1.6 | 0.198 ± 0.016 | −5.88 ± 0.57 |
| SME-3 with resveratrol | 68.8 ± 0.2 | 0.203 ± 0.006 | −7.31 ± 0.81 |
| SME-4 | 205.8 ± 10.8 | 0.536 ± 0.049 | −9.24 ± 0.61 |
| SME-4 with hymecromone | 192.8 ± 1.7 | 0.474 ± 0.033 | −6.54 ± 1.47 |
| SME-4 with resveratrol | 192.5 ± 5.0 | 0.476 ± 0.036 | −4.85 ± 0.30 |
Solubilizing Capability Study
SMEDDS or microemulsions could significantly improve the solubility of poorly water-soluble drugs, especially for lipophilic drugs. To demonstrate the solubilizing capability of the SMEDDS on both nonlipophilic drugs and lipophilic drugs, hymecromone and resveratrol were selected as poorly water-soluble model drugs. Hymecromone, a choleretic and antispasmodic drug, is neither a lipophilic nor a hydrophilic compound with a ClogP value of 1.61 (calculated by ChemBioDraw Ultra 11.0, CambridgeSoft). Its oral bioavailability is just 2.5% due to the low solubility (26). Resveratrol is a naturally occurring polyphenol with various activities, such as anti-oxidant, anti-inflammatory, cardioprotective, and anti-tumor activities. It is a lipophilic compound with the logP of 3.1, belonging to BCS class II (27). Pharmacokinetic studies indicated that the oral bioavailability of resveratrol is almost zero (28). Table III shows the solubilities of hymecromone and resveratrol in all SMEDDS formulations and their components. The solubility of hymecromone in water was just 0.125 ± 0.002 mg/g, and it reached 9.66 ± 1.71, 21.31 ± 3.05, 17.96 ± 0.63, and 19.02 ± 1.57 mg/g in the four SMEDDS formulations, respectively. Among all the components, the solubility of hymecromone in the MCT, ethyl oleate, and castor oil was 5.02 ± 0.14, 1.24 ± 0.13, and 4.21 ± 0.14 mg/g, respectively, while it reached to 27.97 ± 1.83 mg/g in Cremophor RH40 and 24.81 ± 1.38 mg/g in 1, 2-propanediol. The relative higher solubility in surfactant might be due to the formation of hydrogen bonds between oxygen atoms of polyoxyethylene and phenolic hydroxyl of hymecromone. The solubility of resveratrol in water was merely 0.01 mg/g; however, it reached to 60–85 mg/g in the SMEDDS formulations. Among all the components, the solubilization capability was mainly attributed to the surfactant and co-surfactant. Despite the relative lower solubility in oil phase, resveratrol was still solubilized in castor oil more than 1,000-fold than in water.
Table III.
Solubilities of Hymecromone and Resveratrol in SMEDDS Formulations and the Components
| Vehicles | Solubility of hymecromone (mg/g) mean ± SD (n = 3) | Solubility of resveratrol (mg/g) mean ± SD (n = 3) |
|---|---|---|
| MCT | 5.02 ± 0.14 | 2.72 ± 0.25 |
| Ethyl oleate | 1.24 ± 0.13 | 0.86 ± 0.06 |
| Castor oil | 4.21 ± 0.14 | 11.23 ± 0.59 |
| Cremophor RH40 | 27.97 ± 1.83 | 136.60 ± 16.94 |
| 1,2-propanediol | 24.81 ± 1.38 | 91.62 ± 9.14 |
| SME-1 | 9.66 ± 1.71 | 62.54 ± 8.87 |
| SME-2 | 21.31 ± 3.05 | 79.54 ± 7.03 |
| SME-3 | 17.96 ± 0.63 | 83.19 ± 0.33 |
| SME-4 | 19.02 ± 1.57 | 80.44 ± 8.25 |
| Water | 0.125 ± 0.002 | 0.01 |
In the gastrointestinal tract, SMEDDS is diluted to form microemulsions. The solubility of drug in the diluted microemulsions (Smicroemulsion) is a crucial factor affecting the drug release rate; hence, the impact of dilution factor on the drug solubilizing capability of microemulsions was investigated (Table IV). Take SME-2 formulation as an example, the solubility of hymecromone deceased from 21.31 ± 3.05 mg/g to 0.41 ± 0.04, 0.37 ± 0.05, and 0.131 ± 0.004 mg/g, after it was diluted by 10, 20, and 100 times, respectively. Similarly, the solubility of resveratrol decreased from 79.54 ± 7.03 mg/g to 3.37 ± 0.15, 2.95 ± 0.34, and 0.30 ± 0.09 mg/g with the same treatment. The decreased solubilizing capability of microemulsions was mainly caused by the incorporation of co-surfactant into water, as the relationship between co-surfactant concentration and drug solubility is near to logarithmic (29).
Table IV.
Solubility of Hymecromone and Resveratrol in Diluted SME-2 Microemulsions with Different Dilution Factors
| Dilution factor | Solubility of hymecromone (mg/g) mean ± SD (n = 3) | Solubility of resveratrol (mg/g) mean ± SD (n = 3) |
|---|---|---|
| 10 | 1.04 ± 0.10 | 3.37 ± 0.15 |
| 20 | 0.93 ± 0.14 | 2.95 ± 0.34 |
| 100 | 0.33 ± 0.01 | 0.30 ± 0.09 |
Determination of Drug Core/Shell Location in Microemulsions
The drug core/shell location in microemulsions is related to both drug release rate and lipolysis rate (30,31). It is mainly determined by the solubilization capability of each component as well as the interaction between drugs and the components (32,33). The methods to identify the drug core/shell location in microemulsions include fluorescence and NMR spectroscopy methods (34–37). Fluorescence spectroscopy was applied in this study, based on the fact that the fluorescence spectrum of hydrophobic probe was influenced by the polarity of microenvironment.
Figure 1a shows the fluorescence spectra of hymecromone in the phosphate buffer solution, oil phase, micelles, and microemulsions. The fluorescence emission maximum (λem) of hymecromone was 450 nm in the phosphate buffer solution (the polar microenvironment), while it blue-shifted to 380 nm in the MCT solution (the nonpolar microemulsion). Moreover, the fluorescence intensity of hymecromone in MCT was lower than that in PBS solution. The blue-shift of fluorescence spectra and the decrease of fluorescence intensity were mainly caused by the effect of solvent polarity. For the fluorescence spectra of hymecromone in micelles and microemulsions, two emission bands were found at 450 and 380 nm. This indicated that hymecromone was distributed in both polar (450 nm) and nonpolar (380 nm) microenvironments. The distribution coefficient of drug in the polar/nonpolar microenvironment of microemulsions could not be concluded by the fluorescence spectra, however, the major peak centered at 450 nm. This indicated that more hymecromone was distributed in the polar microenvironment, which included the water phase and the hydrophilic group of surfactant. While considering its poor solubility in water, hymecromone was mainly located in the hydrophilic group of surfactant (the shell of microemulsions). The location of hymecromone might be attributed to the hydrogen bonds interaction between drug and surfactant.
Fig. 1.
The fluorescence spectra of hymecromone (a) and resveratrol (b) in water, oil phase, SME-2 microemulsions and micelles
In Fig. 1b, the maximum emission wavelength of resveratrol was 403 nm in the aqueous solution, while it was 380 nm in oil, micelles, and microemulsions. The nonpolarity of solvent resulted in the blue shift of emission spectrum. The fluorescent spectrum of resveratrol in microemulsions was more similar to that in nonpolar solvent other than in water, suggesting that resveratrol is protected from water. Hence, it was concluded that resveratrol was located in the core of microemulsions, which included the hydrophobic palisade layer and the oil phase. The location of resveratrol was mainly caused by its high lipophilicity, which was attributed to the hydrophobic force between resveratrol and the hydrophobic chain.
In Vitro Lipolysis Kinetics of SMEDDS with Different Core/Shell Properties
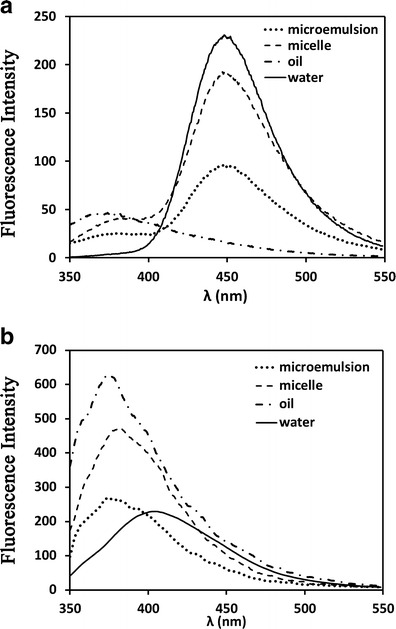
In the gastrointestinal tract, the digestion of oil droplets could be described as follows: firstly, pancreatic lipase is adsorbed to the oil–water interface of oil droplets in the presence of co-lipase, and hydrolyzes 1 mole of triglyceride into 2 moles of fatty acids and 1 mole of 2-monoglycerides. Then, the generated lipolytic products are distributed at the interface inhibiting further digestion by avoiding lipase contacting the oil phase. Finally, the lipolytic products are removed in the form of mixed micelles, vesicles, lamellar, and hexagonal liquid crystalline phases (16,38,39). During the process of digestion, the rate of lipolysis is controlled by factors such as the droplet size, the carbon chain length of lipid, the ratio of surfactant/oil, and the concentration of calcium ion (16,40). In this section, an in vitro lipolysis model was applied to determine the lipolysis rate of SMEDDS with different core/shell properties. The percentages of digested lipids were calculated according to equation (1). For the digestible component of SMEDDS, the lipolysis profiles of MCT, ethyl oleate, and castor oil were firstly compared (Fig. 2a). MCT was completely hydrolyzed during 1 h of digestion, while the percentages of digested ethyl oleate and castor oil were just 13.8% and 23.5%, respectively. The results are in good agreement with previous reports where the rate and extent of digestion of medium-chain lipids were greater than for the long-chain lipids (23,41,42). Among the lipolytic products of three oil phases (caprylic/capric acids, oleic acids, and ricinoleic acids), the medium-chain caprylic/capric acids were more apt to be ionized than long-chain oleic acids and ricinoleic acids at physiological pH (23,40). Therefore, the medium-chain acids were more soluble in the aqueous phase and more prone to form calcium soaps, which means that the clearance rates of medium-chain lipid products from the o/w interface were more rapid. Accordingly, the relative rapid dissociation of medium-chain lipolytic products from the interface lead to the higher extent of digestion of MCT than long-chain ethyl oleate and castor oil.
Fig. 2.
Lipolysis profile of oil solution (a) and SMEDDS formulations (b). The cumulative percentage of lipid digested was calculated by the volume of NaOH titrated
For SMEDDS formulations (seen Fig. 2b), the rate of lipolysis followed the order of SME-1 > SME-2 > SME-3 ≈ SME-4. The lipid component (MCT) of SME-1 and SME-2 was digested nearly 100% and 40%, respectively. In spite of the formation of finer microemulsions from SME-2, the digestion rate of SME-2 was much lower than for SME-1. This situation could be accounted for the higher surfactant/oil ratio. Because the polyoxyethylene chain of nonionic surfactant, e.g., Tween 80, Cremophor EL, or Cremophor RH40, could prevent the adsorption of the pancreatic lipase/co-lipase complex to the oil surface, once it reached to a critical thickness at the interface (39,43). The percentages of digested SME-3 and SME-4 were both less than 10%, which were also lower than the corresponding oil phases. The low digestion rate of the long-chain lipid formulation made it more stable in the gastrointestinal tract and the bioavailability was higher than for the medium- and short-chain lipid formulations (38,39,41).
Effect of Lipolysis on Drug Release from SMEDDS with Different Core/Shell Drug Locations
SMEDDS and microemulsions could significantly improve the dissolution rate of poorly water-soluble drugs and hence improved their bioavailabilities. However, the correlation between in vitro dissolution rate and in vivo bioavailability is poor in many cases. One probable reason is that the in vivo release differs from the in vitro release, as the SMEDDS and microemulsions would be digested in the gastrointestinal tract. In order to investigate the effect of lipolysis on the release of drug from SMEDDS, a modified in vitro drug release model was established by combining the dialysis method with in vitro lipolysis method. Specifically, two factors were taken into consideration: (1) the degree of lipolysis and (2) the drug core/shell location. To overcome the inconvenience of continual titration of NaOH, phosphate buffer solution was selected. Dialysis bag with MWCO 7000 was used to separate drugs from microemulsions and their lipolytic products. Tween 80 (0.5%, w/v) was added to the release medium to maintain the sink conditions.
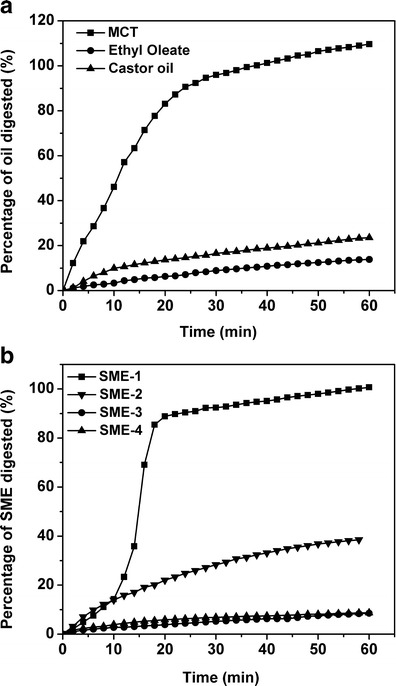
In vitro release profiles of hymecromone from four SMEDDS formulations are shown in Fig. 3. For the no enzyme groups, the percentage of hymecromone released into the release medium was about 50–60% in 6 h and no initial burst release was observed among the four formulations. Despite the different particle sizes of the formed microemulsions, the amounts of dissolved hymecromone from four formulations showed no significant difference. For the enzyme groups, the cumulative release profiles of hymecromone were similar to the groups without enzyme and no significant difference was observed. This indicated that the lipolysis did not affect the release of hymecromone at all regardless of the degree of digestion. During the period of lipolysis, the pH of the release medium ranged from 7.5 to 7.1; hence, the activity of lipase was considered not to be influenced by the slight change of pH.
Fig. 3.
Release profiles of hymecromone from SMEDDS formulations with or without addition of lipase. a SME-1, b SME-2, c SME-3, and d SME-4. Data are expressed as the mean ± SD (n = 3)
Figure 4 shows the release curves of resveratrol from four SMEDDS formulations with or without the presence of pancreatic lipase. The release kinetic was nearly zero-order with a linear relationship between the dissolution time and the amount of released resveratrol. For the no enzyme groups, the cumulative percentages of released resveratrol in 6 h was 8.95 ± 0.44%, 5.95 ± 0.96%, 5.16 ± 0.33%, and 5.33 ± 0.71%, respectively. As the hydrophilic shell was a main barrier of restricting the diffusion of drugs in the core, the dissolution rate of resveratrol from SME-1 was significantly higher than the other formulations due to the lower surfactant/oil ratio. At the same surfactant/oil ratio, SME 2–4 formulations showed no significant difference despite the difference in particle sizes and the type of oil phase used. Hence, the surfactant/oil ratio was a more important factor affecting drug release than particle size and carbon chain length of lipid.
Fig. 4.
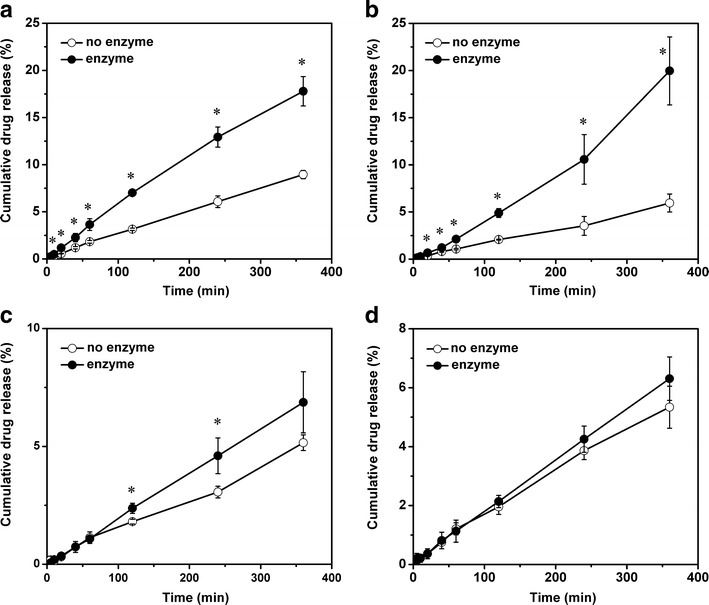
Release profiles of resveratrol from SMEDDS formulations with or without addition of lipase. a SME-1, b SME-2, c SME-3, and d SME-4. Data are expressed as the mean ± SD (n = 3). *p < 0.05
For the enzyme groups, the dissolution rate of resveratrol was increased to a certain extent, depending on the degree of lipolysis. For more digestible SME-1 and SME-2, the dissolution rate of resveratrol was significant increased compared with no enzyme groups. Furthermore, a statistically significantly difference was found even after 10 min’ digestion, where the percentage of lipolysis was about 20%. For long-chain lipid-based SME-3 and SME-4, no obvious difference was found in the release profiles of resveratrol between the enzyme groups and no enzyme groups. But, in fact, a slight increase of dissolution rate was observed after 2 h digestion in SME-3, while no promotion effect was observed for SME-4 during the whole process. At the end of digestion, the samples were collected and centrifuged to determine the concentration of resveratrol in the supernatant. The amount of resveratrol in the supernatant and the release medium was equal to the total amount in SMEDDS before digestion (data not shown), indicating that no precipitation occurred.
Confirming the effect of lipolysis on drug release is essential for understanding the fate of drug in the gastrointestinal tract and predicting the in vitro–in vivo correlation. Some researchers applied ultracentrifugation method to separate the lipolytic products into four phases to study the drug distribution during lipolysis: a pellet, an aqueous phase, an inter phase, and an oil phase (20,22,44). They found that there was a positive correlation between the drug content in aqueous phase and in vivo AUC values. Meanwhile, the drug precipitation presented in the pellet phase could reduce the bioavailability (20,41,45). Some reports compared the drug contents in the aqueous phase of digested with undigested samples, and they found that the poorly water-soluble drugs showed various performances under digestion: the drug content might be improved (e.g., LU 28–179 and probucol), decreased (flupentixol), or unchanged (danazol) compared with the undigested samples (20,21,46). These conflicting results indicate that the effect of lipolysis on the drug dissolution rate from lipid-based formulations was depended on a certain drug-related parameters. In this study, we think the drug core/shell location might be one crucial factor, which influences the drug release rate under digestion.
As shown in Fig. 5, resveratrol was mainly located in the hydrophobic core of microemulsions, while hymecromone was located in the hydrophilic shell. For the microemulsions containing resveratrol (Fig. 5a), the hydrolysis of oil phase could change the microenvironment of the core, which would inevitably “force” resveratrol to release out of the microemulsions. During the process of drug migration from the core to the aqueous phase, resveratrol has to pass through the whole surfactant layer, which is an important barrier of controlling drug release. The generation and removal of the lipolytic products could also alter the arrangement of the surfactant layer at the interface and hence promote the release of resveratrol. In addition, the formed micelles, vesicles, lamellar, and hexagonal liquid crystalline phases had lower solubilities compared with microemulsions because the generated fatty acids and monoglycerides had a decreased solubilization capacity. All the changes above were contributing to the acceleration of the drug release that located in the core; moreover, the increase was related to the degree of digestion. Contrary to the promotion effect on the release of resveratrol, lipolysis did not alter the dissolution rate of hymecromone, even though the lipids had been completely consumed (Fig. 5b). As mainly located in the shell of microemulsions, the release of hymecromone was determined by polyoxyethylene chains of Cremophor RH 40 and co-surfactant. The digestion of oil phase could not change the microenvironment of the hydrophilic shell. Even though the formation of the intermediate phases could alter the interfacial surface area for diffusion, no significant influence on the dissolution rate of hymecromone was found. This might be due to the effect of the interfacial surface area which was negligible compared with other factors such as the solubility and the partition coefficient. Hence, the core/shell location of the drug plays an important role on affecting in vitro–in vivo correlation. The in vivo dissolution rate might be similar to the in vitro dissolution rate of drugs located in the hydrophilic shell while it varied significantly for drugs located in the hydrophobic core. Moreover, in spite of acceleration effect of lipolysis on the release of resveratrol, the exact relationship between the extent of digestion and the amount of drug released is still not clear. Factors, such as solubility, partition coefficient, and the forming of other colloids, need to be further studied to demonstrate the relationship between lipolysis and drug release.
Fig. 5.
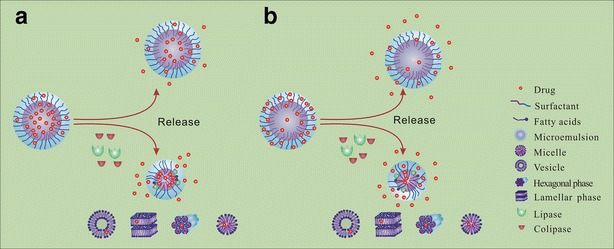
Schematic illustrations showing the effect of lipolysis on the release of drugs from SMEDDS formulations when they located in different parts of microemulsions. a Resveratrol, located in the hydrophobic core; b hymecromone, located in the hydrophilic shell
CONCLUSION
In the present work, four SMEDDS formulations with various core/shell properties were developed based on long-chain lipid or medium-chain lipid as well as different surfactant/oil ratios. In vitro digestion study showed that the carbon chain length of lipid and surfactant/oil ratio were crucial factors affecting the lipolysis rate of SMEDDS. The poorly water-soluble drug hymecromone was located in the hydrophilic shell of diluted microemulsions due to the formation of hydrogen bonds. However, lipophilic resveratrol was distributed in the hydrophobic core of microemulsions. In vitro release study demonstrated that the effect of lipolysis on drug release from SMEDDS was highly associated not only to the properties of formulation but also the drug core/shell location. For the drug located in the shell of microemulsions, the release profiles of hymecromone were not affected at all during the lipolysis process, regardless of the properties of the formulation or the extent of digestion. For the drug located in the core of microemulsions, the dissolution release rates of resveratrol were increased to various degrees depending on the extent of digestion.
ACKNOWLEDGMENTS
The authors are grateful for the financial support of the National Nature Science Foundation of China (grant numbers: 91227126 and 51103157) and National Nature Science Foundation of Liaoning China (grant number: 2013020177).
Contributor Information
Guojun Lv, Email: lgj1802@dicp.ac.cn.
Xiaojun Ma, Email: maxj@dicp.ac.cn.
REFERENCES
- 1.He CX, He ZG, Gao JQ. Microemulsions as drug delivery systems to improve the solubility and the bioavailability of poorly water-soluble drugs. Expert Opin Drug Deliv. 2010;7(4):445–460. doi: 10.1517/17425241003596337. [DOI] [PubMed] [Google Scholar]
- 2.Amidon GL, Lennernas H, Shah VP, Crison JR. A theoretical basis for a biopharmaceutic drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm Res. 1995;12(3):413–420. doi: 10.1023/A:1016212804288. [DOI] [PubMed] [Google Scholar]
- 3.Porter CJ, Charman WN. In vitro assessment of oral lipid based formulations. Adv Drug Deliv Rev. 2001;50:S127–S147. doi: 10.1016/S0169-409X(01)00182-X. [DOI] [PubMed] [Google Scholar]
- 4.Porter CJ, Pouton CW, Cuine JF, Charman WN. Enhancing intestinal drug solubilisation using lipid-based delivery systems. Adv Drug Deliv Rev. 2008;60(6):673–691. doi: 10.1016/j.addr.2007.10.014. [DOI] [PubMed] [Google Scholar]
- 5.Shen H, Zhong M. Preparation and evaluation of self-microemulsifying drug delivery systems (SMEDDS) containing atorvastatin. J Pharm Pharmacol. 2006;58(9):1183–1191. doi: 10.1211/jpp.58.9.0004. [DOI] [PubMed] [Google Scholar]
- 6.Kang BK, Lee JS, Chon SK, Jeong SY, Yuk SH, Khang G, et al. Development of self-microemulsifying drug delivery systems (SMEDDS) for oral bioavailability enhancement of simvastatin in beagle dogs. Int J Pharm. 2004;274(1–2):65–73. doi: 10.1016/j.ijpharm.2003.12.028. [DOI] [PubMed] [Google Scholar]
- 7.Hathout RM, Woodman TJ, Mansour S, Mortada ND, Geneidi AS, Guy RH. Microemulsion formulations for the transdermal delivery of testosterone. Eur J Pharm Sci. 2010;40(3):188–196. doi: 10.1016/j.ejps.2010.03.008. [DOI] [PubMed] [Google Scholar]
- 8.Lv FF, Zheng LQ, Tung CH. Phase behavior of the microemulsions and the stability of the chloramphenicol in the microemulsion-based ocular drug delivery system. Int J Pharm. 2005;301(1–2):237–246. doi: 10.1016/j.ijpharm.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 9.Dalmora ME, Dalmora SL, Oliveira AG. Inclusion complex of piroxicam with beta-cyclodextrin and incorporation in cationic microemulsion. In vitro drug release and in vivo topical anti-inflammatory effect. Int J Pharm. 2001;222(1):45–55. doi: 10.1016/S0378-5173(01)00692-5. [DOI] [PubMed] [Google Scholar]
- 10.Woo JS, Kim TS, Park JH, Chi SC. Formulation and biopharmaceutical evaluation of silymarin using SMEDDS. Arch Pharm Res. 2007;30(1):82–89. doi: 10.1007/BF02977782. [DOI] [PubMed] [Google Scholar]
- 11.Shen Q, Li X, Yuan D, Jia W. Enhanced oral bioavailability of daidzein by self-microemulsifying drug delivery system. Chem Pharm Bull (Tokyo) 2010;58(5):639–643. doi: 10.1248/cpb.58.639. [DOI] [PubMed] [Google Scholar]
- 12.Wu W, Wang Y, Que L. Enhanced bioavailability of silymarin by self-microemulsifying drug delivery system. Eur J Pharm Biopharm. 2006;63(3):288–294. doi: 10.1016/j.ejpb.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 13.Yin YM, Cui FD, Mu CF, Choi MK, Kim JS, Chung SJ, et al. Docetaxel microemulsion for enhanced oral bioavailability: preparation and in vitro and in vivo evaluation. J Control Release. 2009;140(2):86–94. doi: 10.1016/j.jconrel.2009.08.015. [DOI] [PubMed] [Google Scholar]
- 14.Wei JD, Ho HO, Chen CH, Key WT, Chen ET, Sheu MT. Characterisation of fenofibrate dissolution delivered by a self-microemulsifying drug-delivery system. J Pharm Pharmacol. 2010;62(12):1685–1696. doi: 10.1111/j.2042-7158.2010.01182.x. [DOI] [PubMed] [Google Scholar]
- 15.Hong JY, Kim JK, Song YK, Park JS, Kim CK. A new self-emulsifying formulation of itraconazole with improved dissolution and oral absorption. J Control Release. 2006;110(2):332–338. doi: 10.1016/j.jconrel.2005.10.002. [DOI] [PubMed] [Google Scholar]
- 16.Golding M, Wooster TJ. The influence of emulsion structure and stability on lipid digestion. Curr Opin Colloid Interface Sci. 2010;15(1–2):90–101. doi: 10.1016/j.cocis.2009.11.006. [DOI] [Google Scholar]
- 17.Fatouros DG, Bergenstahl B, Mullertz A. Morphological observations on a lipid-based drug delivery system during in vitro digestion. Eur J Pharm Sci. 2007;31(2):85–94. doi: 10.1016/j.ejps.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 18.Fatouros DG, Deen GR, Arleth L, Bergenstahl B, Nielsen FS, Pedersen JS, et al. Structural development of self nano emulsifying drug delivery systems (SNEDDS) during in vitro lipid digestion monitored by small-angle X-ray scattering. Pharm Res. 2007;24(10):1844–1853. doi: 10.1007/s11095-007-9304-6. [DOI] [PubMed] [Google Scholar]
- 19.Warren DB, Anby MU, Hawley A, Boyd BJ. Real time evolution of liquid crystalline nanostructure during the digestion of formulation lipids using synchrotron small-angle X-ray scattering. Langmuir. 2011;27(15):9528–9534. doi: 10.1021/la2011937. [DOI] [PubMed] [Google Scholar]
- 20.Porter CJ, Kaukonen AM, Boyd BJ, Edwards GA, Charman WN. Susceptibility to lipase-mediated digestion reduces the oral bioavailability of danazol after administration as a medium-chain lipid-based microemulsion formulation. Pharm Res. 2004;21(8):1405–1412. doi: 10.1023/B:PHAM.0000036914.22132.cc. [DOI] [PubMed] [Google Scholar]
- 21.Christensen JO, Schultz K, Mollgaard B, Kristensen HG, Mullertz A. Solubilisation of poorly water-soluble drugs during in vitro lipolysis of medium- and long-chain triacylglycerols. Eur J Pharm Sci. 2004;23(3):287–296. doi: 10.1016/j.ejps.2004.08.003. [DOI] [PubMed] [Google Scholar]
- 22.Sek L, Porter CJ, Charman WN. Characterisation and quantification of medium chain and long chain triglycerides and their in vitro digestion products, by HPTLC coupled with in situ densitometric analysis. J Pharm Biomed Anal. 2001;25(3–4):651–661. doi: 10.1016/S0731-7085(00)00528-8. [DOI] [PubMed] [Google Scholar]
- 23.Sek L, Porter CJ, Kaukonen AM, Charman WN. Evaluation of the in-vitro digestion profiles of long and medium chain glycerides and the phase behaviour of their lipolytic products. J Pharm Pharmacol. 2002;54(1):29–41. doi: 10.1211/0022357021771896. [DOI] [PubMed] [Google Scholar]
- 24.Patil P, Joshi P, Paradkar A. Effect of formulation variables on preparation and evaluation of gelled self-emulsifying drug delivery system (SEDDS) of ketoprofen. AAPS Pharm Sci Tech. 2004;5(3):e42. doi: 10.1208/pt050342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Warisnoicharoen W, Lansley AB, Lawrence MJ. Light scattering investigations on dilute nonionic oil-in-water microemulsions. AAPS PharmSci. 2000;2(2):E12. doi: 10.1208/ps020212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Garrett ER, Venitz J, Eberst K, Cerda JJ. Pharmacokinetics and bioavailabilities of hymecromone in human volunteers. Biopharm Drug Dispos. 1993;14(1):13–39. doi: 10.1002/bdd.2510140103. [DOI] [PubMed] [Google Scholar]
- 27.Amri A, Chaumeil JC, Sfar S, Charrueau C. Administration of resveratrol: what formulation solutions to bioavailability limitations? J Control Release. 2012;158(2):182–193. doi: 10.1016/j.jconrel.2011.09.083. [DOI] [PubMed] [Google Scholar]
- 28.Kristl J, Teskac K, Caddeo C, Abramovic Z, Sentjurc M. Improvements of cellular stress response on resveratrol in liposomes. Eur J Pharm Biopharm. 2009;73(2):253–259. doi: 10.1016/j.ejpb.2009.06.006. [DOI] [PubMed] [Google Scholar]
- 29.Pouton CW, Porter CJ. Formulation of lipid-based delivery systems for oral administration: materials, methods and strategies. Adv Drug Deliv Rev. 2008;60(6):625–637. doi: 10.1016/j.addr.2007.10.010. [DOI] [PubMed] [Google Scholar]
- 30.Ljusberg-Wahren H, Seier Nielsen F, Brogård M, Troedsson E, Müllertz A. Enzymatic characterization of lipid-based drug delivery systems. Int J Pharm. 2005;298(2):328–332. doi: 10.1016/j.ijpharm.2005.02.038. [DOI] [PubMed] [Google Scholar]
- 31.Djordjevic L, Primorac M, Stupar M, Krajisnik D. Characterization of caprylocaproyl macrogolglycerides based microemulsion drug delivery vehicles for an amphiphilic drug. Int J Pharm. 2004;271(1):11–19. doi: 10.1016/j.ijpharm.2003.10.037. [DOI] [PubMed] [Google Scholar]
- 32.Constantinides PP. Lipid microemulsions for improving drug dissolution and oral absorption: physical and biopharmaceutical aspects. Pharm Res. 1995;12(11):1561–1572. doi: 10.1023/A:1016268311867. [DOI] [PubMed] [Google Scholar]
- 33.Djordjevic L, Primorac M, Stupar M. In vitro release of diclofenac diethylamine from caprylocaproyl macrogolglycerides based microemulsions. Int J Pharm. 2005;296(1–2):73–79. doi: 10.1016/j.ijpharm.2005.02.014. [DOI] [PubMed] [Google Scholar]
- 34.Nazar MF, Khan AM, Shah SS. Microemulsion system with improved loading of piroxicam: a study of microstructure. AAPS Pharm Sci Tech. 2009;10(4):1286–1294. doi: 10.1208/s12249-009-9328-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li J, Shen X, Gao H. Exploring the locations of different groups of a cationic surface-active 3H-indole probe molecule in AOT-based water-in-oil microemulsions. Chem Phys Lett. 2001;342(5–6):529–535. doi: 10.1016/S0009-2614(01)00640-6. [DOI] [Google Scholar]
- 36.Bermejo R, Tobaruela DJ, Talavera EM, Orte A, Alvarez-Pez JM. Fluorescent behavior of B-phycoerythrin in microemulsions of aerosol OT/water/isooctane. J Colloid Interface Sci. 2003;263(2):616–624. doi: 10.1016/S0021-9797(03)00391-6. [DOI] [PubMed] [Google Scholar]
- 37.Mehta SK, Kaur G, Bhasin KK. Incorporation of antitubercular drug isoniazid in pharmaceutically accepted microemulsion: effect on microstructure and physical parameters. Pharm Res. 2008;25(1):227–236. doi: 10.1007/s11095-007-9355-8. [DOI] [PubMed] [Google Scholar]
- 38.Embleton JK, Pouton CW. Structure and function of gastro-intestinal lipases. Adv Drug Deliv Rev. 1997;25(1):15–32. doi: 10.1016/S0169-409X(96)00488-7. [DOI] [Google Scholar]
- 39.MacGregor KJ, Embleton JK, Lacy JE, Perry EA, Solomon LJ, Seager H, et al. Influence of lipolysis on drug absorption from the gastro-intestinal tract. Adv Drug Deliv Rev. 1997;25(1):33–46. doi: 10.1016/S0169-409X(96)00489-9. [DOI] [Google Scholar]
- 40.McClements DJ, Li Y. Structured emulsion-based delivery systems: Controlling the digestion and release of lipophilic food components. Adv. Colloid Interf. Sci. 2010;159(2):213–228. doi: 10.1016/j.cis.2010.06.010. [DOI] [PubMed] [Google Scholar]
- 41.Porter CJ, Kaukonen AM, Taillardat-Bertschinger A, Boyd BJ, O’Connor JM, Edwards GA, et al. Use of in vitro lipid digestion data to explain the in vivo performance of triglyceride-based oral lipid formulations of poorly water-soluble drugs: studies with halofantrine. J Pharm Sci. 2004;93(5):1110–1121. doi: 10.1002/jps.20039. [DOI] [PubMed] [Google Scholar]
- 42.Dahan A, Hoffman A. Use of a dynamic in vitro lipolysis model to rationalize oral formulation development for poor water soluble drugs: correlation with in vivo data and the relationship to intra-enterocyte processes in rats. Pharm Res. 2006;23(9):2165–2174. doi: 10.1007/s11095-006-9054-x. [DOI] [PubMed] [Google Scholar]
- 43.Christiansen A, Backensfeld T, Weitschies W. Effects of non-ionic surfactants on < i > in vitro</i > triglyceride digestion and their susceptibility to digestion by pancreatic enzymes. Eur J Pharm Sci. 2010;41(2):376–382. doi: 10.1016/j.ejps.2010.07.005. [DOI] [PubMed] [Google Scholar]
- 44.Larsen AT, Sassene P, Mullertz A. In vitro lipolysis models as a tool for the characterization of oral lipid and surfactant based drug delivery systems. Int J Pharm. 2011;417(1–2):245–255. doi: 10.1016/j.ijpharm.2011.03.002. [DOI] [PubMed] [Google Scholar]
- 45.Han SF, Yao TT, Zhang XX, Gan L, Zhu C, Yu HZ, et al. Lipid-based formulations to enhance oral bioavailability of the poorly water-soluble drug anethol trithione: effects of lipid composition and formulation. Int J Pharm. 2009;379(1):18–24. doi: 10.1016/j.ijpharm.2009.06.001. [DOI] [PubMed] [Google Scholar]
- 46.Goddeeris C, Coacci J, Van den Mooter G. Correlation between digestion of the lipid phase of smedds and release of the anti-HIV drug UC 781 and the anti-mycotic drug enilconazole from smedds. Eur J Pharm Biopharm. 2007;66(2):173–181. doi: 10.1016/j.ejpb.2006.10.005. [DOI] [PubMed] [Google Scholar]