

Abstract
The proliferative compartment of the colonic epithelium in vivo is located in the basal crypt where colonic stem cells and transit-amplifying cells reside and fuel the rapid renewal of non-proliferative epithelial cells as they migrate toward the gut lumen. To mimic this tissue polarity, microstructures composed of polydimethylsiloxane (PDMS) microwells and Matrigel micropockets were used to guide a combined 2-dimensional (2D) and 3-dimensional (3D) hybrid culture of primary crypts isolated from the murine colon. The 2D and 3D culture of crypts on a planar PDMS surface was first investigated in terms of cell proliferation and stem cell activity. 3D culture of crypts with overlaid Matrigel generated enclosed, but highly proliferative spheroids (termed colonoids). 2D culture of crypts produced a spreading monolayer of cells, which were non-proliferative. A combined 2D/3D hybrid culture was generated in a PDMS microwell platform on which crypts were loaded by centrifugation into microwells (diameter = 150 μm, depth = 150 μm) followed by addition of Matrigel that formed micropockets locking the crypts within the microwells. Embedded crypts first underwent 3D expansion inside the wells. After the cells filled the microwells, they migrated onto the surrounding surface forming a 2D monolayer in the array regions without Matrigel. This unique 2D/3D hybrid culture generated a continuous, millimeter-scale colonic epithelial tissue in vitro, which resembled the polarized architecture (i.e. distinct proliferative and non-proliferative zones) and geometry of the colonic epithelium in vivo. This work initiates the construction of a “colon-on-a-chip” using primary cells/tissues with the ultimate goal of producing the physiologic structure and organ-level function of the colon.
Introduction
The colon is lined with a single layer of epithelial cells which invade into the underlying mesenchyme to form tubular glands called crypts of Lieberkühn. The proliferative compartments of colonic epithelium are located at the base of these crypts where the colonic stem cells and transit-amplifying cells reside. These cells fuel the rapid renewal (~5 days in mice1) of colonic epithelial cells on the luminal aspect where most of the non-proliferative cells are positioned.2 This polarity of cellular organization is thought to be maintained by a balance of biochemical and biophysical in vivo microenvironments, including gradients of soluble factors (e.g. Wnt, BMP and Notch) across the basal-luminal axis, and biophysical interactions with supporting cells (e.g. pericryptal fibroblasts) and their secreted extracellular matrices.3–5
Culture of live crypts in vitro has been attempted since the 1970s, but it has proven to be extremely difficult to generate a long-term proliferative colonic epithelium in vitro.6, 7 This is thought to be a result of the complexity of in vivo microenvironments that constitute the colonic epithelial stem-cell niche, which has been difficult to recapitulate in vitro. Live intact crypts can be released from animal or human colon specimens by chelating divalent cations and mechanical agitation.8–10 Standard 2D culture of crypts in dishes yields short-term growth of a monolayer of cells.11 3D culture by embedding crypts in collagen gel alone (i.e. without a feeder layer of supporting cells) does not improve crypt survival.9 The loss of proliferative ability of crypt cells under these culture methods suggests the loss of colonic stem cells in vitro. This situation was rectified in 2009 when Hans Clevers and colleagues successfully cultured 3D organoids by inclusion of soluble growth factors (Wnt-3A, R-Spondin 1, noggin and epidermal growth factor [EGF]) in the culture milieu. This breakthrough enabled long-term culture of crypts and stem cells from the small and large intestines.12–16 Embedding isolated crypts or isolated stem cells within a 3D extracellular matrix (ECM) with added growth factors has now been shown to support the survival of stem cells and promote 3D proliferative expansion into epithelial colonoids (defined as colonic organoids without mesenchyme).17 These colonoids contain self-renewing stem cells as well as all differentiated cell types present in crypts: goblet cells (secreting mucus), absorptive colonocytes (absorbing water and electrolytes), and enteroendocrine cells (secreting hormones). While this technique is effective in supporting long-term proliferative growth of colonoids, it does not precisely mimic the in vivo biochemical and biophysical microenvironments of crypts. As a result, the proliferation is unguided and the cell organization and tissue architecture of the colonoids is disorganized without the lumenal-basal cellular architecture.
Microfabrication and microfluidics provide unique in vitro approaches to mimic in vivo microenvironments due to the ability to precisely control the cellular microenvironment both temporally and spatially.18 Substantial advantages have been demonstrated in microdevices to study stem-cell responses to changes in the local microenvironment at the cellular level.19 With the goal of extending these advantages to tissue, or even organ-level, various microdevices, called “organ-on-chips”, have been specifically designed to mimic in vivo architecture and function. The reported “organ-on-chips” have mimicked lung,20 liver,21 heart,22 blood vessel,23 muscle,24 kidney,25 and the gastrointestinal tract26–28 by mimicking a specific feature of the organ microenvironment (e.g. topography, tissue-tissue interface, mechanical movement, shear stress, biochemical gradient). The ambitious goals of “organ-on-chips” are to expand the capabilities of cell culture models and provide high-quality controlled experimental alternatives to animal studies.29–31 Nevertheless, the current “organ-on-chips” remain quite primitive from a biologist’s point-of-view, as many still rely on the use of immortalized cell lines derived from tumors to mimic normal cell physiology. For example, several “gut-on-chips” have used Caco-2 cells derived from a colon carcinoma to simulate the intestinal epithelium.26–28, 32, 33 Although Caco-2 cell lines possess some of the characteristics of differentiated intestinal epithelial cells (e.g. intestinal drug absorption),34 their cancer phenotype poorly reflects normal intestinal physiology and the microarchitecture found in vivo. Thus, one of the future challenges of “organ-on-chips” is to use primary cells derived from normal tissue to form systems representative of in vivo organ systems.30
Our group has attempted to use microdevices to replicate the in vivo microenvironments of primary crypt tissue by capture and culture of crypts in microdevices with the end goal of building a “colon-on-a-chip” to support a living colonic epithelium for in vitro studies in a user-controlled microenvironment.35, 36 In the first study, a freestanding film microfabricated from epoxy photoresist and containing an array of micron-scale capture sites, termed a micromesh (open holes), was used to capture fixed crypts with high efficiency in an ordered and properly oriented fashion.35 Recently, we developed an array of cylindrical microwells with perforated bottoms made from polydimethylsiloxane (PDMS), termed “microstrainers”, to capture and culture colonic crypts as 3D colonoids.35 The arrayed colonoids enabled rapid analysis of a large number of primary colonic tissues during drug exposure. In the current work, we move a step further to recapitulate the in vivo biophysical microenvironment of crypts in a microwell platform to generate colonic epithelium with a polarized architecture (i.e. distinct proliferative and non-proliferative zones) and a geometry that more closely resembles the colonic epithelium in vivo than do colonoids in free-standing culture conditions. To do so, it was anticipated that the microwells would enable a unique combination of 2D/3D hybrid culture system for isolated crypts, as crypts and ECM can be retained inside the microwells. The 2D and 3D culture of crypts on the planar PDMS surface was initially assessed for proliferation and stem-cell activity. Microwell arrays were then fabricated in PDMS to capture crypts which were embedded in the ECM Matrigel. The microwells and Matrigel were designed to mimic the biophysical function of lamina propria in guiding the growth of crypts in vitro. Crypts were cultured on the devices and then examined for 3D expansion within the microwells as well as 2D monolayer formation on the array top surface in the absence of Matrigel. Time-lapse microscopy was used to track the growth of crypts on the microwell platform. A mouse model expressing fluorescent proteins in stem/progenitor and differentiated cells was used for facile identification of these different cell types. Immunofluorescence staining was also used to track the differentiated and proliferative cell zones. The in vitro generated colonic epithelial tissue was fixed, dried and imaged by scanning electron microscope (SEM) to evaluate the morphology of the tissue construct.
Experimental Section
Materials
Fluorescein isothiocyanate–dextran (FITC-dextran, average molecular weight 2,000,000), Y-27632 dihydrochloride (ROCK inhibitor) and N-acetyl-L-cysteine (NAC) were purchased from Sigma Aldrich. The 1002F epoxy photoresist was formulated according to a previous publication.37 PDMS was prepared from the Sylgard 184 silicone elastomer kit (Dow Corning). Advanced DMEM/F-12 medium, EGF recombinant mouse protein, GlutaMAX supplement, penicillin-streptomycin, gentamicin, fetal bovine serum (FBS), and HEPES (1M buffer solution) were obtained from Invitrogen. Growth factor reduced Matrigel was purchased from BD. All other reagents including dithiothreitol (DTT), ethylenediaminetetraacetic acid (EDTA) and paraformaldehyde were from Fisher Scientific.
Fabrication of PDMS microwell arrays
A freestanding PDMS film containing an array of microwells (150 μm in depth, 150 μm in diameter, 200 μm center-to-center spacing) was prepared by three microfabrication steps, the details of which are described in the Electronic Supplementary Information (Fig. S1, ESI). In the first step, a master mold composed of an array of microwells firmly adhered to a glass substrate was fabricated from 1002F photoresist by a one-layer photolithography process (Fig. S1A, ESI). This process also created shallow indentations of alphanumeric characters on the border of the array that labeled the address of each well. In the second step, a PDMS mold was prepared by replicate molding of PDMS on the master mold (Fig. S1B, ESI). The PDMS mold contained an array of large posts (150 μm in height, 150 μm in diameter). In the third step, a PDMS microwell array was fabricated by replicate molding from the PDMS mold (Fig. S1C, ESI). The PDMS microwell had the same geometry as the epoxy microwell on the master mold. The array was glued to a polycarbonate circular chamber by PDMS and cured at 95 °C for 30 min. The chamber had an inner diameter of 12 mm, an outer diameter of 20 mm, and a height of 12.7 mm (Fig. S1E, ESI). The arrays were then placed inside self-seal sterilization pouches (8.9 × 13.3 cm, Henry Schein, # 9004601) and sterilized in a benchtop autoclave (Tuttnauer Brinkmann, Model 3545EP).
Isolation of crypts from mouse colon
Crypts were isolated from CAG-DsRed/Sox9-EGFP mice (6–9 week old) by soaking the excised colon in a chelating buffer (2.0 mM EDTA and 0.5 mM DTT) for 75 min followed by vigorous shaking.38 The CAG-DsRed/Sox9-EGFP mouse is a cross between a bacterial-artificial-chromosome-transgenic mouse in which the enhanced green fluorescent protein (EGFP) is expressed as a function of the Sox9 regulatory region (Sox9-EGFP mouse),39, 40 and the CAG-DsRed mouse (CAG = CMV enhancer plus chicken actin promoter) constitutively expresses the DsRed protein.12, 40, 41 The crypts were used within 30 min after isolation. All experiments were performed in compliance with the relevant laws and institutional guidelines at the University of North Carolina (UNC). All experiments and animal usage were approved by the Institutional Animal Care and Use Committee at UNC.
2D or 3D culture of crypts on a planar PDMS surface
Crypt culture medium (CCM) was formulated according to previous publications with minor modifications.14, 16, 36 CCM was prepared from a mixture of advanced DMEM/F12 medium, Wnt-3A-conditioned medium, R-spondin 2-conditioned medium, and Noggin-conditioned medium at a volumetric ratio of 3:1:1:1, and supplemented with EGF (50 ng/mL), Y27632 ROCK inhibitor (10 μM), NAC (1 mM), GlutaMAX (1×), HEPES (10 mM), penicillin (100 unit/mL), streptomycin (100 μg/mL), and gentamicin (5 μg/mL). The detailed steps to prepare Wnt3A, R-spondin 2 and Noggin-conditioned media are described in the ESI. CCM was prepared in a bulk volume of 500 mL, split into 10-mL aliquots and stored at −80 °C until use.
A planar PDMS film (without microwell structures) was prepared by curing PDMS on a glass slide at 95 °C for 30 min. It was peeled from the glass and glued to the circular chamber in the same way as the PDMS microwell array. The PDMS flat surface (with an area of 113 mm2) was coated with Matrigel (2 vol% in phosphate buffered saline [PBS]) at 4 °C for 24 h, and then rinsed with PBS ×3 prior to being loaded with crypts.
For 2D culture of crypts, isolated crypts were counted using a hemocytometer, and 1,000 crypts were suspended in 1.5 mL CCM and then added to a flat PDMS surface. The crypts landed on the PDMS where their cells adhered to the PDMS surface. The medium was changed every 48 h.
For 3D culture of crypts, isolated crypts were embedded in Matrigel according to previous publications with minor modification.16, 38 1,000 crypts were suspended in 200 μL of cold Matrigel (50 vol% in CCM, 4 °C) and then added to the PDMS surface. After solidification of the Matrigel at 37 °C for 15 min, 1.5 mL of CCM was added to the chamber. The medium was changed every 48 h.
The cell number in the 2D and 3D culture conditions was counted at days 1, 3, 5 and 7. Culture medium was removed and 500 μL 0.25% Trypsin-EDTA (Life Technologies) was added. After incubation at 37 °C for 20 min, colonoids or cell colonies were dissociated into single cells by pipetting, and live-cell numbers were counted using a hemocytometer. Cell numbers are expressed as mean ± standard deviation (n=3).
2D/3D hybrid culture of crypts in microwells
The PDMS microwell arrays were taken from the sterilization pouches and submerged in 10 mL sterile PBS in a 50 mL conical tube. The tube was then centrifuged at 3,000 relative centrifugal force (rcf) for 1 min to remove trapped air bubbles in the wells. The arrays were placed in a Petri dish and coated with Matrigel (2 vol% in PBS) at 4 °C for 24 h. The coated arrays were rinsed with PBS ×3 prior to being loaded with crypts. A defined number of crypts (1,000) were suspended in 0.5 mL CCM and added to the array. The array was then placed in a 50 mL sterile conical tube which was plugged with a 35 mL cured PDMS plug. The tube was centrifuged at 1,000 rcf for 1 min to load the crypts into the microwells. The majority of crypts remained out of wells and were removed by aspirating CCM from the array, followed by rinsing with 200 μL CCM ×3. After the last rinse, 200 μL CCM was added to the array followed by incubation in a 4 °C refrigerator for 10 min. The CCM was aspirated from the array, and 200 μL of cold Matrigel (50 vol% in CCM, 4 °C) was added to the array and again incubated in a 4 °C refrigerator for 10 min. The array was then placed in a cold 50 mL conical tube (plugged with 35 mL cured PDMS plug, 4 °C) and centrifuged at 3,000 rcf for 1 min to bring the Matrigel into the microwells. The sample was tilted, and aspiration of liquid Matrigel from the array generated isolated Matrigel pockets embedding the crypts. After this step, the device was placed at 37 °C for 15 min to solidify the Matrigel. CCM (1.5 mL) was then added to the chamber for crypt culture. Medium was changed every 24 h.
Microscopic imaging
The crypts and arrays were imaged using a Nikon Eclipse TE300 inverted epifluorescence microscope equipped with DAPI/FITC/Texas Red/CY5 filter sets. Fluorescence images were taken daily up to 7 days. The percentage of crypts possessing EGFP expression was manually counted. The surface area and area coverage of the tissue was quantified by Image J. The data are expressed as mean ± standard deviation. Time-lapse imaging of cell expansion from isolated crypts was conducted with an Olympus inverted microscope with a cooled CCD camera (Photometrix Cool Snap HQ2; Roper Scientific) using a Micro-Manager hardware control interface. 3D images of crypts embedded in solidified Matrigel pockets (mixed with 100 μg/mL FITC-dextran) on the array were obtained using an Olympus spinning disk confocal microscope FITC/Texas Red filter sets. To preserve the 3D structure, the generated colonic epithelial tissues were fixed with 4% paraformaldehyde for 20 min, rinsed with PBS ×3, dehydrated in a graded ethanol series, and dried with a critical point dryer (Tousimis Semidri PVT-3). To view the structure of the tissues, the samples were transferred from PDMS microwells to adhesive tape (Scotch® Brand). The dried tissue on the PDMS microwells, the transferred tissue on the tape, and the PDMS microwells were then inspected by SEM (FEI Quanta 200 ESEM, FEI Company).
Immunofluorescence
The freshly isolated crypts, in vitro cultured 3D colonoids (7 days in culture), 2D monolayers (7 days in culture), and 2D/3D hybrid cultured tissues (4 days in culture) were fixed with 4% paraformaldehyde for 20 min, followed by permeabilization with 0.5% Triton X-100 for 20 min. Immunofluorescence staining was performed using the following primary antibodies: rabbit anti-mucin2 (Muc2, 1:200, Santa Cruz, #SC-15334), rabbit anti-chromogranin A (CGA, 1:1000, Bioss, #bs-0539R), and mouse anti-carbonic anhydrase II (CA-II, 1:500, Santa Cruz, #SC-48351). The secondary antibodies were donkey anti-rabbit or mouse antibodies conjugated with NL637 (1:200, Santa Cruz). DNA was stained with Hoechst 33342 (1 μg/mL, Sigma Aldrich, #B2261). The stained crypts and colonoids were imaged using the Nikon Eclipse TE300 microscope described above.
Results and Discussion
Colonic crypts are polarized tissues with distinct proliferative and non-proliferative zones
Crypts possess a polarized architecture (i.e. distinct proliferative and non-proliferative zones) and a mushroom-shaped geometry as depicted in the simple schematic shown in Fig. 1A. Colonic epithelial stem cells reside at the crypt base at a distance from the harsh luminal environment. The stem cells give rise to transit-amplifying cells (dividing progenitors, some of them already partially differentiated) that move in a tight cohort upward along the basal-luminal axis.42 The polarized architecture was demonstrated in crypts isolated from a CAG-DsRed/Sox9-EGFP mouse model after staining with differentiation markers and DNA (Fig. 1B). The freshly isolated mouse crypts had a diameter at the luminal end of 100 ± 23 μm, a basal diameter of 50 ± 10 μm and a length of 241 ± 49 μm (n = 20). EGFP was expressed at the basal end of the crypts. Since EGFP was expressed under the Sox9 promoter and Sox9 expression is restricted to intestinal stem and progenitor cells (but not to the differentiated colonic epithelium),39 EGFP was used as an indicator of cell proliferation. As cells migrate toward the upper crypt regions, cells terminally differentiate into three major types of epithelial cells: goblet cells, absorptive colonocytes and enteroendocrine cells (Fig. 1B). Goblet cells (Muc2+), which secrete mucus to protect and lubricate the colon, are located at the luminal end of the crypts. Enteroendocrine cells (CGA+), which release hormones or peptides to control important physiological functions of the colon, are present in low numbers and appear randomly distributed in the crypts. Colonic enterocytes (CA-II+), which uptake water and ions from the solid waste in the colon, are located in the luminal crypt end and along the luminal gut surface. As the cells reach the gut surface, the cells undergo apoptosis and are sloughed from the epithelium.
Figure 1.
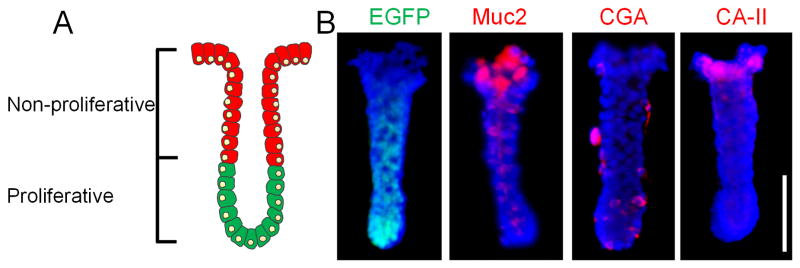
Tissue polarity of colonic crypts. (A) A simplified schematic of colonic crypts possessing distinct proliferative (green cells) and non-proliferative (red cells) zones. (B) EGFP (green) fluorescence and immunofluorescence staining of Muc2, CGA and CA-II (red). Nuclei were stained with Hoechst 33342 (blue). Scale bar = 100 μm.
In vitro 2D culture of crypts on a planar PDMS is non-proliferative, while 3D culture is proliferative
The goal of the current study was to use microwell structures fabricated from PDMS to enable a hybrid 2D/3D in vitro culture of crypts with improved architecture relative to that of the standard colonoid culture system. Before studying the 2D/3D hybrid culture in microwells, the differences between the 2D and 3D culture of crypts on a planar PDMS surface were compared. A CAG-DsRed/Sox9-EGFP mouse model was used to monitor and quantify proliferative, undifferentiated cells which express both red and green fluorescent proteins (DsRed+/EGFP+; yellow-green) and non-proliferative, differentiated cells (DsRed+/EGFP−; red) by fluorescence microscopy.
2D culture of crypts on a PDMS surface was performed in a similar manner to standard 2D cell culture (Fig. 2A). The cells in contact with PDMS began to adhere to the PDMS surface within a few hours. By 24 h, 93 ± 2% crypts placed on the PDMS surface formed small patches of cells in a monolayer (n = 6 fields of view) with cells not in contact with the PDMS detaching and shedding into the medium where they were removed during medium exchange. The number of cells from the monolayers formed was counted and found to slowly decrease over time, from 7,400 ±2,000 at day 1 to 6,500 ±1,500 at day 7 (Fig. 2C). This loss of proliferative capability in the in vitro 2D culture suggests the loss of stem-cell activity and the loss of Sox9EGFP in the monolayers supported this interpretation. All of the monolayer cell regions (100 ± 0%) lost EGFP expression by day 2 (Fig. 2A and 2D). Together, these results indicated that the cells comprising the 2D monolayer were not proliferative, even in the presence of the soluble growth factors Wnt, R-Spondin, Noggin and EGF in the CCM. It is likely that both soluble factors and insoluble biophysical signals (e.g. physical contact with ECM components) are required to maintain intestinal stem-cell proliferative capacity.
Figure 2.

Comparison between 2D and 3D culture of crypts on or over a PDMS planar surface. (A) 2D culture. (B) 3D culture. (A–B) Top panels are schematics of the culture system. Bottom panels are brightfield (left) and overlaid images of EGFP and DsRed fluorescence (right) of crypt fate over time. (C) Cell number vs. time in 2D and 3D culture systems. Both contained the same number of crypts (1,000) at time point 0. (D) EGFP expression vs. time for crypts cultured in 2D and 3D. (E) EGFP (green) fluorescence image and immunofluorescence staining of Muc2, CGA and CA-II (red) for 2D monolayers (left panel) and 3D colonoids (right panels). Nuclei were stained with Hoechst 33342 (blue). Time in culture was 7 days. Scale bars = 200 μm for images in A, B and F.
Both soluble factors and insoluble biophysical signals are present in the 3D culture technique developed by Clevers and colleagues.12–16 To assess the 3D culture of crypts with an underlying PDMS surface, crypts were plated on the PDMS surface and encapsulated in Matrigel. Insoluble biophysical signals may come from Matrigel (the main component being laminin) that mimic the basement membrane underlying the crypt base.12 As expected, a majority of crypts underwent continuous expansion forming 3D colonoids (Fig. 2B). At 24 h, 91±4% of crypts formed 3D colonoids, and only 5±5% formed 2D monolayer (n = 6 fields of view). The cells in the luminal portion of the original crypt rapidly died while the crypt base containing the stem cells persisted in culture and developed into colonoids (Fig. 2B). Colonoids continued to grow and by day 7 possessed an enclosed central lumen and surrounding buds. The number of cells increased over time, from 11,200 ±1,500 at day 1 to 108,700 ±2,000 at day 7 (Fig. 2C). The cell number at day 7 in 3D culture was 16.5 times higher than that in 2D culture system. the difference in cell numbers between the 3D and 2D cultures was statistically significant based on a t-test (p = 0.0002). EGFP expression was preserved in most of colonoids up to day 7, the longest time measured (Fig. 2B). The slow decrease of the percentage of colonoids expressing EGFP (Fig. 2D) was likely due to the mosaic nature of the transgenic mouse model.43 The difference in EGFP expression between the 2D and 3D culture at day 7 was statistically significant (p = 0.002). The above data suggested that the crypts were proliferative in 3D culture, which is consistent with previous publications in which the colonoids were cultured under similar in vitro conditions for up to 1 year.12–16
The morphological difference of 2D and 3D culture was clearly shown in the culture of a high density of crypts (a total of 2,000 crypts per surface) at day 7 (Fig. S2, ESI) where the 2D culture formed a continuous monolayer of cells while the 3D culture generated isolated colonoids. The cell composition was also different for the 2D and 3D cultures (Fig. 2E). As stated above, the 2D monolayer lost all EGFP expression, while the majority of 3D colonoids displayed robust EGFP expression (Fig. S2, ESI). In both the 2D and 3D culture, cells were present that expressed the differentiation markers Muc2, CGA and CA-II, indicating the presence of the differentiated cell types (goblet cells, absorptive colonocytes and enteroendocrine cells). At day 7, 62 ± 15% of 3D colonoids expressed EGFP, 100 ± 0% expressed Muc2, 92 ± 6% expressed CGA, and 84 ± 13% expressed CA-II (n = 10 fields of view). However, for the 2D monolayer at day 7, 0 ± 0% expressed EGFP, 100 ± 0% expressed Muc2, 75 ± 16% expressed CGA, and 81 ± 14% expressed CA-II (n = 10 fields of view). These data suggest that the major difference between the 2D and 3D culture systems is the continued presence of the stem and progenitor cells in the 3D culture and their loss in 2D culture.
Compared to crypts in vivo, the cells in both the 2D and 3D culture systems lacked clear proliferative and differentiated compartments. The various cell types were randomly dispersed in both the 3D colonoids and 2D monolayers (Fig. 2D). In addition, the elongated, mushroom-shaped geometry of the original crypt was lost. This was likely the result of the current 2D and 3D culture systems which lack both the structural and chemical cues to mimic the in vivo microenvironments. It was reasoned that if 2D and 3D geometries could be combined in the appropriate manner to culture individual crypts, a tissue with a distinct proliferative (3D) and differentiated (2D) compartments might be generated. Thus, it was the goal of the current study to propose and test a new 2D/3D hybrid culture system aided by microengineered features.
Design of a microwell platform to enable 2D/3D hybrid culture of colonic epithelial cells in vitro
In vivo, crypts reside on a lamina propria or connective tissue which provides soluble and insoluble signals for cell survival and renewal (Fig. 3A-i). Detaching crypts from the lamina propria allows isolation of individual crypts (Fig. 3A-ii), but apoptosis is rapidly induced due to the absence of these signals.44 In order to partially mimic the biophysical in vivo microenvironment, a microwell approach was proposed to combine the 2D and 3D formats for culture of individual colonoids in vitro (Fig. 3A-iii & iv). This format was selected with the goal of forming 3-D colonoids within the microwells and a 2-D cell layer on the upper device surface. The colonoids with proliferating stem cells within the microwells might then act as a supply of differentiated, post-mitotic cells for the upper device surface. This would create a proliferative cell zone akin to the lower portion of an intact crypt while a differentiated cell zone similar to the luminal gut surface formed outside of the microwells, mimicking the proliferation/differentiation zones formed in vivo. Since PDMS supported colonic cell growth in both a 2D and 3D format, a PDMS microwell array was fabricated. The microwells were 150 μm in diameter and height (Fig. 3B) to capture isolated crypts (typical dimensions of basal diameter-50 μm, luminal diameter-100 μm, and length-241 μm). The efficiency of loading crypts into the wells was the major consideration in choosing the 150-μm diameter wells, as it was not possible to load crypts into 50-μm diameter wells due to the high aspect ratio (about 5) of the crypts. The array contained 900 wells which were divided into 9 sections, each containing 100 wells. To track the growth of crypts, each section was labeled by capital letters, and the rows and columns of wells were labeled by lowercase letters and numbers (Fig. 3B).
Figure 3.

Microwell arrays for combined 2D/3D culture of crypts. (A) Schematic of the 2D/3D culture strategy. (i) The colonic epithelium in vivo is composed of crypt subunits each in contact with a lamina propria. (ii) Individual crypts are isolated from a colon. (iii) Crypts are loaded into PDMS microwells and encased within Matrigel micropockets. (iv) In vitro culture of crypts in microwells generates colonic epithelium resembling the polarized architecture of crypts in vivo. (B) SEM image of the PDMS microwell array. The wells were labeled with shallow alphanumeric characters. (C) A close-up SEM image of a disrupted section of a microwell. (D–E) Brightfield (D) and SEM image (E) of a microwell with a loaded crypt. (F) Brightfield image showing 28 out of 49 wells loaded with crypts. (G) DsRed fluorescence image of F. (H) Fluorescence confocal image of Matrigel pockets formed in the microwell array. (I–J) Shown is a standard fluorescence image (I) and a confocal reconstructed Z slice (J) of crypts encapsulated within isolated Matrigel pockets in the microwell array. The panels display overlaid red/green fluorescence images. The Matrigel was mixed with 100 μg/mL fluorescein-dextran in images H through I. Scale bars = 100 μm for images in B–J.
A suspension of crypts was used to plate crypts into the microwell array. It was found that sedimentation by gravity was ineffective in loading the crypts into the microwells; therefore, centrifugation at 1,000 rcf for 1 min was used to load the crypts (Fig. 3D and 3E). Theoretically, a Poisson distribution can be used to estimate the distribution of crypts on the array. At a ratio of crypt/well = 2/1, Poisson distribution predicts that 14% of the wells will be empty, 27% of wells will capture single crypts, and 59% of wells will contain 2 or more crypts. Nevertheless, experimental data demonstrated a large deviation from this simple theoretical model with 66 ± 8% wells empty, 32 ± 8% wells loaded with single crypts, and 2 ± 2% wells with 2 or more crypts (n = 3). This was likely due to the high aspect ratio of the crypts. Loading efficiency could be improved by increasing the crypt/well ratio. At crypt/well = 5/1, 55 ± 9% wells were loaded with crypts (n = 3 arrays) (Fig. 3F and G). Some wells were loaded with more than one crypt which was deemed acceptable, since it was observed that two crypts within a single microwell always fused into a single colonoid during culture. A further improvement in loading efficiency may be achievable by optimizing the crypt/well ratio and geometry of wells, or by sequentially loading with crypts.
To prevent apoptosis of the freshly captured crypts and enable 3-D colonoid growth within the microwells, the crypts were enclosed within a Matrigel plug (Fig. 3A-iii). The array with captured crypts was overlaid with cold liquid Matrigel. Centrifugation was used to improve loading the microwells with the viscous liquid Matrigel. Aspirating excess Matrigel from the array left isolated Matrigel pockets in the microwells. Incubation of the array at 37 °C caused the Matrigel to gel, encasing the crypts. Matrigel premixed with fluorescein-dextran enabled analysis of the contents of the microwells by fluorescence confocal microscopy which confirmed that the crypts were fully embedded in isolated Matrigel pockets with the same height and diameter as the microwells (Fig. 3H–J). A visible Matrigel layer was absent from the upper surface of the array. CCM was then added to the microwell array to supply nutrients and growth factors followed by culture.
Generation of a millimeter-scale epithelium in vitro during continued 2D/3D culture of colonoids on the microarray
Within the microwells, 95 ± 4% crypts (n=6 fields of view) underwent 3D expansion to form colonoids over a 48-h period (Fig. 4A), and the colonoids displayed a similar spherical morphology as that seen during 3D culture (Fig. 2B). 36 ± 8% of the colonoids filled their entire well by 48 h. The colonoids expressed EGFP fluorescence indicating that stem/progenitor cells were present. No colonoids were observed growing on the top surface of the array suggesting that when the expanding mass of cells reached the top surface of the microwells where Matrigel was no longer present, 3D expansion ceased. Expansion of the cells out of the microwells transited to a 2D monolayer ((Fig. 4C, Video #1, ESI). The 2D monolayer outside of the wells did not express EGFP suggesting the loss of stem/progenitor cells in the monolayer (Fig. 4B). The 3D-2D transition started as early as 48 h when 7.1 ± 5.3% of colonoids formed a 2D/3D hybrid structure (n = 3 arrays). The tissue area coverage was quantified by Image J and clearly showed the progressive generation of a continuous colonic epithelial tissue in the 2D/3D hybrid culture system (Fig. S3). Most of the 3D-2D transition occurred at day 2–5 when the surface coverage rapidly increased from 36 ± 4% (day 2) to 77 ± 7% (day 5). A continuous tissue was formed after day 6 when 3D colonoids in adjacent microwells were connected by a 2D monolayer. Since EGFP+ cells were present within the microwells but not outside of the microwells, it was likely that proliferative cells within the microwells were the cell source for the expansion of the 2D monolayer across the device top-surface. During time lapse movies of the growing epithelium, the expanding 2D monolayers from adjacent microwells fused to from a continuous tissue (Fig. 4D, Video #2, ESI). At day 8, a 1.7 mm2 continuous epithelial-like layer was generated that was transferred to tape for SEM imaging (Fig. 4E). The tissue possessed a crypt-like 3-D geometry connected by a cellular monolayer (Fig. 4F). Cells in this 2D/3D culture system were maintained up to 10 days.
Figure 4.

In vitro generation of millimeter-scale, colonic epithelium with a polarized architecture from primary tissue. (A) Culture of colonoids in a PDMS microwell. A crypt was loaded into a microwell on the array. By 48 h, a 3D colonoid formed that filled the microwell. By 96 h, a 2D/3D hybrid culture was formed. The top panels are brightfield images while the bottom panels are overlaid red/green fluorescence images. (B) Overlaid fluorescence image of the 2D/3D hybrid culture at 96 h (green = EGFP, red = Muc2 stained by immunofluorescence, blue = nuclei stained with Hoechst 33342). (C–D) SEM image of colonic epithelial tissues while still on the PDMS microwell array. The 2D/3D hybrid culture was imaged at 96 h (C) and 144 h (D). (E) SEM images of a 1.7 mm2 section of the in vitro-generated colonic epithelial tissue transferred to a tape so that the tissue is viewed from its underside. (F) A close-up of the underside of the tissue shown in (E). Scale bars = 200 μm. The Matrigel plug is not visualized in the SEM images since its protein concentration (~4 mg/mL) is too low to maintain a solid structure during fixation.
To further evaluate the 2D/3D hybrid culture system, control experiments were performed by culturing crypts in microwells without Matrigel or with a 1.8-mm-deep Matrigel layer overlying the microwells (Fig. S4, ESI). Under both conditions, all growth factors were supplied in the CCM. In the absence of Matrigel, crypts within microwells spread into monolayers inside the microwells and lost EGFP fluorescence by day 4 much like the 2D culture (Fig. S4A, ESI). In the presence of Matrigel filling the microwells and forming a contiguous overlying layer, crypts underwent 3D expansion first inside the wells, and then continued their 3D expansion outside of wells into the Matrigel layer (Fig. S4B, ESI). Only in the 2D/3D hybrid culture did colonoids form a unique geometry similar to that of crypts (Fig. S4C, ESI) supporting that the microarchitecture of the PDMS microwells combined with the Matrigel pockets guided crypt-like growth.
Conclusion
A millimeter-scale colonic epithelial tissue was generated in vitro by using primary tissue capable of proliferation and differentiation. Isolated crypts were cultured under conditions in which only a monolayer (2D) formed or under conditions in which a 3D growth was possible within Matrigel. Cells expressing Sox9 (EGFR+) or stem cells were readily identified within the 3D tissue regions. In contrast, the 2D culture regions possessed only non-proliferative cells. The combined 2D/3D hybrid culture of colonoids was realized by loading crypts into the microwells and then embedding them within isolated Matrigel pockets. This 2D/3D hybrid culture generated a continuous primary colonic epithelial tissue in vitro with distinct proliferative and non-proliferative zones that recapitulated some features of in vivo growth. The most prominent feature was a distinct stem cell zone found only within the Matrigel pocket and a distinct nonproliferative cell region surrounding these stem cell zones. Within the Matrigel pockets, each colonoid formed a fully enclosed structure as rather than the open-lumen crypt structures present in native tissue. A future goal is to develop a tissue construct containing open lumen crypts representing a more physiologic colon-on-chip system. Additional enhancements, such as imposing gradients of factors that mimic the biochemical microenvironments of the epithelium may permit formation of such a tissue. This 2D/3D hybrid culture technique should be readily extendable to other polarized organ systems possessing distinct proliferative and differentiated cell zones, such as the small intestine and stomach.
Supplementary Material
Acknowledgments
This research was supported by the NIH (EB012549, EB013803, DK091427 and DK034987) and the University Cancer Research Fund (UCRF). We acknowledge the Chapel Hill Analytical and Nanofabrication Laboratory for access to the critical point drying and SEM facilities.
References
- 1.Barker N, van de Wetering M, Clevers H. Genes & Development. 2008;22:1856–1864. doi: 10.1101/gad.1674008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fuchs E, Chen T. Embo Reports. 2013;14:39–48. doi: 10.1038/embor.2012.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brittan M, Wright NA. Gut. 2004;53:899–910. doi: 10.1136/gut.2003.025478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kosinski C, Li VSW, Chan ASY, Zhang J, Ho C, Tsui WY, Chan TL, Mifflin RC, Powell DW, Yuen ST, Leung SY, Chen X. Proc Natl Acad Sci U S A. 2007;104:15418–15423. doi: 10.1073/pnas.0707210104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yen TH, Wright NA. Stem Cell Rev. 2006;2:203–212. doi: 10.1007/s12015-006-0048-1. [DOI] [PubMed] [Google Scholar]
- 6.Eastwood GL, Trier JS. Gastroenterology. 1973;64:375–382. [PubMed] [Google Scholar]
- 7.Autrup H, Barrett LA, Jackson FE, Jesudason ML, Stoner G, Phelps P, Trump BF, Harris CC. Gastroenterology. 1978;74:1248–1257. [PubMed] [Google Scholar]
- 8.Booth C, Patel S, Bennion GR, Potten CS. Epithelial Cell Biol. 1995;4:76–86. [PubMed] [Google Scholar]
- 9.Whitehead RH, Brown A, Bhathal PS. In Vitro Cellular & Developmental Biology. 1987;23:436–442. doi: 10.1007/BF02623860. [DOI] [PubMed] [Google Scholar]
- 10.Seidelin JB, Horn T, Nielsen OH. Am J Physiol-Gastroint Liver Physiol. 2003;285:G1122–G1128. doi: 10.1152/ajpgi.00533.2002. [DOI] [PubMed] [Google Scholar]
- 11.Quaroni A. Gastroenterology. 1989;96:535–536. doi: 10.1016/0016-5085(89)91584-9. [DOI] [PubMed] [Google Scholar]
- 12.Sato T, Vries RG, Snippert HJ, van de Wetering M, Barker N, Stange DE, van Es JH, Abo A, Kujala P, Peters PJ, Clevers H. Nature. 2009;459:262–U147. doi: 10.1038/nature07935. [DOI] [PubMed] [Google Scholar]
- 13.Sato T, van Es JH, Snippert HJ, Stange DE, Vries RG, van den Born M, Barker N, Shroyer NF, van de Wetering M, Clevers H. Nature. 2011;469:415–418. doi: 10.1038/nature09637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sato T, Stange DE, Ferrante M, Vries RG, Van Es JH, Van den Brink S, Van Houdt WJ, Pronk A, Van Gorp J, Siersema PD, Clevers H. Gastroenterology. 2011;141:1762–1772. doi: 10.1053/j.gastro.2011.07.050. [DOI] [PubMed] [Google Scholar]
- 15.Jung P, Sato T, Merlos-Suarez A, Barriga FM, Iglesias M, Rossell D, Auer H, Gallardo M, Blasco MA, Sancho E, Clevers H, Batlle E. Nature Medicine. 2011;17:1225–1227. doi: 10.1038/nm.2470. [DOI] [PubMed] [Google Scholar]
- 16.Yui SR, Nakamura T, Sato T, Nemoto Y, Mizutani T, Zheng X, Ichinose S, Nagaishi T, Okamoto R, Tsuchiya K, Clevers H, Watanabe M. Nature Medicine. 2012;18:618–623. doi: 10.1038/nm.2695. [DOI] [PubMed] [Google Scholar]
- 17.Stelzner M, Helmrath M, Dunn JCY, Henning SJ, Houchen CW, Kuo C, Lynch J, Li LH, Magness ST, Martin MG, Wong MH, Yu J N. I. H. I. S. C. Consortiu, . Am J Physiol-Gastroint Liver Physiol. 2012;302:G1359–G1363. doi: 10.1152/ajpgi.00493.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kovarik ML, Gach PC, Ornoff DM, Wang YL, Balowski J, Farrag L, Allbritton NL. Analytical Chemistry. 2012;84:516–540. doi: 10.1021/ac202611x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gupta K, Kim DH, Ellison D, Smith C, Kundu A, Tuan J, Suh KY, Levchenko A. Lab Chip. 2010;10:2019–2031. doi: 10.1039/c004689b. [DOI] [PubMed] [Google Scholar]
- 20.Huh D, Matthews BD, Mammoto A, Montoya-Zavala M, Hsin HY, Ingber DE. Science. 2010;328:1662–1668. doi: 10.1126/science.1188302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee SA, No DY, Kang E, Ju J, Kim DS, Lee S. Lab Chip. 2013;13:3529–3537. doi: 10.1039/c3lc50197c. [DOI] [PubMed] [Google Scholar]
- 22.Grosberg A, Alford PW, McCain ML, Parker KK. Lab Chip. 2011;11:4165–4173. doi: 10.1039/c1lc20557a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zheng Y, Chen JM, Craven M, Choi NW, Totorica S, Diaz-Santana A, Kermani P, Hempstead B, Fischbach-Teschl C, Lopez JA, Stroock AD. Proc Natl Acad Sci U S A. 2012;109:9342–9347. doi: 10.1073/pnas.1201240109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wilson K, Das M, Wahl KJ, Colton RJ, Hickman J. Plos One. 2010;5:e11042. doi: 10.1371/journal.pone.0011042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jang KJ, Suh KY. Lab Chip. 2010;10:36–42. doi: 10.1039/b907515a. [DOI] [PubMed] [Google Scholar]
- 26.Kim HJ, Huh D, Hamilton G, Ingber DE. Lab Chip. 2012;12:2165–2174. doi: 10.1039/c2lc40074j. [DOI] [PubMed] [Google Scholar]
- 27.Ramadan Q, Jafarpoorchekab H, Huang CB, Silacci P, Carrara S, Koklu G, Ghaye J, Ramsden J, Ruffert C, Vergeres G, Gijs MAM. Lab Chip. 2013;13:196–203. doi: 10.1039/c2lc40845g. [DOI] [PubMed] [Google Scholar]
- 28.Sung JH, Yu JJ, Luo D, Shuler ML, March JC. Lab Chip. 2011;11:389–392. doi: 10.1039/c0lc00273a. [DOI] [PubMed] [Google Scholar]
- 29.Huh D, Hamilton GA, Ingber DE. Trends in Cell Biology. 2011;21:745–754. doi: 10.1016/j.tcb.2011.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sung JH, Esch MB, Prot JM, Long CJ, Smith A, Hickman JJ, Shuler ML. Lab Chip. 2013;13:1201–1212. doi: 10.1039/c3lc41017j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huh D, Torisawa YS, Hamilton GA, Kim HJ, Ingber DE. Lab Chip. 2012;12:2156–2164. doi: 10.1039/c2lc40089h. [DOI] [PubMed] [Google Scholar]
- 32.Kim HJ, Ingber DE. Integr Biol. 2013;5:1130–1140. doi: 10.1039/c3ib40126j. [DOI] [PubMed] [Google Scholar]
- 33.Wang L, Murthy SK, Fowle WH, Barabino GA, Carrier RL. Biomaterials. 2009;30:6825–6834. doi: 10.1016/j.biomaterials.2009.08.046. [DOI] [PubMed] [Google Scholar]
- 34.Artursson P, Palm K, Luthman K. Adv Drug Deliv Rev. 2001;46:27–43. doi: 10.1016/s0169-409x(00)00128-9. [DOI] [PubMed] [Google Scholar]
- 35.Wang YL, Dhopeshwarkar R, Najdi R, Waterman ML, Sims CE, Allbritton N. Lab Chip. 2010;10:1596–1603. doi: 10.1039/b927316f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang Y, Ahmad AA, Shah PK, Sims CE, Magness ST, Allbritton NL. Lab Chip. 2013;13:4625–4634. doi: 10.1039/c3lc50813g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pai JH, Wang Y, Salazar GT, Sims CE, Bachman M, Li GP, Allbritton NL. Anal Chem. 2007;79:8774–8780. doi: 10.1021/ac071528q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sato T, Stange DE, Ferrante M, Vries RGJ, van Es JH, van den Brink S, van Houdt WJ, Pronk A, van Gorp J, Siersema PD, Clevers H. Gastroenterology. 2011;141:1762–1772. doi: 10.1053/j.gastro.2011.07.050. [DOI] [PubMed] [Google Scholar]
- 39.Formeister EJ, Sionas AL, Lorance DK, Barkley CL, Lee GH, Magness ST. Am J Physiol-Gastroint Liver Physiol. 2009;296:G1108–G1118. doi: 10.1152/ajpgi.00004.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gracz AD, Ramalingam S, Magness ST. Am J Physiol-Gastroint Liver Physiol. 2010;298:G590–G600. doi: 10.1152/ajpgi.00470.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schepers AG, Snippert HJ, Stange DE, van den Born M, van Es JH, van de Wetering M, Clevers H. Science. 2012;337:730–735. doi: 10.1126/science.1224676. [DOI] [PubMed] [Google Scholar]
- 42.Crosnier C, Stamataki D, Lewis J. Nat Rev Genet. 2006;7:349–359. doi: 10.1038/nrg1840. [DOI] [PubMed] [Google Scholar]
- 43.Leneuve P, Colnot S, Hamard G, Francis F, Niwa-Kawakita M, Giovannini M, Holzenberger M. Nucleic Acids Res. 2003;31:e21. doi: 10.1093/nar/gng021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dufour G, Demers MJ, Gagne D, Dydensborg AB, Teller IC, Bouchard V, Degongre I, Beaulieu JF, Cheng JQ, Fujita N, Tsuruo T, Vallee K, Vachon PH. Journal of Biological Chemistry. 2004;279:44113–44122. doi: 10.1074/jbc.M405323200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.