

Abstract
One of the most important uses of whole-genome expression data is for the discovery of new genes with similar function to a given list of genes (the query) already known to have closely related function. We have developed an algorithm, called the gene recommender, that ranks genes according to how strongly they correlate with a set of query genes in those experiments for which the query genes are most strongly coregulated. We used the gene recommender to find other genes coexpressed with several sets of query genes, including genes known to function in the retinoblastoma complex. Genetic experiments confirmed that one gene (JC8.6) identified by the gene recommender acts with lin-35 Rb to regulate vulval cell fates, and that another gene (wrm-1) acts antagonistically. We find that the gene recommender returns lists of genes with better precision, for fixed levels of recall, than lists generated using the C. elegans expression topomap.
The genome sequences of several animals have now been determined, revealing that the majority of genes have never been studied before (C. elegans Sequencing Consortium 1998; Myers et al. 2000; Lander et al. 2001; Venter et al. 2001). A key goal is to use high-throughput approaches to elucidate the function of large numbers of genes in parallel, in order to map gene functions onto the genome sequence. One of the most powerful methods to annotate the function of genes is to cluster sets of genes based on their expression profiles from microarray experiments (Eisen et al. 1998). Genes that show tight levels of coexpression not only in one microarray experiment, but in a large number of diverse experiments are likely to function together (Eisen et al. 1998; Brown and Botstein 1999).
A previous report compiled data from a large number of C. elegans DNA microarray experiments, and then used a variant of multidimensional scaling to generate a gene expression terrain map (a topomap; Kim et al. 2001). In this topomap, gene clusters are represented as different mountains on a two-dimensional plane; the height of the mountain indicates the local density of genes in the plane. Of the 44 different mountains in the gene expression topomap, 38 have so far been found to be enriched for genes expressed in specific tissues or involved in particular biological functions. For example, several mountains (mounts 7, 11, 18, and 20) are highly enriched for germ line genes, mount 16 is enriched for muscle genes, mount 1 is enriched for both muscle and neuronal genes, and mount 15 is enriched for aging-regulated genes (Kim et al. 2001). The clustering depicted by the topomap is a powerful resource that has been used to suggest potential biological functions for genes that had not been studied previously (Lund et al. 2002; Piano et al. 2002; Roy et al. 2002; Walhout et al. 2002; S. Mango, pers. comm.; A. Villeneuve, data not shown). Similar approaches have been used in yeast to find coexpressed genes from a compendium of many yeast DNA microarray experiments (Eisen et al. 1998; Hughes et al. 2000; Ihmels et al. 2002).
The multidimensional scaling algorithm used to generate the gene expression topomap (Kim et al. 2001) used every microarray experiment to generate a solution mapping each gene against every other gene. In many cases, however, one is not interested in the entire genome but only in genes that are coexpressed with a particular set of genes of interest. In these cases, it is not necessary to consider interactions between all pairs of genes (global clustering), but only interactions that are relevant to the genes of interest (targeted clustering). In this paper, we develop such a targeted clustering algorithm (called the gene recommender) that may be better suited to the predominant use of the gene expression database. The gene recommender takes a query list of genes, finds experiments in which those genes appear to be coregulated, and then uses only those experiments to generate a ranked list of coexpressed genes. If the data values for one experiment are very noisy, then the query values for that experiment are unlikely to be very similar, and that experiment will be filtered out.
In addition to noise, we must contend with multifunctional genes. Suppose that all of our query genes are expressed in muscle and that some, but not all, of these genes are also expressed in neurons. We would then expect the entire query list to be coregulated in experiments relevant to muscle expression but to be split in experiments related to neurons. Informed by the query list, the gene recommender algorithm would likely include the muscle experiments, exclude the neuronal experiments, and thus produce a hit list consisting of new candidate muscle genes but not neuronal genes. A global clustering approach such as the method used to construct the gene expression topomap would be expected to place a multifunctional gene with just one of its relevant groups, either muscle or neuronal. The problem addressed by the gene recommender closely resembles that of recommending movies, books, or web documents similar to those in a given list. Commonalities among these problems of diverse origin are outlined at http://www-stat.stanford.edu/~owen/transposable.
We tested the gene recommender using a query of five C. elegans genes involved in the retinoblastoma (Rb) complex. The gene recommender produced a short list of genes that are highly coexpressed with the five Rb query genes: three known to interact with Rb, five involved in the chromatin structure or the cell cycle (functions similar to those of Rb), and two that we show to have functions related to lin-35 Rb in RNA interference experiments. We compared the performance of the gene recommender algorithm to that of the gene expression topomap, and found that the gene recommender produced candidate lists that were shorter and more concentrated with the query genes.
RESULTS
We present a search algorithm called gene recommender to find new genes that are coexpressed with a given set of genes using data from a large number of C. elegans microarray experiments. The expression data set consists of 553 DNA microarray hybridizations, including a diverse set of experiments that profile expression changes during development, aging, following stress, in various mutants, and under various growth conditions (Kim et al. 2001). Of these experiments, 178 used partial DNA microarrays (containing 11,917 genes; 63% of the 18,967 genes from the C. elegans genome) and the remaining 375 experiments used DNA microarrays containing nearly all of the genes in the genome (17,871 genes, 94%; Reinke et al. 2000; Jiang et al. 2001). Two genes whose expression levels increase and decrease together are considered to be coregulated, even if their absolute expression levels differ markedly. To capture this notion of coregulation via a simple correlation measure, we first applied a rank-based normalization to the raw data. We rank the expression values of each gene across the experiments from its most induced expression level (+1) to that gene's most repressed expression level (-1; see Methods). Subsequent computations are performed on the normalized data.
The gene recommender algorithm takes a list of query genes and scores each gene in the genome based on how similar its expression profile is to the expression profiles of the query genes. We use the term “cassette” to refer to a group of genes with a common function of interest. When some, but perhaps not all, of the cassette genes are known to us, we can use the known members as a query to the gene recommender and obtain a rank ordering of all genes. High-ranking genes are then strong candidates for membership in the cassette. The highest-ranking genes constitute a hit list analogous to the high-ranking web pages produced by a search engine. In both cases, there is usually not a sharp demarcation between relevant and irrelevant hits.
The gene recommender first assigns a numerical score, called a Z score, to each experiment measuring the extent to which the query genes tend to cluster within that experiment (Methods). The high-scoring experiments are taken to be the ones that are most relevant to the query. The low-scoring ones may be irrelevant to the query and detrimental to the search for new genes. We use only the high-scoring experiments to rank genes according to their correlation with the query genes. To find the threshold score separating high-scoring from low-scoring experiments, we re-compute the gene ranking using a variety of thresholds and then select a threshold for which the query genes come closest to the top of the list (Methods).
We tested the performance of the gene recommender algorithm using four query lists (Table 1; http://pmgm2.stanford.edu/~kimlab/cassettes). The queries were: five genes related to the retinoblastoma complex, 41 major sperm protein (MSP) genes, six synaptonemal complex genes, and six meiotic recombination/DNA repair genes (Table 1). These query lists were chosen somewhat arbitrarily, except that we already knew that the genes showed strong regulation in at least some DNA microarray experiments (Kim et al. 2001). We anticipate that the gene recommender algorithm will work well for other query lists generated by the C. elegans research community, as long as at least some of the DNA microarray experiments show strong coregulation of the query genes; the Supplemental Web site includes a search engine for researchers to input their own queries. Furthermore, we anticipate that gene recommender will perform well not only for C. elegans data, but for data from any organism with a reasonably extensive gene expression database; the Supplemental Web site also includes the code for the gene recommender algorithm so that it could be used to analyze gene expression data from other organisms.
Table 1.
Query Genes
| Retinoblastomaa | MSP | Synaptonemal complex | Recombination/repair | ||
|---|---|---|---|---|---|
| lin-35 | msp-36 | msp-10 | Y50E8A.B | syp-2 | spo-11 |
| lin-53 | msp-55 | msp-1 | Y59E9AR.1 | syp-1 | him-14 |
| hda-1 | msp-40 | msp-38 | Y59E9AR.7 | syp-3 | msh-5 |
| lin-9 | msp-142 | M199.2 | Y59H11AM.D | F41H10.10 | rad-50 |
| lin-36 | msp-49 | msp-31 | ZK1248.17 | F57C9.5 | mre-11 |
| C36H8.1 | msp-32 | ZK1248.4 | him-3 | rad-51 | |
| msp-74 | msp-33 | msp-113 | |||
| msp-3 | msp-57 | ZK1251.6 | |||
| msp-77 | msp-53 | msp-65 | |||
| msp-19 | T13F2.10 | msp-59 | |||
| msp-45 | msp-78 | msp-50 | |||
| F58A6.9 | Y116A8A.2 | msp-51 | |||
| msp-142 | Y116A8A.7 | ZK546.3 | |||
| msp-63 | Y39G10BM.E | msp-152 | |||
| msp82 | |||||
MSP, major sperm protein
Each column lists the WormBase gene identifiers corresponding to each gene included in the query list
For the sake of brevity, we only describe the results from the Rb query here; the gene recommender performed well with the other queries (presented on the Supplemental Web site). The retinoblastoma complex is a transcription factor complex that is conserved from worms to humans, and is involved in regulating the cell cycle (Dyson 1998). Our Rb query list contained five genes (Table 1). lin-35 encodes the C. elegans retinoblastoma protein, and loss-of-function mutations in this gene result in a class B synthetic multivulva phenotype (Lu and Horvitz 1998). lin-53 also has a class B synthetic multivulva mutant phenotype and encodes an ortholog of RbAP48, which binds to Rb in mammalian cells (Lu and Horvitz 1998). lin-9 and lin-36 have the same mutant phenotype as lin-35 (Ferguson and Horvitz 1989). hda-1 encodes histone deacetylase, which is known to bind Rb in mammalian cells (Solari and Ahringer 2000).
Coregulation of the Rb Query List in DNA Microarray Experiments
In order to be useful input for the gene recommender algorithm, a set of query genes must be coregulated in at least a subset of microarray experiments. To evaluate whether or not the genes in our Rb query list show coregulation in the microarray experiments, we first compared the level of coregulation of the Rb query list to lists of random genes of the same size; to do this, we plotted the experimental ZE scores for the Rb and control queries. The ZE scores were designed to have roughly Student's t distribution on k-1 degrees of freedom for queries of k genes, under some simplifying assumptions. For large queries, the distribution is nearly normal. For small queries such as the Rb query, we expect a heavier than normal histogram qualitatively like the one in Figure 1. The experiment ZE scores obtained using the Rb query list were much higher than those using random query lists (Fig. 1). For example, there were 30 experiments with a ZE score greater than 15 using the Rb query genes, whereas there were rarely any experiments with a ZE score larger than this using a random set of five genes (Fig. 1). This result shows that there is strong coregulation of the Rb genes in the DNA microarray data set. We obtained similar evidence for coregulation of genes for the other query lists (Supplemental Web site).
Figure 1.

Some DNA microarray experiments show coregulation of the genes in the Rb query. Histogram of experiment scores obtained using the retinoblastoma query (black bars) or using 100 random queries of the same size (white bars). The arrow indicates the maximum experiment score obtained across all the randomizations. A substantial fraction of the experiment Z scores obtained using the retinoblastoma query were higher than the maximum Z score obtained from random queries.
A second method to show that there is strong coregulation of the query genes in the microarray data is to determine that each of the genes in the query has a high score in leave-one-out experiments. For each query, one of the genes in the query was left out, the algorithm was rerun on the remaining genes, and the rank obtained by the gene that was left out was then scored. The ranks are scored as percentiles with 100 corresponding to the gene most similar to the query and 0 corresponding to the least similar. As we would expect, the histogram for random queries is nearly uniform over the range from 0% to 100% (Fig. 2). In contrast, the histogram for the real queries has a very large spike between the 95th and 100th percentile and a very small number of genes with much lower scores. This result shows that the genes in the query lists are coregulated, and that the gene recommender algorithm can accurately identify genes based on their level of coregulation.
Figure 2.

Leave-one-out cross-validation. Histograms of percentile ranks obtained after removing a gene from a query list of genes, building a cassette around the remaining query genes, and then scoring the held-out gene. Open bars show the histogram obtained from random queries ranging in size from 4 to 50; black bars show the histogram obtained from all the queries used in this study. The inset is an expanded view of the highest-scoring genes.
An Rb Hit List Generated by the Gene Recommender
We then used gene recommender to identify other genes that are coexpressed with the five genes in the Rb query list. The gene recommender used 320 of the 553 experiments, including every experiment in which none of the five query genes were missing. The gene recommender was able to cluster the five Rb query genes in a small group of 13 genes (shown in red in Table 2). As a control, the gene recommender did not succeed in clustering query genes into comparably small groups when it used five random samples of five genes (Table 3). This finding is consistent with the random queries having smaller experiment scores (Fig. 2), and indicates that the new candidate genes for the Rb query identified by the gene recommender are unlikely to be due to chance.
Table 2.
The Rb Hit List

Red denotes a gene from the query, blue denotes two genes known to interact with Rb not included in the query, and green denotes genes involved in the cell cycle or chromatin modeling.
SG is the gene score.
ZG is the gene score SG divided by an estimate of its standard deviation.
L is the likelihood ratio computed as the probability the gene received the score SG given it was drawn from the query's distribution over the probability the gene received the score SG given it was drawn from the background distribution.
Table 3.
Rb Query Has Higher Experiments Z Scores Than Random Queries
| Z*a | Rbb | r5ac | r5b | r5c | r5d | r5e |
|---|---|---|---|---|---|---|
| 0 | 3 | 19 | 82 | 116 | 197 | 48 |
| 1 | 3 | 8 | 31 | 116 | 163 | 88 |
| 2 | 4 | 3 | 63 | 359 | 69 | 328 |
| 3 | 8 | 3 | 81 | 23 | 332 | |
| 4 | 8 | 13 | 103 | 29 | 500 | |
| 5 | 13 | 61 | 191 | 149 | 421 | |
| 6 | 11 | 41 | 234 | 210 | 537 | |
| 7 | 8 | 44 | 344 | 228 | 537 | |
| 8 | 4 | 106 | 196 | 399 | ||
| 9 | 4 | 140 | 164 | |||
| 10 | 3 | 254 | 164 | |||
| 11 | 3 | |||||
| 12 | 3 | |||||
| 13 | 4 | |||||
| 14 | 4 |
Z* is a threshold used to select experiments
The number of genes not in the query that score higher than the median gene in the query, using an experiment threshold of Z*
Results for five randomly-generated queries of five genes each. The blank entries appear for cases where fewer than five experiments scored higher than Z*. No gene scoring was done in such cases
Among the top 20 ranked genes in the hit list generated using the Rb query, five are from the Rb query set itself. Three of the remaining 15 genes are also known to interact with Rb. In hindsight, they could reasonably have been included in the query group. Similarly, the top genes in the hit list generated by the gene recommender for the MSP query included some MSP genes that had been overlooked (Supplemental Table 2). The gene at the top of the Rb hit list, dpl-1, has a mutant phenotype similar to that of lin-35 Rb in worms and encodes a protein similar to mammalian DP1, a known Rb-binding protein (Lu and Horvitz 1998). The next candidate gene, K12D12.1, encodes topoisomerase II, which is known to bind to the Rb protein in mammalian cells (Bhat et al. 1999). The eighteenth gene on the hit list (mcm-7) encodes a protein involved in regulating DNA replication whose mammalian ortholog binds to Rb (Sterner et al. 1998).
Of the remaining 12 candidate genes, there are currently no data directly confirming whether or not they interact with the Rb complex. However, it is interesting that this set of genes is highly enriched for genes involved in regulation of chromatin structure and the cell cycle, which are functions related to those of the Rb complex. F55A3.7 encodes a protein similar to S. cerevisiae general chromatin factor Spt16p (Rowley et al. 1991). R06F6.1 encodes a protein similar to human histone hairpin-binding protein, which binds to histone mRNA and regulates its translation (Schaller et al. 1997). smc-4 encodes a component of a condensing complex required for normal mitotic chromosome architecture (Hagstrom et al. 2002). Ce Bub1 encodes a protein similar to yeast Bub1p, which is a serine/threonine protein kinase required for cell-cycle arrest in response to loss of microtubule function (Oegema et al. 2001). plk-1 encodes a member of the polo-like kinase family, which are involved in activation of the anaphase-promoting complex in late mitosis (Chase et al. 2000).
To examine how the genes in the Rb pathway are regulated, we analyzed the expression of the top 20 genes from the Rb hit list in published microarray experiments (Fig. 3). Expression of these genes is enriched in the germ line, and is stronger in oocytes than in sperm (Reinke et al. 2000). During development, the genes in the Rb hit list are expressed at highest levels in eggs and in adults (Jiang et al. 2001), and are induced during exit from the dauer stage (Wang and Kim 2003). As expected, the genes of the Rb hit list show high levels of coregulation to each other, and are most abundantly expressed under conditions with high levels of cell growth and division, such as in embryos and in adults with actively dividing germ cells.
Figure 3.

Expression profiles of genes in the Rb hit list. The germ line experiments compared expression in wild-type animals to glp-4 mutants lacking a germ line, and in mutants making only sperm to mutants making only oocytes (Reinke et al. 2000). The development experiments compared expression in whole wild-type worms throughout development and in hermaphrodites versus males (Jiang et al. 2001). The dauer experiments compared expression as animals exit the dauer stage following feeding in a timecourse experiment (Wang and Kim 2003). Rb query genes are shown in red. Scale shows level of expression.
RNAi Analysis
To analyze the function of the genes in the Rb hit list, we used RNA interference (RNAi) to induce their loss-of-function phenotypes. During C. elegans vulval development, the Rb complex and a redundant pathway both regulate cell divisions (Ferguson and Horvitz 1989; Lu and Horvitz 1998). Genes in the Rb pathway are referred to as class B synMuv genes, and genes in the redundant pathway are referred to as class A synMuv genes. Mutants defective in both pathways (class A and class B) have a multivulva phenotype, but mutants defective in only one pathway are wild-type (Ferguson and Horvitz 1989).
We used RNAi to specifically inhibit expression of the top 51 genes in the Rb hit list (Table 2 and Supplemental Web site). We induced RNAi by feeding bacterial strains that express dsRNA corresponding to genes in the Rb hit list. We tested the RNAi phenotype for each candidate gene in wild-type worms, a class A mutant strain (lin-8[n111]), and a class B mutant strain (lin-9[n112]). In these experiments, we found that the RNAi treatment did not induce a mutant phenotype in many cases, indicating that this assay may miss some new genes with a class B synMuv phenotype. Specifically, 30 of the 51 genes in the gene recommender hit list were previously known to have phenotypes that we would score as mutant in our RNAi assay (Supplemental Table 1). Of these 30, our RNAi experiments agreed with the previous mutant phenotype for seven genes, showed a weaker mutant phenotype for six genes, and exhibited no mutant phenotype for 17 genes. The RNAi experiments included four out of the five genes included in the Rb query. lin-35(RNAi) and lin-53(RNAi) had the expected class B synMuv phenotype. lin-36(RNAi) and hda-1(RNAi) appeared wild-type in all strains, even though loss-of-function mutations in these two genes result in a class B synMuv phenotype (Ferguson and Horvitz 1989; Solari and Ahringer 2000).
Of the three genes known to encode Rb-binding proteins, one (dpl-1) had a class B synMuv phenotype in RNAi experiments, consistent with previous results, whereas the other two (K12D12.1 and mcm-7) showed no mutant phenotype using any of the strains (Ceol and Horvitz 2001).
Among the remaining 43 genes that we tested, two showed a phenotype indicating that they interact with the lin-35 Rb pathway. RNAi analysis of the 42nd gene in the Rb list (JC8.6) elicited a synMuv phenotype very similar to that of lin-35 Rb (Fig. 4). JC8.6 encodes a protein similar to mammalian tesmin and Arabidopsis TSO1. Although little is known about the function of tesmin, TSO1 plays a role in plant meristem cell division (Sugihara et al. 1999; Hauser et al. 2000; Song et al. 2000). RNAi analysis of the 35th gene in the Rb list (wrm-1) indicates that it might act to antagonize the Rb pathway. Specifically, wrm-1(RNAi) resulted in embryonic lethality that was suppressed by loss-of-function lin-35 or lin-9 mutations (Table 4). wrm-1 encodes a β-catenin that functions in an embryonic Wnt signaling pathway (Rocheleau et al. 1999).
Figure 4.

JC8.6(RNAi) results in a Muv phenotype similar to lin-35 Rb(RNAi). (A) JC8.6(RNAi), (B) JC8.6(RNAi) lin-8(n111), (C) JC8.6(RNAi) lin-9(n112), (D) lin-35 Rb(RNAi) lin-8(n111). Arrows point to the vulva in A and C, and to pseudovulvae in B and D. Adults were fed bacteria expressing JC8.6 dsRNA, and the phenotypes of their progeny were scored. Scale bar is 20 μM.
Table 4.
lin-8 Suppresses wrm-1 Lethality
|
% viability of wrm-1 (RNAi)
|
|||
|---|---|---|---|
| RNAi feedinga | Wild-type | lin-8 (n111)b | lin-9(n112) |
| 8-16 h | 17 (n = 36) | 76 (n = 28) | 14 (n = 21) |
| 16-24 h | 0.2 (n = 413) | 25 (n = 342) | 0 (n >300) |
Length of time that adult animals were fed bacteria expressing wrm-1 ds-RNA
In addition to lin-8, we tested two other class A genes (lin-38 and lin-15A). lin-38(n751) but not lin-15(n767) could suppress wrm-1 (RNAi) lethality
Gene Recommender Generates a More Specific Hit List Than Does the Gene Expression Topomap
One advantage of targeted clustering over global clustering algorithms is that experiments that do not contribute useful clustering information can be removed; for example, experiments that do not show coordinate regulation of the query list. Either noise or multifunctionality of some query genes can lead to an experiment's removal. To demonstrate the advantages of targeted clustering, we compared hit lists generated by the gene recommender to some hit lists generated using the gene expression topomap. To derive a hit list from the topomap, we located the query genes on the topomap, and then ranked all genes according to their distance from the centroid (average) of the query gene locations.
First, we compared the hit lists produced by the gene recommender and the topomap using a method borrowed from information retrieval. If we knew the complete set of genes in the genome associated with the Rb pathway (the true Rb cassette), then we could compute the precision and recall for any hit list. Precision is the fraction of true Rb genes in a hit list out of the total number of genes in that hit list. Recall is the fraction of true Rb genes in the hit list out of all of the true Rb genes in the genome. There is a precision–recall tradeoff, because increasing the size of a hit list usually lowers precision, but cannot lower recall.
For us, precision is more important than recall. Higher precision means a greater chance that subsequent experiments will confirm predictions made by the gene recommender. In contrast, high recall is important when one is more interested in finding all or almost all genes relevant to a query.
Because the true status of whether a gene interacts with the Rb complex is usually unknown, we judge the precision of a hit list by the proportion of the Rb query genes near the top of the list. Specifically, we construct hit lists containing just enough of the highest-ranking genes to obtain a given number of query genes, such that a shorter list is evidence of a more precise result. The true precision cannot be worse than our estimate, but it could be better due to true unknown Rb genes in the list. When comparing algorithms, small differences in estimated precision could arise from our inability to count true Rb genes that were not in the query. On the assumption that Rb genes are rare, large differences in estimated precision, like those shown below, cannot plausibly be due to uncounted true Rb genes.
The set of five Rb query genes are localized in a broad area of mount 11 in the gene expression topomap. This broad area includes not only the five Rb query genes, but also 337 other genes (at 100% recall). In comparison, the top 13 genes from the gene recommender contained all five of the Rb query genes. To capture at least two Rb query genes requires the top six genes of the gene recommender list, but requires the top 138 genes from the topomap list (50% recall; Table 5). In addition to the Rb query list, the gene recommender provided a shorter list of candidate genes for each of the four sets of query genes compared to the list generated by the gene expression topomap (Supplemental Web site). These results demonstrate that the clusters generated by the gene recommender algorithm are tighter than those created by the gene expression topomap.
Table 5.
The Gene Recommender Is More Precise Than the Gene Expression Topomap
|
Size
|
Precisione
|
||||||
|---|---|---|---|---|---|---|---|
| Query | Na | Gen. Rec.b | Topomapc | Randd | Gen. Recall | Topomap | Random |
| Retinoblastoma | 5 | 6 | 138 | 160 | 50% | 2% | 2% |
| Recombination/Repairf | 6 | 57 | 1271 | 85 | 5% | 0% | 4% |
| Synap complex | 6 | 4 | 246 | 85 | 75% | 1% | 4% |
| MSP | 43 | 32 | 225 | 4017 | 69% | 10% | 1% |
MSP, major sperm protein
Number of genes in the query
Number of genes found by the gene recommender at 50% recall
Number of genes found by the gene expression topomap at 50% recall
Number of genes found by the gene recommender from a set of random genes at 50% recall
Percent of genes in the list at 50% recall that are from the query list
This group is known to be comprised of two distinct subgroups, and hence resulted in a hit list with lower precision
If a candidate gene showed tighter clustering with genes in a second pathway or if there were more experiments showing clustering with a second pathway, then a global clustering approach would include the candidate gene together with the genes in the second pathway instead of with the query genes. However, the gene recommender could cluster the candidate gene along with the genes in the Rb complex, because it only scores interactions with the query genes. We determined whether the gene recommender found genes that were missed by the gene expression topomap. Among the top 15 candidate genes in the gene recommender hit list, 11 are clustered along with the Rb genes in mount 11 on the gene expression topomap. However, four (K12D12.1, T16G12.5, F55A3.7, and drp-1) do not cluster with the Rb genes at all but are clustered together in mount 5 (Kim et al. 2001). This result suggests that these four genes are also coregulated with genes in another pathway, and that this coregulation led to their clustering in mount 5 rather than mount 11.
DISCUSSION
The entire C. elegans research community is aided by functional genomics approaches, such as whole-genome expression profiling, global RNAi analysis, and high-throughput yeast two-hybrid analysis (Kim 2001). For example, the gene expression topomap has been used by a wide variety of labs to gain biological insight. Several groups have used the topomap to study new genes that are coexpressed with genes in the germ line or pharynx (Piano et al. 2002; Walhout et al. 2002; S. Mango, pers. comm.; A. Villeneuve, data not shown). The topomap has been used to show that genes that are coexpressed together are clustered on the chromosome, possibly due to the effects of chromatin domains on gene expression (Roy et al. 2002). The topomap was used to show that genes in mount 15 are regulated by both aging and the dauer state, possibly identifying genes involved in a common mechanism between these two processes (Lund et al. 2002). Similarly, we make the gene recommender available as a tool for the C. elegans research community.
As many researchers use microarray data as the basis for designing genetic experiments, it is critical to develop better algorithms in order to better discern inherent biological patterns. Better algorithms in this setting have greater specificity and result in fewer false positives, thus saving time and expense in follow-up experiments.
Here, we present an algorithm called a gene recommender to identify genes that are coexpressed with a given set of genes of interest. For identifying new genes coexpressed with a known set of genes, this algorithm has a number of advantages over global approaches used previously. First, the gene recommender selects for microarray experiments that are informative (i.e., showing tight coregulation of the query genes) and ignores uninformative experiments that would otherwise add noise to the calculations. As a result, it generates hit lists that are shorter and more concentrated with query genes than lists generated by the gene expression topomap. Second, the gene recommender can find interactions for genes that are in multiple clusters. Genes that are multifunctional, that are expressed at different times during development, or that are expressed in different tissues may interact with multiple different pathways. Global clustering approaches such as hierarchical clustering or multidimensional scaling would place such multifunctional genes into the strongest cluster and would thus lose interactions with other clusters. For example, the Rb hit list generated by the gene recommender included four genes (K12D12.1, T16G12.5, F55A3.7, and drp-1) that were not clustered with Rb by the gene expression topomap, but were instead clustered together along with other genes in mount 5.
While our work was in progress, a similar strategy (termed the signature algorithm) was developed independently by Ihmels et al. (2002), and a strategy using fuzzy clustering was developed by Gasch and Eisen (2002). Both of these clustering algorithms allow one gene to be placed in several different gene clusters, similar to the gene recommender algorithm presented here. Both the gene recommender and the signature algorithm have the ability to choose a subset of experiments to cluster genes, based on the set of input query genes. These two algorithms may group sets of genes that are coregulated in a small set of experiments, but not in the entire set of experiments. In contrast, the hierarchical clustering and perhaps even fuzzy clustering algorithms cluster genes based on correlations using the whole set of experiments, and may have difficulty finding genes that cluster only in a small number of experiments.
In addition to the examples shown here, the gene recommender can be used by the research community to find interacting genes, both in C. elegans and for other organisms once gene expression databases have been assembled (http://pmgm2.stanford.edu/~kimlab/cassettes). For expository purposes, we have taken a conservative approach using only fixed queries. Users may prefer to iteratively modify their list of query genes. When the gene recommender produces a list identifying a gene that was inadvertently left off of the query list (such as dpl-1 from the Rb query), the user could add such candidates to the query list and re-run the gene recommender program. Similarly, when a leave-one-out or other analysis suggests that a gene in the query does not belong, that gene can be deleted from the query and the recommender can be re-run. However, such ad hoc modifications risk overfitting the data; for example, if one adds genes to the query because they cluster tightly, then a leave-one-out analysis of the modified list would not be meaningful because the new genes must necessarily cluster. Even such ad hoc queries can be convincingly confirmed by experimentation, such as with RNAi knockdown experiments.
In some cases, a single query list might be divided into subgroups that have distinct gene expression profiles. For example, the six genes in the meiotic recombination/DNA repair query could be split into a repair group and a recombination group. In some instances, it might be useful to run the gene recommender on the separate groups instead of the entire list; using the entire list could cause both subgroups to be averaged together, resulting in a loss of specificity.
For the analysis discussed here, we found that queries ranging from five to 41 genes were reliably distinct from queries of the same size using random genes. But we found that queries using only three genes were oftentimes not distinct from random queries. These conclusions are specific to the data set used in the present study, and thus these findings regarding usable sizes of queries may change as more C. elegans expression data are added or for calculations on expression from other organisms. By performing the controls reported here, one can evaluate whether the hit list generated by the gene recommender using a real query is significantly different from a control using random data.
New Genes That Interact With the Rb Complex
For the case of the Rb complex, we found several new genes that appear to interact with Rb in regulating the cell cycle, a finding of some biological significance. The Rb protein complex has been extensively analyzed in worms, flies, mice, and humans (Dyson 1998). In C. elegans, genes that encode components of the Rb complex are involved in specifying vulval cell fates, and these genes have been found and analyzed in a large number of genetic experiments regarding vulval development (Ferguson and Horvitz 1989; Kornfeld 1997). Among the top 15 candidate genes, three (dpl-1, K12D12.1, and mcm-7) encode proteins similar to mammalian proteins that bind to Rb (Sterner et al. 1998; Bhat et al. 1999; Ceol and Horvitz 2001). We also used RNAi to show that two genes (wrm-1 and JC8.6) interact with the genes in the Rb pathway; neither gene was previously known to do so. JC8.6 acts along with Rb, whereas wrm-1 acts to antagonize the Rb pathway. JC8.6 encodes a tesmin-related protein, which is involved in cell division in Arabidopsis (Hauser et al. 2000; Song et al. 2000). In summary, RNAi analysis of candidates provided by the gene recommender identified two new genes that interact with the lin-35 Rb genetic pathway, even though this pathway has previously undergone considerable genetic analysis. In addition to these two, the Rb hit list may contain other candidates that interact with the Rb genetic pathway; these genes might have been missed because the RNAi experiments did not induce a mutant phenotype or because they act redundantly (two mutations plus a class A mutation might be needed to produce a multivulva phenotype).
wrm-1 encodes a β-catenin that functions to transduce a Wnt signal in the C. elegans embryo (Rocheleau et al. 1999). Previous work has focused on the roles of wrm-1 and lin-35 in separate tissues and had not revealed any functional interaction between them; wrm-1 is involved in a Wnt signaling pathway in the early embryo, and lin-35 Rb is involved in regulating vulval cell fates in the second larval stage (Lu and Horvitz 1998; Rocheleau et al. 1999). It is interesting that the gene recommender found a strong correlation between these two genes based on microarray data involving RNA extracted from entire worms. Correlation using RNA from whole worms indicates that the expression of these two genes are correlated in multiple tissues throughout development, suggesting that both these genes have interrelated functions that are widespread throughout development.
Neither WRM-1 activity nor Rb activity are thought to be directly controlled by transcriptional regulation: WRM-1 is a homolog of β-catenin, which is released into the nucleus as a result of Wnt signaling (Willert and Nusse 1998), and Rb is regulated by phosphorylation during the cell cycle (Dyson 1998). Nevertheless, the gene recommender found them to be coexpressed based on gene expression data. The coexpression of WRM-1 and Rb indicates that the Wnt and Rb pathways are both present at similar developmental times and tissues in order to enable these patheleau ways to regulate each other.
There are examples of other Wnt signaling pathways that are known to antagonize the Rb pathway. In mouse breast cancers, Wnt acts as an oncogene by turning on a pathway involving β-catenin, whereas Rb is a tumor suppressor, indicating that these two genes act oppositely to regulate cancer growth (Nusse et al. 1984; Dyson 1998). In worms, another β-catenin homolog (bar-1) acts to induce vulval cell fates, whereas lin-35 Rb acts in the synMuvB pathway to repress these fates (along with the synMuvA pathway; Eisenmann et al. 1998; Lu and Horvitz 1998). These observations are further evidence that Rb and Wnt signaling are coupled, each acting to counteract the other to jointly regulate cell division.
METHODS
Algorithm
Within the basic outline of our algorithm there is scope for variation. The choices we made were influenced by several factors. First, we wanted our method to be usable even with a large amount of missing data. Second, we put greater priority on precision than recall. Third, we put greater priority on finding cassette members not in the query list than on identifying possibly incorrect members of the query list. Some other choices were made for statistical simplicity and computational efficiency. For example, some of the simple statistics we use have distributions that are easily analyzed in ideal settings, giving a rough guide to, and a benchmark for, their behavior on real data. We think that the choices we made are appropriate, but we do not claim that any set of choices is compelling.
Normalization
We begin with an n by p matrix of real values Yij in which missing values are identified. Each row corresponds to a gene and each column to an experiment.
The data are normalized by taking their ranks within rows. Let pi be the number of non-missing values among Yij for j=1,..., p. Let Rij be the rank of Yij among the non-missing values in Yi1,...,Yip. That is, Rij=1 for the smallest non-missing value, 2 for the second smallest, and so on. The transformed values are
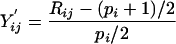 |
that have very nearly the uniform distribution on the interval (-1,1). Our data set has very few tied values. For data with many ties, we recommend first averaging the ranks of tied values. If the 9′th, 10′th and 11′th smallest values are equal, they should all get rank 10.
To simplify notation, we now suppose that Yij are themselves the rank transformed values previously denoted by Y′ij. Rank transformation has several advantages. First, it diminishes the effects of outliers. Second, the data values for each gene have mean zero and variance 1/3. As a consequence, the correlation between the (ranked) expression levels of genes i and i′ is linearly related to the sum Σj YijYi′j, as is the Euclidean distance between the vectors of expression levels.
The non-missing data in each row could be replaced by quantiles for distributions other than U(-1,1). For example, normal scores Φ-1 (Rij/[pi+1]) would space out the extreme ranks more than uniform scores.
Experiment Scoring
We use Q to designate the set of genes in the query. The biological factors underlying coexpression of the query genes might only be applicable within a subset of the experiments in a data set. If the values Yij for i∈Q are similar, then experiment j is likely to be informative regarding the joint behavior of these genes. On the other hand, if the query values Yij for i∈Q vary by about the same amount as the non-query values, then including experiment j in gene scoring may add unwanted noise.
For the j′th experiment, let ȲQ,j be the average of the non-missing values among Yij for i∈Q and let V̂Q,j be the sample variance of those values. Then the score for experiment j is
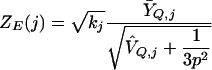 |
1 |
where kj is the number of non-missing values among Yij for fixed j and all i∈Q. The score itself is taken to be missing if kj is too small. The minimum value of kj is usually 5, but when the number |Q| of genes in Q is below 5, then |Q| becomes the lower limit on kj.
The informative experiments are considered to be those with ZE(j) far from zero. This score combines a preference for experiments with extreme expression levels (very large or very small ȲQ,j) with a preference for tight clustering of expression levels (small V̂Q,j). There is a small possibility that V̂Q,j = 0. If we consider rank r out of p to represent a rank uniformly distributed between r-1/2 and r+1/2, then we could reasonably add a variance of 1/12 to any rank. In scaling ranks to the interval (-1,1), this variance changes to 1/(3p2), which appears in the denominator of ZE(j).
As a rough guide, if experiment j is completely irrelevant to Q, then we would expect the values Yij to be a random sample of k values without replacement from the uniform distribution on (-1 and 1). The distribution of ZE(j) is then very nearly Student's t on k-1 degrees of freedom (assuming k≪n), which in turn is close to N(0,1) unless k is very small.
Gene Scoring
Let ε be a set of experiments deemed relevant to the query genes Q. Then the score for gene i is SG(i), the mean of ȲQ,j ×Yij, over experiments j∈Q for which both ȲQ,j and Yij are not missing. If too few such j are available, then SG(i) is itself missing. All of the Yij and ȲQ,j are between -1 and 1, and so therefore is SG(i).
There is some randomness in whether Yij will cluster near -1 or 1, due to “sign noise” in the original expression data. If two experimenters make different choices for which sample goes in the red versus green channels, then those experimenters' expression measurements will tend to have the same magnitude but opposite signs. This sign noise can have a severe effect on correlations but tends to have less effect on the uncentered correlation.
The score SG(i) is our measure of the extent to which gene i matches the query Q. We also compute a Z score
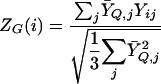 |
2 |
The sums in (2) are over experiments j∈ε for which neither ȲQ,j nor Yij are missing. For nonquery genes, i∉Q, if Yij are randomly assigned independently of ȲQ,j, then ZG(i) has approximately an N(0,1) distribution.
We use SG as our guide to biological significance and ZG as a rough guide to statistical significance. In the absence of missing data, the ratio ZG(i)/SG(i) is the same for all genes i. When there are many missing values and their number varies from gene to gene, then |ZG| tends to be much larger for genes with fewer missing values.
Threshold Selection
An important decision in the search for genes related to Q is the number of experiments to include in the relevant set ε. The gene score is a sum of contributions from experiments of which some may be irrelevant to the functioning of the genes in the query Q.
While it is intuitively reasonable that including irrelevant and noisy experiments can degrade the performance of gene scoring, there is not a good statistical argument to select a priori a Z-score above which an experiment will improve the search performance, and below which an experiment will degrade the search. The combined effect of several not-quite significant experiments may be informative, because lack of significance is a lack of proven relevance, and not necessarily a lack of relevance. Our approach is to explore a small grid of threshold values. We select the threshold Z to minimize the number of genes i Q that score higher than the median score of genes i Q. Our rationale is that a good set of experiments should bring the known members of Q to the top of the list. Our interest in specificity and belief that there may only be a small number of true unknown Q members lead us to prefer a small number of non-Q genes near the top of the list. Though the threshold was chosen to bring the original query to the top of the list, we see from Figure 2 that the query genes get high ranks even in leave-one-out experiments.
Comparing Hit Lists
The result of a targeted clustering produces a ranked list of genes. The ranked list can then be truncated to produce a hit list. Recall is the fraction of genes from the cassette included in the list. Precision is the fraction of the hit list's genes that also belong to the cassette. Since we don't know which genes actually belong to the cassette, we fix the fraction of query genes captured in the hit list. If the size of the cassette is comparable to the size of the query, then the fraction of query genes will be a good approximation to a measure of recall.
Likelihood Ratios
In addition to ranking the genes by decreasing score, we also compute a measure indicating the likelihood that the assigned score came from the query distribution versus the background distribution. For each gene in the genome i we compute the log-likelihood ratio:
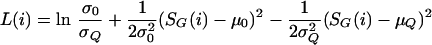 |
where μ0 and σ0 are the mean and standard deviation computed from the SG(i) scores of the background distribution, and μQ and σQ are the mean and standard deviation computed from the SG(i) scores of the query genes. We report the likelihood ratio eL(i) for each gene. We include all nonquery genes into the background distribution. Because some genes may have been placed erroneously into the query set, we only include query genes into the query distribution if their SG(i) scores in the 90th percentile among all genes. These ratios should be used with caution, because the normal approximation makes the likelihood ratios nonmonotonic with respect to SG(i). Extremely high-scoring genes can have smaller likelihood ratios than genes with correspondingly smaller scores. However this causes little problem, because the likelihood ratios are used to find an intuitive cutoff between the background and query distributions where the ratios are usually monotonically increasing in SG(i). We also count and record the number of non-Q genes, if any, scoring higher than all Q members as well as the number scoring higher than at least one Q member.
Similar Algorithms
Our method is related to a body of research called feature selection in which a set of informative features are sought that optimally classify sets of observations. Feature selection has been applied primarily in cases where at least two classes of observations (e.g., positive and negative) are known. The problem we address here is different because only a partial list of related genes is provided as input, and thus a clear distinction between class boundaries cannot be assumed. The feature selection task here seeks experiments that group the given genes together, as opposed to those that distinguish some genes from others. Friedman and Meulman (2002) define metrics for finding clusters on preferentially selected sets of attributes. However, the application for their approach is replacing the distance metric used in a global clustering of the data. Our work is also similar to the recent semisupervised approaches in which pre-established classes of genes are scored based on several different measures computed from expression data (Pavlidis et al. 2002). Yeung et al. (2001) use a variability-based measure on held-out experiments to decide whether a cluster is real. Mateos et al. (2002) attempted to define the learnability of a gene class using multilayer neural networks trained on microarray data. Their goals were to investigate how well pre-established classes of genes could be distinguished from the rest of the genes in the genome. These methods assign a score to a set of genes that reflects how well the group can be distinguished from the rest of the genome based on their expression profiles. Our work differs in that it seeks out new candidates for inclusion in the given set. Lastly, a recent method developed by Ihmels et al. (2002) also attempts to find new candidates to a given query set. Though the details of the algorithms are different, the gene recommender and signature algorithms both are similar to recommender systems and search engines. Both methods select a set of informative experiments prior to scoring genes.
RNAi Technique
NGM agar with 1mM IPTG and 50 μg/mL ampicillin was inoculated with bacteria for each targeted gene (Fraser et al. 2000). Three to five L4 worms were placed on each RNAi plate, and F1 progeny were scored for survival and multivulval phenotype.
Competing Interests Statement
The authors declare that they have no competing financial interests.
Acknowledgments
We thank Laura Lazzeroni for helpful discussions. This work was supported by grants from NIH GMS and NCRR to S.K.K. and by the NSF (DMS-0072445) to A.B.O.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.1125403.
Footnotes
[Supplemental material is available online at www.genome.org.]
References
- Bhat, U.G., Raychaudhuri, P., and Beck, W.T. 1999. Functional interaction between human topoisomerase IIα and retinoblastoma protein. Proc. Natl. Acad. Sci. 96: 7859-7864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, P.O. and Botstein, D. 1999. Exploring the new world of the genome with DNA microarrays. Nat. Genet. 21: 33-37. [DOI] [PubMed] [Google Scholar]
- The C. elegans Sequencing Consortium. 1998. Genome sequence of the nematode C. elegans: A platform for investigating biology. Science 282: 2012-2018. [DOI] [PubMed] [Google Scholar]
- Ceol, C.J. and Horvitz, H.R. 2001. dpl-1 DP and efl-1 E2F act with lin-35 Rb to antagonize Ras signaling in C. elegans vulval development. Mol. Cell 7: 461-473. [DOI] [PubMed] [Google Scholar]
- Chase, D., Serafinas, C., Ashcroft, N., Kosinski, M., Longo, D., Ferris, D.K., and Golden, A. 2000. The polo-like kinase PLK-1 is required for nuclear envelope breakdown and the completion of meiosis in Caenorhabditis elegans. Genesis 26: 26-41. [DOI] [PubMed] [Google Scholar]
- Dyson, N. 1998. The regulation of E2F by pRB-family proteins. Genes & Dev. 12: 2245-2262. [DOI] [PubMed] [Google Scholar]
- Eisen, M.B., Spellman, P.T., Brown, P.O., and Botstein, D. 1998. Cluster analysis and display of genome-wide expression patterns. Proc. Natl. Acad. Sci. 95: 14863-14868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenmann, D.M., Maloof, J.N., Simske, J.S., Kenyon, C., and Kim, S.K. 1998. The β-catenin homolog BAR-1 and LET-60 Ras coordinately regulate the Hox gene lin-39 during Caenorhabditis elegans vulval development. Development 125: 3667-3680. [DOI] [PubMed] [Google Scholar]
- Ferguson, E.L. and Horvitz, H.R. 1989. The multivulva phenotype of certain Caenorhabditis elegans mutants results from defects in two functionally redundant pathways. Genetics 123: 109-121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser, A.G., Kamath, R.S., Zipperlen, P., Martinez-Campos, M., Sohrmann, M., and Ahringer, J. 2000. Functional genomic analysis of C. elegans chromosome I by systematic RNA interference. Nature 408: 325-330. [DOI] [PubMed] [Google Scholar]
- Friedman, J.H. and Meulman, J.J. 2002. Clustering objects on susbets of attributes. Technical Report, Stanford University, Statistics.
- Gasch, A.P. and Eisen, M.B. 2002. Exploring the conditional coregulation of yeast gene expression through fuzzy k-means clustering. Genome Biol. 3: RESEARCH0059.1-RESEARCH0059.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagstrom, K.A., Holmes, V.F., Cozzarelli, N.R., and Meyer, B.J. 2002. C. elegans condensin promotes mitotic chromosome architecture, centromere organization, and sister chromatid segregation during mitosis and meiosis. Genes & Dev. 16: 729-742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauser, B.A., He, J.Q., Park, S.O., and Gasser, C.S. 2000. TSO1 is a novel protein that modulates cytokinesis and cell expansion in Arabidopsis. Development 127: 2219-2226. [DOI] [PubMed] [Google Scholar]
- Hughes, T.R., Marton, M.J., Jones, A.R., Roberts, C.J., Stoughton, R., Armour, C.D., Bennett, H.A., Coffey, E., Dai, H., He, Y.D., et al. 2000. Functional discovery via a compendium of expression profiles. Cell 102: 109-126. [DOI] [PubMed] [Google Scholar]
- Ihmels, J., Friedlander, G., Bergmann, S., Sarig, O., Ziv, Y., and Barkai, N. 2002. Revealing modular organization in the yeast transcriptional network. Nat. Genet. 31: 370-377. [DOI] [PubMed] [Google Scholar]
- Jiang, M., Ryu, J., Kiraly, M., Duke, K., Reinke, V., and Kim, S.K. 2001. Genome-wide analysis of developmental and sex-regulated gene expression profiles in Caenorhabditis elegans. Proc. Natl. Acad. Sci. 98: 218-223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, S.K. 2001. Http://C. elegans: Mining the functional genomic landscape. Nat. Rev. Genet. 2: 681-689. [DOI] [PubMed] [Google Scholar]
- Kim, S.K., Lund, J., Kiraly, M., Duke, K., Jiang, M., Wylie, B.N., and Davidson, G.S. 2001. A gene expression map for C. elegans. Science 293: 2087-2092. [DOI] [PubMed] [Google Scholar]
- Kornfeld, K. 1997. Vulval development in Caenorhabditis elegans. Trends Genet. 13: 55-61. [DOI] [PubMed] [Google Scholar]
- Lander, E.S., Linton, L.M., Birren, B., Nusbaum, C., Zody, M.C., Baldwin, J., Devon, K., Dewar, K., Doyle, M., FitzHugh, W., et al. 2001. Initial sequencing and analysis of the human genome. Nature 409: 860-921. [DOI] [PubMed] [Google Scholar]
- Lu, X. and Horvitz, H.R. 1998. lin-35 and lin-53, two genes that antagonize a C. elegans Ras pathway, encode proteins similar to Rb and its binding protein RbAp48. Cell 95: 981-991. [DOI] [PubMed] [Google Scholar]
- Lund, J., Tedesco, P., Duke, K., Wang, J., Kim, S.K., and Johnson, T.E. 2002. Transcriptional profile of aging in C. elegans. Curr. Biol. 12: 1566-1573. [DOI] [PubMed] [Google Scholar]
- Mateos, A., Dopazo, J., Jansen, R., Tu, Y., Gerstein, M., and Stolovitzky, G. 2002. Systematic learning of gene functional classes from DNA array expression data by using multilayer perceptrons. Genome Res. 12: 1703-1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers, E.W., Sutton, G.G., Delcher, A.L., Dew, I.M., Fasulo, D.P., Flanigan, M.J., Kravitz, S.A., Mobarry, C.M., Reinert, K.H., Remington, K.A., et al. 2000. A whole-genome assembly of Drosophila. Science 287: 2196-2204. [DOI] [PubMed] [Google Scholar]
- Nusse, R., van Ooyen, A., Cox, D., Fung, Y.K., and Varmus, H. 1984. Mode of proviral activation of a putative mammary oncogene (int-1) on mouse chromosome 15. Nature 307: 131-136. [DOI] [PubMed] [Google Scholar]
- Oegema, K., Desai, A., Rybina, S., Kirkham, M., and Hyman, A.A. 2001. Functional analysis of kinetochore assembly in Caenorhabditis elegans. J. Cell. Biol. 153: 1209-1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlidis, P., Lewis, D.P., and Noble, W.S. 2002. Exploring gene expression data with class scores. Pac. Symp. Biocomput. : 474-485. [PubMed]
- Piano, F., Schetter, A.J., Morton, D.G., Gunsalus, K.C., Reinke, V., Kim, S.K., and Kemphues, K.J. 2002. Gene clustering based on RNAi phenotypes of ovary-enriched genes in C. elegans. Curr. Biol. 12: 1959-1964. [DOI] [PubMed] [Google Scholar]
- Reinke, V., Smith, H.E., Nance, J., Wang, J., Van Doren, C., Begley, R., Jones, S.J., Davis, E.B., Scherer, S., Ward, S., et al. 2000. A global profile of germline gene expression in C. elegans. Mol. Cell 6: 605-616. [DOI] [PubMed] [Google Scholar]
- Rocheleau, C.E., Yasuda, J., Shin, T.H., Lin, R., Sawa, H., Okano, H., Priess, J.R., Davis, R.J., and Mello, C.C. 1999. WRM-1 activates the LIT-1 protein kinase to transduce anterior/posterior polarity signals in C. elegans. Cell 97: 717-726. [DOI] [PubMed] [Google Scholar]
- Rowley, A., Singer, R.A., and Johnston, G.C. 1991. CDC68, a yeast gene that affects regulation of cell proliferation and transcription, encodes a protein with a highly acidic carboxyl terminus. Mol. Cell. Biol. 11: 5718-5726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy, P.J., Stuart, J.M., Lund, J., and Kim, S.K. 2002. Chromosomal clustering of muscle-expressed genes in Caenorhabditis elegans. Nature 418: 975-979. [DOI] [PubMed] [Google Scholar]
- Schaller, A., Martin, F., and Muller, B. 1997. Characterization of the calf thymus hairpin-binding factor involved in histone pre-mRNA 3′ end processing. J. Biol. Chem. 272: 10435-10441. [DOI] [PubMed] [Google Scholar]
- Solari, F. and Ahringer, J. 2000. NURD-complex genes antagonize Ras-induced vulval development in Caenorhabditis elegans. Curr. Biol. 10: 223-226. [DOI] [PubMed] [Google Scholar]
- Song, J.Y., Leung, T., Ehler, L.K., Wang, C., and Liu, Z. 2000. Regulation of meristem organization and cell division by TSO1, an Arabidopsis gene with cysteine-rich repeats. Development 127: 2207-2217. [DOI] [PubMed] [Google Scholar]
- Sterner, J.M., Dew-Knight, S., Musahl, C., Kornbluth, S., and Horowitz, J.M. 1998. Negative regulation of DNA replication by the retinoblastoma protein is mediated by its association with MCM7. Mol. Cell. Biol. 18: 2748-2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugihara, T., Wadhwa, R., Kaul, S.C., and Mitsui, Y. 1999. A novel testis-specific metallothionein-like protein, tesmin, is an early marker of male germ cell differentiation. Genomics 57: 130-136. [DOI] [PubMed] [Google Scholar]
- Venter, J.C., Adams, M.D., Myers, E.W., Li, P.W., Mural, R.J., Sutton, G.G., Smith, H.O., Yandell, M., Evans, C.A., Holt, R.A., et al. 2001. The sequence of the human genome. Science 291: 1304-1351. [DOI] [PubMed] [Google Scholar]
- Walhout, A.J., Reboul, J., Shtanko, O., Bertin, N., Vaglio, P., Ge, H., Lee, H., Doucette-Stamm, L., Gunsalus, K.C., Schetter, A.J., et al. 2002. Integrating interactome, phenome, and transcriptome mapping data for the C. elegans germline. Curr. Biol. 12: 1952-1958. [DOI] [PubMed] [Google Scholar]
- Wang, J. and Kim, S.K. 2003. Global analysis of dauergene expression in Caenorhabditis elegans. Development 130: 1621-1634. [DOI] [PubMed] [Google Scholar]
- Willert, K. and Nusse, R. 1998. β-catenin: A key mediator of Wnt signaling. Curr. Opin. Genet. Dev. 8: 95-102. [DOI] [PubMed] [Google Scholar]
- Yeung, K.Y., Haynor, D.R., and Ruzzo, W.L. 2001. Validating clustering for gene expression data. Bioinformatics 17: 309-318. [DOI] [PubMed] [Google Scholar]
WEB SITE REFERENCES
- http://pmgm2.stanford.edu/~kimlab/cassettes; details of the gene recommender and a Web interface to our software and data.
- http://www-stat.stanford.edu/~owen/transposable; articles and links comparing data analysis of DNA expression, recommender engines, search engines, and educational testing.