

Abstract
Many important cellular functions are implemented by protein complexes that act as sophisticated molecular machines of varying size and temporal stability. Here we demonstrate quantitatively that protein complexes in the yeast Saccharomyces cerevisiae are comprised of a core in which subunits are highly coexpressed, display the same deletion phenotype (essential or nonessential), and share identical functional classification and cellular localization. This core is surrounded by a functionally mixed group of proteins, which likely represent short-lived or spurious attachments. The results allow us to define the deletion phenotype and cellular task of most known complexes, and to identify with high confidence the biochemical role of hundreds of proteins with yet unassigned functionality.
Large-scale mass-spectrometric studies in Saccharomyces cerevisiae provide a compendium of protein complexes (Alberts 1998; Hartwell et al. 1999) that are considered to play a key role in carrying out yeast functionality (Gavin et al. 2002; Ho et al. 2002). Although vastly informative, such libraries offer information only on the composition of a protein complex at a given time and developmental or environmental condition. In addition, mass spectrometry is unable to distinguish those subunits that carry the key functional modules (i.e., the core) of the complex from those structural subunits that represent short-lived modulatory or spurious associations (Von Mering et al. 2002). Repeated individual purifications coupled with, for example, crystallographic or cryo-electron microscopy characterization of each of these complexes could offer a more precise picture (Frank 2001; Abbott 2002), but such approaches on a large scale are unavailable at present. Yet extensive data sets on the essentiality, cellular localization, and functional role of individual proteins, together with their corresponding gene expression, may allow us to develop an insight into the organization of protein complexes, and to provide a new perspective on the role of the various protein subunits.
RESULTS
We start by demonstrating that the cellular role and essentiality of a protein complex may largely be determined by a small group of protein subunits that display a high mRNA coexpression pattern, belong to the same functional class, and share the same deletion phenotype and cellular localization. For each i and j protein pair of an experimentally identified N protein complex, we calculated their corresponding mRNA coexpression coefficient (Eisen et al. 1998), φij, that approximates the average coexpression coefficient of protein i with all other subunits of the complex (Futcher et al. 1999). We determined CiD separately from global microarray data obtained on individual gene deletion mutants (Winzeler et al. 1999; Hughes et al. 2000), and CiC from time kinetic data obtained on the yeast cell cycle (Cho et al. 1998; Spellman et al. 1998). The average correlation coefficient for each of the protein subunits of six large complexes (from Gavin et al. 2002) is shown in the first columns of Figure 1. We find that a significant fraction of the protein subunits display a large, positive average mRNA coexpression coefficient with each other, indicating their potential functional relatedness to the other subunits within the complex. This result is in agreement with earlier findings of correlation between protein-protein interaction and transcriptional profiles (Ge et al. 2001; Grigoriev 2001; Mrowka et al. 2001; Jansen et al. 2002; Kemmeren et al. 2002). Some subunits, however, possess close to zero or even a negative correlation coefficient with the other subunits, indicating that they are not consistently coexpressed with the other subunits within the complex.
Figure 1.

Characterizing three essential and nonessential complexes. (A, column I) mRNA coexpression patterns for three large complexes identified in Gavin et al. (2002). For each protein subunit (identified at the bottom of each panel), we show the average correlation coefficient for their corresponding relative mRNA expression level with all other subunits based on the microarray data obtained on gene deletion mutants (CD, top plot; Winzeler et al. 1999; Hughes et al. 2000), and cell cycle measurements (CC, bottom plot; Cho et al. 1998; Spellman et al. 1998). We denote by red the known essential proteins and by black the nonessential proteins. (Column II) Cross-correlation plot obtained by plotting for each protein i within the three selected complexes the cell cycle correlation coefficient CiC on the horizontal axis, and the gene deletion correlation coefficient CiD on the vertical axis. Each symbol corresponds to a single gene product (protein), the color reflecting its known deletion phenotype (red: essential; black: nonessential). The shaded area separates the highly coexpressed core proteins, the boundaries of the area being given by CiC = C̄C - σC and CiD = C̄D - σD. (Column III) The same coexpression plot as in column II, but the symbols are color-coded based on the functional classification of the corresponding proteins. The green symbols denote gene products that belong to the majority regarding their known functional role (Complexes 365 and 360: green proteins simultaneously belong to protein fate and subcellular localization; Complex 363: transcription) unfilled symbols denote proteins with unknown functional role; and the blue symbols denote those subunits that do not share the functional classification with the majority. (Column IV) Coexpression plot with proteins colored based on their known cellular localization. Green symbols denote those with the same subcellular localization, which is nucleus for all three complexes. Blue symbols denote proteins whose localization differs from the majority, and unfilled symbols represent those with unknown cellular localization. In columns II-IV, we used a two-dimensional representation to demonstrate that the essentiality-, functional-classification-, and cellular-localization-based separation is simultaneously present by using two widely different transcriptional data sets. A control plot with only the Cho et al. (1998) cell cycle data is shown in the Supplemental material. (B) The same as A, but for three complexes with predominantly nonessential subunits. In column II, we used red squares to denote those essential proteins that are part of the core of other essential complexes. In column III, the green symbols represent proteins participating in synthesis. In column IV, the green symbols denote proteins localized in the mitochondria for all three complexes.
The internal correlations among the subunits of a protein complex are best revealed using a two-dimensional representation, plotting for each protein i the correlation coefficient CiD on one axis and CiC on the other. On such a plot, we color code each protein using essentiality information based on single gene deletions (Fig. 1, column II), on the proteins' functional role (Fig. 1, column III), and their known cellular localization (Fig. 1, column IV), based on information compiled by the MIPS database (Mewes et al. 2002). Such plots indicate the existence of two types of protein complexes, which we refer to as essential (Fig. 1A) and nonessential (Fig. 1B) complexes. For essential complexes, we observe a relatively clear separation between the many essential and few nonessential protein subunits. For example, in the three complexes shown in Figure 1A, essential proteins aggregate in the high coexpression region of the mRNA coexpression phase space. A similar separation is observed for the nonessential complexes as well (Fig. 1B), where nonessential proteins aggregate in the high coexpression region. Finally, although most proteins belong to several functional classes, we find that for each complex displayed in Figure 1, the vast majority of the highly coexpressed proteins share the same functional class and subcellular localization (Fig. 1, columns III and IV).
To quantify the observed essentiality-, functional role- and cellular localization based separation we denote by C̄D and C̄C the average coexpression coefficient, obtained by averaging CiD and CiC over all proteins within a given complex, and by σD or σC the standard deviation around the average. We assume that all protein subunits i for which 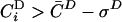 and
and 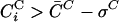 are part of the core of the protein complex. The protein subunits satisfying this condition are those depicted in the shaded areas in Figure 1, allowing us to separate the core proteins from those that show only weak correlation with the other components of the complex. As Figure 1 shows, we find that the core is characterized by a surprising degree of functional, essentiality, and localization homogeneity: For example, of the 40 proteins within the core of the complexes shown in Figure 1A, 38 are essential. In addition, all core subunits share the same functional classification and cellular localization. Similarly, for the three complexes shown in Figure 1B, of the 49 core proteins only one is an essential protein; only four proteins with known functional role do not share the function of the majority; and all proteins share their cellular localization with the majority within the core. As the Supplemental material demonstrates, where we list similar plots for 132 additional complexes, an essentiality-, function-, and localization-based homogeneity of the core is a generic property of most protein complexes.
are part of the core of the protein complex. The protein subunits satisfying this condition are those depicted in the shaded areas in Figure 1, allowing us to separate the core proteins from those that show only weak correlation with the other components of the complex. As Figure 1 shows, we find that the core is characterized by a surprising degree of functional, essentiality, and localization homogeneity: For example, of the 40 proteins within the core of the complexes shown in Figure 1A, 38 are essential. In addition, all core subunits share the same functional classification and cellular localization. Similarly, for the three complexes shown in Figure 1B, of the 49 core proteins only one is an essential protein; only four proteins with known functional role do not share the function of the majority; and all proteins share their cellular localization with the majority within the core. As the Supplemental material demonstrates, where we list similar plots for 132 additional complexes, an essentiality-, function-, and localization-based homogeneity of the core is a generic property of most protein complexes.
The relatively unambiguous segregation of the essential and nonessential proteins within the complexes indicates that protein complexes may be categorized according to the deletion phenotype of the majority of their core subunits. Here we consider a specific complex essential if ≥60% of the core proteins with the known deletion phenotype are essential, and nonessential if ≥60% of the core subunits are nonessential. We find that of the 383 complexes identified by Gavin et al. (2002) with three or more protein subunits, 174 are essential, 155 are nonessential, and only 54 do not show a clear classification based on the deletion phenotype of the core. Yet, a closer inspection indicates the majority of these 54 complexes are in fact nonessential. Indeed, most essential proteins found in the core of the ambiguous complexes participate in the core of other unambiguously essential complexes (see square symbols in column II of Fig. 1B), indicating that their essentiality likely stems from their association with other essential complexes. When not considering these subunits, we find that 35 of the 54 complexes with previously unclear classification are in fact nonessential. We also expect that the remaining 19 unclassified complexes could be also unambiguously classified as nonessential once a more complete list of all essential complexes becomes available. The Supplemental material provides detailed predictions on the characteristics of all complexes identified by Gavin et al. (2002), Ho et al. (2002), and those collected in the MIPS database (Mewes et al. 2002). In addition, when we computationally simulate subunit compositions identical in numbers with those identified experimentally by Gavin et al. (2002), but whose composition is selected randomly from the yeast proteome, we derive only 9 essential complexes (Fig. 2A), indicating that the experimentally identified complex ensemble is highly nonrandom and is biased toward essential complexes. As a specific example in the Supplemental material, we show a negative control set of Figure 1, with randomly selected proteins, indicating the absence of functional- and essentiality-based separation of the core and halo proteins.
Figure 2.

Characterization of the protein complex ensemble and schematic illustration of the internal organization of protein complexes. (A) The number of complexes in the Gavin et al. (2002) data set that are found to be essential (red), nonessential (black), and of unknown (white) deletion phenotype. Next to each column we show the number of corresponding complexes if the proteins were randomly distributed in the various complexes, indicating the highly nonrandom character of the complex composition and essentiality. For this, each protein subunit of the known Gavin et al. (2002) complexes is replaced with proteins randomly selected from the yeast proteome. (B) The size dependence of essentiality in protein complexes. The plot shows the fraction of essential subunits within the complex (vertical axis) as a function of the number of protein subunits in the complexes (horizontal axis). (C) The predicted functional classification of the complexes identified by Gavin et al. (2002), showing separately the number of essential and nonessential complexes found in each functional class. (D) The predicted cellular localization of the identified essential and nonessential complexes. A full list of predictions for each complex is shown in the Supplemental material. (E) We find that 43% of the protein complexes possess a core comprised of highly coexpressed proteins, that are all essential and belong to the same functional class, indicating that they represent the functional building blocks of the complex. Such core is shown schematically as tightly locked P1-P5 proteins. Mass spectroscopic methods inevitably identify other proteins as well with those complexes. Yet, we find that these halo proteins (P6-P9) show a small coexpression pattern with the core, and are both phenotypically and functionally mixed, indicating that they likely represent proteins that display only temporal or spurious attachment to the complex. (F) Approximately 46% of the complexes have a core composed of predominantly nonessential proteins (P1-P5), surrounded again by a halo of proteins with mixed essentiality and functional classification (P6-P8). These complexes likely are not essential for cell growth; therefore, all core proteins are uniformly nonessential. The few essential proteins found predominantly in the halo of such nonessential complexes often simultaneously take part in the core of other essential complexes, explaining the origin of their essentiality. For example, the P1 protein, which is part of the core of the essential complex shown in E, could also attach to the surface of the nonessential complex shown in F. Therefore, the essentiality of P1* is derived not from its role in complex F, but from its role in the essential complex E.
The results also indicate a relatively uneven distribution of the essential complexes in different functional categories and localization classes. Indeed, we find that the majority of protein complexes are responsible for subcellular localization and transcription (Fig. 2C), and are located in the nucleus and cytoplasm (Fig. 2D). This is consistent with the known bias of mass-spectrometry approaches toward nuclear proteins (Von Mering et al. 2002). Interestingly, in the nucleus, the essential complexes outnumber the nonessential complexes, a bias that is inverted in the cytoplasm-associated complexes. Finally, we find a weak, but positive correlation between the size of the complex and its essentiality: The larger the complex, the more likely that its core is essential (Fig. 2B). For example, only 45% of the complexes identified by Gavin et al. (2002) with 10 or less proteins are essential. This fraction increases to 100% for complexes with more than 40 subunits.
DISCUSSION
Many biological functions are carried out by the integrated activity of highly interacting cellular components, referred to as functional modules. Here we investigated the properties of one type of such modules; the protein complexes found in S. cerevisiae. Our results indicate that many of the identified protein complexes possess an invariant core, in which the biochemical role of each protein subunit is irreplaceable, and is seamlessly integrated into a higher-level function of the whole complex. In turn, the deletion phenotype of each core protein is determined by the role of the complex in the organism. If the given complex is essential for cell growth, the deletion of any core protein disrupts the complex's functional integrity, and subsequently renders the cell unviable (Fig. 2E). If, however, the cell is able to tolerate the loss of a complex's function, none of its specific core subunits are essential (Fig. 2F). The core is generally surrounded by several “halo” proteins that typically do not share a common deletion phenotype, functional classification, or cellular localization with the core subunits (Fig. 2E,F). This indicates that they likely represent temporal attachments, some acting as modifiers of the complex's function, whereas others are functionally unrelated proteins that spuriously attach to the surface of the core proteins (Von Mering et al. 2002).
Our ability to identify the core, together with the observed essentiality-, functional-, and localization-based homogeneity of the core, allows a more precise identification of those subunits for which a possible cellular function can be inferred (Gavin et al. 2002; Ho et al. 2002; Supplemental material). Indeed, participation in a specific complex can be considered as source of functional classification. Our results indicate, however, that such functional assignment can be made with high confidence only for the core proteins. To turn our findings into a predictive tool, we identified all proteins that belong to the core of a large complex, and have either an unknown functional classification or one whose present functional annotation differs from the majority of the other core proteins in the complex. This identification allowed us to assign functional prediction to 869 core proteins listed in Tables II, IV, and VI in the Supplemental material.
The segregation of protein complexes into essential and nonessential ones offers a new perspective on the organizational level at which a protein's deletion phenotype is determined. Based on data, it is evident that to a high degree a protein's phenotypic essentiality is determined by the role it plays in ensuring the integrity of vital molecular complexes, thus elevating essentiality from the property of an individual protein (Jeong et al. 2001) to a characteristic of the protein complex. In agreement with this proposition, we find that almost 47% (508) of all known essential yeast proteins (1085) are part of the core of complexes identified by Gavin et al. (2002), despite the fact that the total number of proteins in these complexes represent only 20% (1363) of all yeast proteins (6316). Presumably, a complete list of protein complexes could associate an even larger fraction of essential proteins with such essential complexes. This internal organization is consistent with the notion of stable or unstable protein complexes (Jansen et al. 2002), and the dynamical coexpression of selected open reading frames (Alter et al. 2000; Holter et al. 2000; Ge et al. 2001). Understanding the dynamics of the complex genetic networks (Hasty et al. 2001; Solé and Satorras 2002) potentially responsible for synchronizing the expression of the core subunits is now a prime challenge.
METHODS
Protein Complexes
We used the complete list of protein complexes identified by Gavin et al. (2002; Table S1 in Gavin et al.), by Ho et al. (2002; Table S1 in Ho et al.), and the MIPS database (http://mips.gsf.de). We focused on complexes of three or more proteins, which limits us to 384, 585, and 144 complexes for the three databases, respectively. Each complex is identified based on the order number given in the original databases, and reproduced in the Supplemental material. The deletion phenotype data for individual ORFs were downloaded from http://www-deletion.stanford.edu (version June 2002), whereas the functional classification and cellular localization of the individual proteins were obtained from the MIPS database (as of June 2002).
Coexpression Patterns
The global mRNA expression data for yeast were downloaded from http://www.rii.com/tech/pubs/cell_hughes.htm (Hughes et al. 2000), limiting our analysis to the genomic expression program of 287 single-gene-deletion mutant S. cerevisiae strains grown under identical cell culture conditions as wild-type yeast cells. A similar analysis was performed on the cell cycle data sets (Cho et al. 1998; Spellman et al. 1998). For each protein belonging to a given complex, we determined φij, following Eisen et al. (1998). The obtained φijC encodes the coexpression matrix based on the cell cycle data, and φijD based on the deletion data sets. The coefficient
 |
where N denotes the number of proteins in the studied complex, quantifies the average coexpression with the rest of the proteins within the complex, was determined for each protein subunit of all known complexes. The typical values of the correlation coefficient range between -0.5 and 0.5. Note that for pairwise protein-protein interactions, occasionally higher correlation coefficients are observed (Ge et al. 2001; Grigoriev 2001; Mrowka et al. 2001; Jansen et al. 2002; Kemmeren et al. 2002), a difference rooted in the fact that Ci reflects the average correlation with all other complex subunits, some proteins contributing with small or negative values.
Functional Prediction
We assign to each complex the functional role (cellular localization) shared by the majority of the core proteins. The confidence level of each prediction is based on the percentage of the core proteins known to belong to the selected functional class. Next, we identify all core proteins that either do not have a known functional classification, or whose functional classification does not agree with the predicted functional role of the protein complex core in which they participate. For these proteins, based on the association with the core, we assign the functional role/cellular localization as predicted by the complex's role. Halo proteins are not included in this prediction process, as they do not display the functional and phenotype homogeneity seen in the core.
Acknowledgments
Research at Northwestern University and the University of Notre Dame was supported by the Department of Energy and by the National Institutes of Health.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.1073603. Article published online before print in October 2003.
[Supplemental material is available online at www.genome.org.]
References
- Abbott, A. 2002. Proteomics: The society of proteins. Nature 417: 894-896. [DOI] [PubMed] [Google Scholar]
- Alberts, B. 1998. The cell as a collection of protein machines: Preparing the next generation of molecular biologists. Cell 92: 291-294. [DOI] [PubMed] [Google Scholar]
- Alter, O., Brown, P.O., and Botstein, D. 2000. Singular value decomposition for genome-wide expression data processing and modeling. Proc. Natl. Acad. Sci. 97: 10101-10106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho, R.J., Campbell, M.J., Winzeler, E.A., Steinmetz, L., Conway, A., Wodicka, L., Wolfsberg, T.G., Gabrielian, A.E., Landsman, D., Lockhart, D.J., et al. 1998. A genome-wide transcriptional analysis of the mitotic cell cycle. Mol. Cell 2: 65-73. [DOI] [PubMed] [Google Scholar]
- Eisen, M.B., Spellman, P.T., Brown, P.O., and Botstein, D. 1998. Cluster analysis and display of genome-wide expression patterns. Proc. Natl. Acad. Sci. 95: 14863-14868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank, J. 2001. Cryo-electron microscopy as an investigative tool: The ribosome as an example. Bioessays 23: 725-732. [DOI] [PubMed] [Google Scholar]
- Futcher, B., Latter, G.I., Monardo, P., McLaughlin, C.S., and Garrels, J.I. 1999. A sampling of the yeast proteome. Mol. Cell. Biol. 19: 7357-7368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavin, A.C., Bösche, M., Krause, R., Grandi, P., Marzioch, M., Bauer, A., Schultz, J., Rick, J.M., Michon, A.M., Cruciat, C.M., et al. 2002. Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature 415: 141-147. [DOI] [PubMed] [Google Scholar]
- Ge, H., Liu, Z., Church, G.M., and Vidal, M. 2001. Correlation between transcriptome and interactome mapping data from Saccharomyces cerevisiae. Nat. Genet. 29: 482-486. [DOI] [PubMed] [Google Scholar]
- Grigoriev, A. 2001. A relationship between gene expression and protein interactions on the proteome scale: Analysis of the bacteriophage T7 and the yeast Saccharomyces cerevisiae. Nucleic Acids Res. 29: 3513-3519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartwell, L.H., Hopfield, J.J., Leibler, S., and Murray, A.W. 1999. From molecular to modular cell biology. Nature 402: C47-C52. [DOI] [PubMed] [Google Scholar]
- Hasty, J., McMillen, D., Isaacs, F., and Collins, J.J. 2001. Computational studies of gene regulatory networks: In numero molecular biology. Nat. Rev. Genet. 2: 268-279. [DOI] [PubMed] [Google Scholar]
- Ho, Y., Gruhler, A., Heilbut, A., Bader, G.D., Morre, L., Adams, S.L., Millar, A., Taylor, P., Bennett, K., Boutilier, K., et al. 2002. Systematic identification of protein complexes in Saccharomyces cerevisiae by mass spectrometry. Nature 415: 180-183. [DOI] [PubMed] [Google Scholar]
- Holter, N.S., Mitra, M., Maritan, A., Cieplak, M., Banavar, J.R., and Fedoroff, N.V. 2000. Fundamental patterns underlying gene expression profiles: Simplicity from complexity. Proc. Natl. Acad. Sci. 97: 8409-8414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes, T.R., Marton, M.J., Jones, A.R., Roberts, C.J., Stoughton, R., Armour, C.D., Bennett, H.A., Coffey, E., Dai, H., He, Y.D., et al. 2000. Functional discovery via a compendium of expression profiles. Cell 102: 109-126. [DOI] [PubMed] [Google Scholar]
- Jansen, R., Greenbaum, D., and Gerstein, M. 2002. Relating whole-genome expression data with protein-protein interactions. Genome Res. 12: 37-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong, H., Mason, S.P., Barabási, A.-L., and Oltvai, Z.N. 2001. Lethality and centrality in protein networks. Nature 411: 41-42. [DOI] [PubMed] [Google Scholar]
- Kemmeren, P., van Berkum, N.L., Vilo, J., Bijma, T., Donders, R., Brazma, A., and Holstege, F.C.P. 2002. Protein interaction verification and functional annotation by integrated analysis of genome-scale data. Mol. Cell 9: 1133-1143. [DOI] [PubMed] [Google Scholar]
- Mewes, H.W., Frishman, D., Güldener, U., Mannhaupt, G., Mayer, K., Mokrejs, M., Morgenstern, B., Münsterkötter, M., Rudd, S., and Weil, B. 2002. MIPS: A database for genomes and protein sequences. Nucleic Acids Res. 30: 31-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mrowka, R., Patzak, A., and Herzel, H. 2001. Is there a bias in proteome research? Genome Res. 11: 1971-1973. [DOI] [PubMed] [Google Scholar]
- Solé, R.V. and Satorras, R.P. 2002. Complex networks in genomics and proteomics. In Handbook of graphs and networks: From the genome to the internet (eds. S. Bormholdt and H.G. Schuster), pp. 145-167. Wiley-VHC, Berlin, Germany.
- Spellman, P.T., Sherlock, G., Zhang, M.Q., Iyer, V.R., Anders, K., Eisen, M.B., Brown, P.O., Botstein, D., and Futcher, B. 1998. Comprehensive identification of cell cycle-regulated genes of the yeast Saccharomyces cerevisiae by microarray hybridization. Mol. Biol. Cell 9: 3273-3297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Von Mering, C., Krause, R., Snel, B., Cornell, M., Oliver, S.G., Fields, S., and Bork, P. 2002. Comparative assessment of large-scale data sets of protein protein interactions. Nature 417: 399-403. [DOI] [PubMed] [Google Scholar]
- Winzeler, E.A., Shoemaker, D.D., Astromoff, A., Liang, H., Anderson, K., Andre, B., Bangham, R., Benito, R., Boeke, J.D., Bussey, H., et al. 1999. Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science 285: 901-906. [DOI] [PubMed] [Google Scholar]
WEB SITE REFERENCES
- http://mips.gsf.de; MIPS database.
- http://www.rii.com/tech/pubs/cell_hughes.htm; global mRNA expression data.
- http://www-deletion.stanford.edu; deletion phenotype data.