

Abstract
Emerging information implies that the Ron receptor tyrosine kinase may play a role in the inflammatory response. However, the manner in which this receptor contributes to the response is not well understood. In the present studies, we investigated the role of the Ron receptor in the acute lung inflammatory response. Wild-type and mutant mice lacking the tyrosine kinase domain of Ron (Ron TK−/−) were subjected to acute lung injury induced by intranasal administration of bacterial lipopolysaccharide (LPS). Wild-type mice showed increased lung injury after LPS administration, as determined by the leakage of albumin into the lung and by histopathological changes. Ron TK−/− mice had more than twice the amount of albumin leak and much greater thickening of the alveolar septae. Lipopolysaccharide administration caused neutrophil recruitment into the lungs, as measured by myeloperoxidase. However, Ron TK−/− mice had much higher baseline levels of myeloperoxidase, which did not increase further after LPS. Lung injury in wild-type mice occurred with activation of the transcription factor, nuclear factor κB (NF-κB), and subsequent increases in intrapulmonary generation of tumor necrosis factor α. In TK−/− mice, there was far less IκB-α and IκB-β protein and greater activation of NF-κB. This was associated with substantially increased production of tumor necrosis factor α and the nitric oxide (NO) by-product, nitrite. The data suggest that the Ron receptor tyrosine kinase plays an important regulatory role in acute inflammatory lung injury by suppressing signals leading to activation of NF-κB.
Keywords: Inflammation, transcription factors, cytokines, hepatocyte growth factor–like protein, receptor tyrosine kinase
INTRODUCTION
Multiple organ failure is the most common cause of death in critically ill patients, and acute lung injury is a primary component of this clinical scenario. Development of acute lung injury and/or acute respiratory distress syndrome typically occurs secondary to an initial systemic insult, such as sepsis, trauma, or ischemia/reperfusion injury (1–3). Using experimental animal models, much has been learned regarding the pathogenesis of acute lung injury, including mediator pathways and cellular effector mechanisms (4–7). It is clear that the inflammatory response induced by such insults triggers complement activation and activation of tissue macrophages. Activated pulmonary macrophages generate the early-response cytokines, tumor necrosis factor α (TNF-α) and interleukin 1 (IL-1), which propagate the inflammatory response throughout the lung by stimulating the expression of vascular cell adhesion molecules and chemokines (8–11). These secondary mediators facilitate the pulmonary recruitment of neutrophils which, with activated macrophages, contribute to lung injury through the release of oxidants and proteases (12, 13).
The regulation of early-response cytokines required for the acute lung inflammatory response has also been well studied and found to be largely under the control of the transcription factor, nuclear factor κB (NF-κB) (5). During lung injury, ligand binding and activation of a number of receptor systems lead to activation of NF-κB in lung cells. These include receptors for TNF-α, IL-1, and lipopolysaccharide (LPS), all of which are relevant to inflammatory lung injury. However, other mechanisms may augment or suppress the signaling of these receptors in ways which may fine tune the signaling response. With regard to NF-κB activation, there is evidence to suggest that the Ron receptor tyrosine kinase may regulate the response to inflammatory mediators.
Ron is a member of the Met family of receptor tyrosine kinases and is widely expressed in epithelial cells, hematopoietic stem cells, and macrophages (14–17). The ligand for Ron is hepatocyte growth factor–like protein, HGFL (also known as macrophage-stimulating protein). The HGFL is primarily produced by hepatocytes and is secreted into the circulation as an inactive precursor and at relatively high levels (~400 ng/mL) (18). At sites of injury, endogenous proteases cleave the inactive pro-HGFL into an active, disulfide-linked heterodimeric form that can bind to and activate Ron (18, 19). Mice with targeted deletions in Ron have augmented responses in models of delayed-type hypersensitivity, endotoxin-induced acute liver failure, and lung injury induced by metal exposure (20–24). In vitro, HGFL binding to Ron suppresses peritoneal macrophage production of inflammatory mediators induced by LPS and interferon γ (IFN-γ) (25–28). One of these studies suggests that the inhibitory effects of Ron may occur through suppression of NF-κB (26). Although this in vitro study suggests that Ron may suppress peritoneal macrophage activation via an NF-κB–dependent mechanism, it is unknown whether this mechanism is operant in vivo or whether it is a relevant regulatory pathway during fulminant inflammatory injury. Because NF-κB activation is critical for the induction and propagation of acute inflammatory lung injury, the present studies sought to determine whether Ron is an important regulatory component of the inflammatory response culminating in acute lung injury.
MATERIALS AND METHODS
Mice
The mice used in this experiment contained a targeted disruption of the tyrosine kinase domain of the Ron receptor tyrosine kinase and have been described previously (23, 24). Briefly, the experimental mice produce a truncated form of Ron which contains only the extracellular and transmembrane domains of the protein plus five amino acids of the cytoplasmic domain, thus abolishing Ron intracellular signaling. These mice are designated Ron TK−/−, whereas their controls, designated as Ron TK+/+, express the wild-type Ron protein. Both genotypes are on a C57BL/6 background. Male mice (8–12 weeks of age) were used in all experiments.
LPS-induced acute lung injury
Mice were anesthetized with isoflurane and were administered 200 μg Escherichia coli LPS (serotype 0111:B4; Sigma-Aldrich, St. Louis, Mo) intra-nasally in 40 μL sterile saline. At the times indicated, mice were killed by carbon dioxide inhalation and either cannulated through the trachea with a blunted 20-gauge needle for lavage or the lungs were isolated and excised. This project was approved by the University of Cincinnati Animal Care and Use Committee and conforms to the National Institutes of Health guidelines.
Bronchoalveolar lavage
Bronchoalveolar lavage (BAL) fluids were collected by instilling and withdrawing 1 mL of sterile phosphate-buffered saline three times from the lungs via an intratracheal cannula. The BAL fluids were centrifuged, and supernatants were assayed by enzyme-linked immunosorbent assay (ELISA) for murine albumin (Bethyl Laboratories, Montgomery, Tex), as an indicator of pulmonary vascular leak, and the cytokines/chemokines IFN-γ, IL-6, monocyte chemoattractant protein 1 (MCP-1), macrophage inflammatory protein 2 (MIP-2), and TNF-α (R&D Systems, Minneapolis, Minn).
Electrophoretic mobility shift assay
Nuclear extracts of whole lung tissues were prepared by the method of Deryckere and Gannon (29). Protein concentrations were determined by bicinchoninic acid assay with trichloroacetic acid precipitation using bovine serum albumin as a reference standard (Pierce, Rockford, Ill). Double-stranded NF-κB consensus oligonucleotide (5′-AGTGAGGGGACTTTCCCAGGC-3′; Promega Corp., Madison, Wis) was end labeled with γ[32P] adenosine triphosphate (3,000 Ci/mmol at 10 mCi/mL; Amersham, Arlington Heights, Ill). Binding reactions containing equal amounts of protein (20 μg for whole lung extracts) and 35 fmol (~50,000 cpm, Cherenkov counting) of oligonucleotide were performed for 30 min in binding buffer (4% glycerol, 1 mmol/L MgCl2, 0.5 mmol/L EDTA [pH 8.0], 0.5 mmol/L dithiothreitol, 50 mmol/L sodium chloride [NaCl], 10 mmol/L Tris [pH 7.6], 50 μg/mL poly(dI•dC); Pharmacia, Piscataway, NJ). Reaction volumes were held constant to 15 μL. Reaction products were separated in a 4% polyacrylamide gel and analyzed by autoradiography.
Myeloperoxidase assay
Lung myeloperoxidase (MPO) content was assessed by methods similar to those of Schierwagen et al (30). Lung tissue (100 mg) was homogenized in 2 mL of buffer A (3.4 mmol/L KH2HPO4 and 16 mmol/L Na2HPO4 [pH 7.4]). After centrifugation for 20 min at 10,000g, the pellet was resuspended in 10 volumes of buffer B (43.2 mmol/L KH2HPO4, 6.5 mmol/L Na2HPO4, 10 mmol/L EDTA, and 0.5% hexadecyltrimethylammonium [pH 6.0]) and sonicated for 10 s. After heating for 2 h at 60° C, the supernatant was reacted with 3,3,5,5-tetramethylbenzidine (Sigma Chemical Co., St. Louis, Mo), and optical density was determined at 655 nm.
Western blot analysis
Lungs were homogenized in lysis buffer (10 mmol/L HEPES [pH 7.9], 150 mmol/L NaCl, 1 mmol/L EDTA, 0.6% NP-40, 0.5 mmol/L phenylmethanesulfonyl fluoride, 1 μg/mL leupeptin, 1 μg/mL aprotinin, 10 μg/mL soybean trypsin inhibitor, and 1 μg/mL pepstatin) on ice. Homogenates were sonicated and centrifuged at 5,000 rpm to remove cellular debris. Protein concentrations were determined as described for nuclear extracts. Samples were separated in a denaturing 10% polyacrylamide gel and transferred to a 0.1-μm-pore nitrocellulose membrane. Nonspecific binding sites were blocked with Tris-buffered saline (40 mmol/L Tris [pH 7.6], 300 mmol/L NaCl) containing 5% nonfat dry milk for 12 h at 4°C. Membranes were then incubated with anti–IκB-α or anti–IκB-β (Santa Cruz Biotechnology, Santa Cruz, Calif) in Tris-buffered saline with 0.1% Tween 20. The detection system used secondary antibodies conjugated to alkaline phosphatase (Vector Laboratories, Burlingame, Calif) and the enhanced chemifluorescence reagent (GE Healthcare Bio-Sciences Corp, Piscataway, NJ).
Nitrite assay
Nitrite, a stable by-product of NO, was measured from whole lung tissue homogenates. Lung lysates (250 μg) were added to 100 μL Griess reagent (1% sulfanilamide and 0.1% naphthylethylene diaminehydrochloride in 2.5% H3PO4; Promega Corp.). The absorbance of the resulting colorimetric reaction was measured at 540 nm. All samples were assayed in triplicate.
Statistical and image analyses
All data are expressed as mean ± SEM. Data were analyzed with a one-way analysis of variance with subsequent Student-Newman-Keuls test. Differences were considered significant when P < 0.05. Quantitation of bands in Western analyses was performed with AlphaEase FC software from Alpha Innotech Corporation (San Leandro, Calif).
RESULTS
Mice receiving intrapulmonary administration of LPS typically develop acute lung injury, characterized by fluid and protein leakage into the alveolar space and inflammation of the alveolar septae. Wild-type mice displayed significant protein leakage into the alveoli, as determined by albumin ELISA of BAL fluids 4 h after LPS administration (Fig. 1A). At the same time point, Ron TK−/− mice had greater than twofold more albumin in BAL fluids than those for wild-type mice (Fig. 1A). Similarly, inflammation and alveolar thickening observed by lung histology in wild-type mice receiving LPS were much more severe in the Ron TK−/− mice (Fig. 1B). Inasmuch as the albumin levels were not significantly different until 4 h after LPS exposure, we chose to analyze lung histology at a slightly later time frame of 6 h. Wild-type mice receiving phosphate-buffered saline had normal lung architecture (Fig. 1B, upper left panel). Ron TK−/− mice also had normal lung architecture (Fig. 1B, upper right panel) with the exception of the presence of inflammatory cell clusters around areas of the vascular endothelium and airways, as described previously (22). At 6 h after LPS administration, wild-type mice displayed thickening of the alveolar septae and noticeable neutrophilic infiltrates (Fig. 1B, lower left panel). Ron TK−/− mice had more marked changes (Fig. 1B, lower right panel), consistent with the albumin data, suggesting more lung injury in Ron TK−/− mice.
Fig. 1. Lung injury in wild-type (TK+/+) and Ron tyrosine kinase-deficient (TK−/−) mice after intranasal administration of LPS.

A, The levels of albumin were measured in BAL fluids as an index for vascular leakage. Results are expressed as mean ± SEM with n = 3 to 5 mice per group. *P < 0.05 compared with TK+/+ treated mice. B, Histological analysis of stained lung tissues obtained from TK+/+ and TK−/− mice treated with saline (sham) or LPS. Sections from each lung of the TK−/− mice exhibited acute pneumonitis defined by inflammatory cells in the airspaces compared with the TK+/+ mice. Arrows point to inflammatory cells in the airspaces of the TK−/− lung section.
A previous study from our laboratory reported that there are cellular clusters in the lungs of untreated Ron TK−/− mice which are not observed in wild-type mice (22). These clusters seem adjacent to large blood vessels and large airway epithelium in the TK−/− lungs but are not observed in the alveolar spaces or outside these clustered areas. In addition, immunohistochemical analyses demonstrated that the cell clusters in the TK−/− lungs are composed of neutrophils, macrophages, and lymphocytes. Consistent with these findings, biochemical assessment of MPO, a surrogate marker of neutrophil accumulation, was elevated in Ron TK−/− mice receiving saline compared with their wild-type counterparts (Fig. 2). At 4 h after LPS administration, lung MPO increased in wild-type mice (Fig. 2). In Ron TK−/− mice, LPS did not cause any further increase in MPO, but a large number of inflammatory cells are then observed throughout the alveolar compartment in the TK−/− lungs compared with wild-type mice receiving LPS and untreated TK−/− lungs (Figs. 1 and 2).
Fig. 2. Lung neutrophil accumulation in wild-type (TK+/+) and Ron tyrosine kinase–deficient (TK−/−) mice after LPS-induced lung injury.

The MPO activity in whole lungs was measured in units (U) per gram weight as a marker for neutrophil influx. Results are expressed as mean ± SEM with n = 7 mice per group. *P < 0.05 compared with the 0-h TK+/+ control, #P < 0.05 compared with the 4-h TK+/+ mice.
Inasmuch as inflammatory lung injury in this and similar models has been shown to be heavily dependent on activation of the transcription factor, NF-κB (31, 32), we next sought to determine whether Ron may regulate lung injury through effects on NF-κB. Lung extracts were obtained from mice receiving saline or LPS during a time course of injury. In wild-type mice, there was rapid activation of NF-κB induced by LPS (Fig. 3A). In Ron TK−/− mice, there was rapid and sustained NF-κB activation, which was much greater than in wild-type mice (Fig. 3A). Based on competition and antibody supershift analyses, binding to the NF-κB consensus sequence was specific, as the protein complex was not competed by a 100-fold excess of a nonspecific sequence, and the NF-κB complexes consisted of p65/p50 NF-κB subunits, respectively (Fig. 3B). Consistent with these results were Western blot analyses of lung lysates probed for the IκB proteins, IκB-α, and IκB-β. In the Ron TK−/− mice, there was evidence of a significant loss of IκB-α and IκB-β proteins within 2 h of LPS administration (Fig. 3C). Figure 3D provides the quantitation of bands from the Western blot analyses in Figure 3C and confirms the loss of IκB proteins in the TK−/− mice compared with controls.
Fig. 3. Effects of Ron signaling on LPS-induced activation of NF-κ B and degradation of I κB proteins.

Whole lung extracts were obtained at 0, 1, 2, or 4 h after intranasal administration of LPS into wild-type (TK+/+) or TK−/− mice. A, Analysis of whole lung nuclear extracts for NF-κB–DNA binding by electrophoretic mobility shift assay. B, Competition and supershift analyses for NF-κB–DNA binding by electrophoretic mobility shift assays. Lung lysates from two independent treatments taken at 0 or 2 h after LPS exposure were used. The protein/DNA complexes were competed with either a 100-fold excess of specific (S) or nonspecific (N) competitor. In addition, antibodies against the p65 or p50 subunits of NF-κB were used. A shift of the specific protein/DNA complexes was observed with antibody addition. No shifts were observed with other antibodies to different NF-κB complexes (data not shown). C, Analysis of IκB protein expression in whole lung lysates. Western blot analysis of IκB-α (top) and IκB-β (middle) protein in TK+/+ and TK−/− lungs. The Western membranes were also reprobed with actin (bottom) to serve as a loading control. Data in A, B, and C are representative of two separate and independent experiments from at least six mice. D, Quantitation of the amount of IκB proteins in C. Densitometry was used to determine the amount of IκB protein. The values represent averages of the two independent samples per time point normalized to the actin control. The values presented are with the 0-h time point normalized to 1.
Increases in NF-κB activation corresponded to increases in TNF-α protein expression in BAL fluids. In wild-type mice, there was a progressive increase in TNF-α production in the lung after LPS administration (Fig. 4). However, whereas Ron TK−/− mice expressed similar amounts of TNF-α as wild-type mice 1 h after LPS administration, at 2 or 4 h after LPS, Ron TK−/− produced nearly twice as much TNF-α as wild-type mice (Fig. 4). We also observed significantly less IL-6 and MCP-1 in BAL fluids from Ron TK−/− mice 2 h after LPS administration (Table 1). No differences were observed between wild-type and Ron TK−/− mice in BAL levels of IFN-γ or MIP-2 at any time point (Table 1).
Fig. 4. The TNF-α levels in BAL fluid of TK+/+ and TK−/− mice after LPS-induced lung injury.

The BAL fluids were collected at the indicated times after LPS administration and were analyzed by ELISA. Results are expressed as mean ± SEM with n = 6 mice per group. *P < 0.05 compared with TK+/+ mice.
Table 1.
Temporal cytokine and chemokine production in TK+/+ and TK −/− BAL fluid after LPS-induced lung injury
| Genotype | Cytokine (ng/mL) | 0 h | 1 h | 2 h | 4 h |
|---|---|---|---|---|---|
| TK+/+ | IL-6 | 0.002 ± 0.002 | 0.65 ± 0.022 | 2.41 ± 0.35 | 1.05 ± 0.37 |
| MIP-2 | 0.34 ± 0.082 | 8.6 ± 3.7 | 7.9 ± 0.56 | 4.6 ± 1.01 | |
| IFN-γ | 0.053 ± 0.007 | 0.035 ± 0.014 | 0.047 ± 0.0076 | 0.063 ± 0.010 | |
| MCP-1 | 0.02 ± 0.001 | 0.047 ± 0.018 | 0.373 ± 0.057 | 0.087 ± 0.038 | |
| TK−/− | IL-6 | 0.05 ± 0.05 | 0.47 ± 0.16 | 1.31 ± 0.23* | 1.17 ± 0.124 |
| MIP-2 | 0.30 ± 0.14 | 5.5 ± 0.79 | 7.3 ± 1.47 | 4.8 ± 0.48 | |
| IFN-γ | 0.088 ± 0.006 | 0.038 ± 0.020 | 0.030 ± 0.011 | 0.044 ± 0.010 | |
| MCP-1 | 0.04 ± 0.032 | 0.042 ± 0.007 | 0.182 ± 0.060* | 0.280 ± 0.232 |
P < 0.05 compared with the wild-type treated group at the same time point.
Previous studies have shown that inducible nitric oxide synthase (iNOS) is upregulated during LPS-induced lung injury and that overproduction of NO via this enzyme is detrimental to lung function (33–35). The iNOS gene is also regulated by NF-κB (36). To determine if there was increased production of NO in Ron TK−/− mice, we assessed lung tissue levels of the stable by-product of NO, nitrite. In wild-type mice, lung nitrite levels did not change significantly after LPS administration (Fig. 5). However, in Ron TK−/− mice, lung nitrite levels were 2.5- to 5-fold greater than wild-type mice at every time point assessed (Fig. 5).
Fig. 5. Ron signaling reduces the production of nitrite in lung tissue after LPS-induced lung injury.
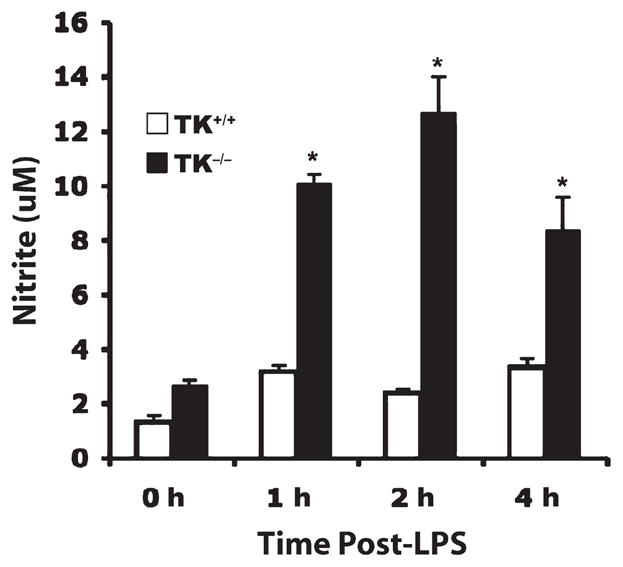
Whole lung extracts were isolated at the indicated time points after LPS exposure and were analyzed for nitrite, a stable metabolite of NO. Results are expressed as mean ± SEM with n = 4 to 6 mice per group. *P < 0.05 compared with TK+/+ mice.
DISCUSSION
The data presented herein demonstrate an important regulatory role of the Ron receptor tyrosine kinase in the acute inflammatory response to intrapulmonary LPS. Injury in this model is thought to be primarily mediated by alveolar macrophage activation by LPS, resulting in the release of proinflammatory mediators, such as TNF-α and IL-1 (37). Other studies have shown that TNF-α propagates the inflammatory response throughout the lung by activating the transcription factor, NF-κB, resulting in upregulation of proinflammatory cytokines, chemokines, and adhesion molecules (5, 7). Our present data suggest that the Ron receptor tyrosine kinase functions to suppress signals leading to activation of NF-κB in vivo. We observed that degradation of IκB proteins and subsequent activation of NF-κB were far greater in mice lacking the tyrosine kinase domain of Ron, indicating that Ron signaling is an important regulatory control mechanism for inflammation in this setting. Our findings of augmented NF-κB activation in Ron TK−/− mice are consistent with in vitro studies of Ron in peritoneal macrophages and engineered macrophage cell lines. The latter studies have shown that ligand activation of Ron suppresses peritoneal macrophage production of inflammatory mediators induced by LPS and IFN-γ through suppression of NF-κB (25–28). Other studies have shown that Ron cross talks with other cell surface receptors and regulates their signal transduction pathways (38–40). It remains to be determined in the current setting if Ron associates with other receptors, such as the toll-like receptor 4 or TNF receptors, to elicit its regulatory effects. Our studies provide strong evidence that Ron-mediated suppression of signals leading to activation of NF-κB occurs in a global fashion in the lung.
The increased activation of NF-κB we observed in Ron TK−/− mice was associated with increased amounts of TNF-α and the NO by-product, nitrite, in the lung. Increased TNF-α expression in these mice would be expected to contribute to the increased lung injury by promoting inflammation. Enhanced NO production has also been linked to acute lung injury. The iNOS, like TNFα, is a proinflammatory gene that is controlled in part by NF-κB and known to be upregulated in acute lung injury models (36, 41). Studies examining the role of iNOS in acute lung injury have suggested that the NO generated from this enzyme contributes significantly to lung injury (33, 35). Our finding of increased nitrite in the lungs of Ron TK−/− mice is consistent with the notion that increased NF-κB activation in these mice leads to increased iNOS expression and activation resulting in greater production of NO, contributing to lung injury in these mice.
Another finding of this study was that Ron TK−/− mice had lower levels of IL-6 in BAL fluids after LPS administration than did wild-type controls. Differences in IL-6 and MCP-1 were found 2 h after LPS administration, at a time when the inflammatory response is in a period of intense amplification. Based on studies in similar models of acute lung inflammatory injury and the modest changes observed here, the changes may be important. However, IL-6 has been shown to confer highly protective effects (42–44). The mechanism by which IL-6 is thought to regulate acute lung injury is through the modulation of proinflammatory cytokine expression. Animals in which IL-6 was neutralized had increased pulmonary expression of TNF-α and greater inflammatory injury than control animals (43). Thus, prevention of Ron signaling may reduce the release of this protective soluble mediator at a time when inflammation is spreading throughout the lung parenchyma.
Similar to a previous report from our laboratory (22), we observed the presence of inflammatory cell infiltrates in the lungs of unmanipulated TK−/− mice. These infiltrates seem to cluster around large blood vessels and airway epithelium. Our previous report demonstrated that these infiltrates contained neutrophils, T cells, and macrophages (22). Because much of the lung injury induced in the current model is caused by activated macrophages and neutrophils, and given that our measurements of lung MPO content found no changes in this parameter after administration of LPS in Ron TK−/− mice, it seems that these cell types are already present in the lungs of Ron TK−/− mice. This suggests that Ron signaling may regulate normal leukocyte trafficking through the lung and that defects in Ron signaling, as occurs in Ron TK−/− mice, result in immune cell retention in otherwise normal lung parenchyma.
The results of this study demonstrate a regulatory role for the Ron receptor tyrosine kinase in acute lung injury induced by intrapulmonary administration of LPS. Removal of Ron signaling, by deletion of the tyrosine kinase domain, results in increased activation of NF-κB leading to greatly enhanced production of TNF-α and NO and substantially increased lung injury. Further studies examining how Ron couples to other receptors and/or how it integrates with other signal transduction pathways are warranted to determine the upstream mechanism(s) by which Ron elicits its regulatory effects on the acute inflammatory response.
Acknowledgments
This study was supported by the National Institutes of Health (grant no. DK073552 to S.E.W., grant no. CA100002 to S.E.W., and grant no. HL72552 to A.B.L.), and the Shriners Hospitals for Children (grant no. 8950 to S.E.W. and grant no. 8720 to A.B.L.).
References
- 1.Bass TL, Miller PK, Campbell DB, Russell GB. Traumatic adult respiratory distress syndrome. Chest Surg Clin N Am. 1997;7:429–442. [PubMed] [Google Scholar]
- 2.Fantini GA, Conte MS. Pulmonary failure following lower torso ischemia: clinical evidence for a remote effect of reperfusion injury. Am Surg. 1995;61:316–319. [PubMed] [Google Scholar]
- 3.Hudson LD, Milberg JA, Anardi D, Maunder RJ. Clinical risks for development of the acute respiratory distress syndrome. Am J Respir Crit Care Med. 1995;151(2 pt 1):293–301. doi: 10.1164/ajrccm.151.2.7842182. [DOI] [PubMed] [Google Scholar]
- 4.Abraham E. Neutrophils and acute lung injury. Crit Care Med. 2003;31(4 suppl):S195–S199. doi: 10.1097/01.CCM.0000057843.47705.E8. [DOI] [PubMed] [Google Scholar]
- 5.Fan J, Ye RD, Malik AB. Transcriptional mechanisms of acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2001;281:L1037–L1050. doi: 10.1152/ajplung.2001.281.5.L1037. [DOI] [PubMed] [Google Scholar]
- 6.Guo RF, Ward PA. Role of C5a in inflammatory responses. Annu Rev Immunol. 2005;23:821–852. doi: 10.1146/annurev.immunol.23.021704.115835. [DOI] [PubMed] [Google Scholar]
- 7.Lentsch AB, Ward PA. Regulation of experimental lung inflammation. Respir Physiol. 2001;128:17–22. doi: 10.1016/s0034-5687(01)00260-2. [DOI] [PubMed] [Google Scholar]
- 8.Lo SK, Everitt J, Gu J, Malik AB. Tumor necrosis factor mediates experimental pulmonary edema by ICAM-1 and CD18-dependent mechanisms. J Clin Invest. 1992;89:981–988. doi: 10.1172/JCI115681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shanley TP, Warner RL, Ward PA. The role of cytokines and adhesion molecules in the development of inflammatory injury. Mol Med Today. 1995;1:40–45. doi: 10.1016/1357-4310(95)80019-0. [DOI] [PubMed] [Google Scholar]
- 10.Strieter RM, Kunkel SL, Keane MP, Standiford TJ. Chemokines in lung injury: Thomas A. Neff lecture. Chest. 1999;116(1 suppl):103S–110S. doi: 10.1378/chest.116.suppl_1.103s. [DOI] [PubMed] [Google Scholar]
- 11.Warren JS, Kunkel SL, Cunningham TW, Johnson KJ, Ward PA. Macrophage-derived cytokines amplify immune complex-triggered O2−. Responses by rat alveolar macrophages. Am J Pathol. 1988;130:489–495. [PMC free article] [PubMed] [Google Scholar]
- 12.Petty TL. Protease mechanisms in the pathogenesis of acute lung injury. Ann N Y Acad Sci. 1991;624:267–277. doi: 10.1111/j.1749-6632.1991.tb17025.x. [DOI] [PubMed] [Google Scholar]
- 13.Schraufstatter IU, Revak SD, Cochrane CG. Proteases and oxidants in experimental pulmonary inflammatory injury. J Clin Invest. 1984;73:1175–1184. doi: 10.1172/JCI111303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gaudino G, Follenzi A, Naldini L, Collesi C, Santoro M, Gallo KA, Godowski PJ, Comoglio PM. Ron is a heterodimeric tyrosine kinase receptor activated by the HGF homologue MSP. EMBO J. 1994;13:3524–3532. doi: 10.1002/j.1460-2075.1994.tb06659.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Iwama A, Okano K, Sudo T, Matsuda Y, Suda T. Molecular cloning of a novel receptor tyrosine kinase gene, STK, derived from enriched hematopoietic stem cells. Blood. 1994;83:3160–3169. [PubMed] [Google Scholar]
- 16.Iwama A, Wang MH, Yamaguchi N, Ohno N, Okano K, Sudo T, Takeya M, Gervais F, Morissette C, Leonard EJ, et al. Terminal differentiation of murine resident peritoneal macrophages is characterized by expression of the STK protein tyrosine kinase, a receptor for macrophage-stimulating protein. Blood. 1995;86:3394–3403. [PubMed] [Google Scholar]
- 17.Quantin B, Schuhbaur B, Gesnel MC, Doll’e P, Breathnach R. Restricted expression of the ron gene encoding the macrophage stimulating protein receptor during mouse development. Dev Dyn. 1995;204:383–390. doi: 10.1002/aja.1002040405. [DOI] [PubMed] [Google Scholar]
- 18.Leonard EJ. Biological aspects of macrophage-stimulating protein (MSP) and its receptor. Ciba Found Symp. 1997;212:183–191. discussion 192–187. [PubMed] [Google Scholar]
- 19.Waltz SE, McDowell SA, Muraoka RS, Air EL, Flick LM, Chen YQ, Wang MH, Degen SJ. Functional characterization of domains contained in hepatocyte growth factor–like protein. J Biol Chem. 1997;272(48):30526–30537. doi: 10.1074/jbc.272.48.30526. [DOI] [PubMed] [Google Scholar]
- 20.Correll PH, Iwama A, Tondat S, Mayrhofer G, Suda T, Bernstein A. Deregulated inflammatory response in mice lacking the STK/Ron receptor tyrosine kinase. Genes Funct. 1997;1:69–83. doi: 10.1046/j.1365-4624.1997.00009.x. [DOI] [PubMed] [Google Scholar]
- 21.Leonis MA, Toney-Earley K, Degen SJ, Waltz SE. Deletion of the Ron receptor tyrosine kinase domain in mice provides protection from endotoxin-induced acute liver failure. Hepatology. 2002;36(5):1053–1060. doi: 10.1053/jhep.2002.36822. [DOI] [PubMed] [Google Scholar]
- 22.Mallakin A, Kutcher LW, McDowell SA, Kong S, Schuster R, Lentsch AB, Aronow BJ, Leikauf GD, Waltz SE. Gene expression profiles of Mst1r-deficient mice during nickel-induced acute lung injury. Am J Respir Cell Mol Biol. 2006;34:15–27. doi: 10.1165/rcmb.2005-0093OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McDowell SA, Mallakin A, Bachurski CJ, Toney-Earley K, Prows DR, Bruno T, Kaestner KH, Witte DP, Melin-Aldana H, Degen SJ, et al. The role of the receptor tyrosine kinase Ron in nickel-induced acute lung injury. Am J Respir Cell Mol Biol. 2002;26:99–104. doi: 10.1165/ajrcmb.26.1.4621. [DOI] [PubMed] [Google Scholar]
- 24.Waltz SE, Eaton L, Toney-Earley K, Hess KA, Peace BE, Ihlendorf JR, Wang MH, Kaestner KH, Degen SJ. Ron-mediated cytoplasmic signaling is dispensable for viability but is required to limit inflammatory responses. J Clin Invest. 2001;108:567–576. doi: 10.1172/JCI11881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen YQ, Fisher JH, Wang MH. Activation of the Ron receptor tyrosine kinase inhibits inducible nitric oxide synthase (iNOS) expression by murine peritoneal exudate macrophages: phosphatidylinositol-3 kinase is required for Ron-mediated inhibition of iNOS expression. J Immunol. 1998;161:4950–4959. [PubMed] [Google Scholar]
- 26.Liu QP, Fruit K, Ward J, Correll PH. Negative regulation of macrophage activation in response to IFN-gamma and lipopolysaccharide by the STK/Ron receptor tyrosine kinase. J Immunol. 1999;163:6606–6613. [PubMed] [Google Scholar]
- 27.Morrison AC, Wilson CB, Ray M, Correll PH. Macrophage-stimulating protein, the ligand for the stem cell–derived tyrosine kinase/Ron receptor tyrosine kinase, inhibits IL-12 production by primary peritoneal macrophages stimulated with IFN-gamma and lipopolysaccharide. J Immunol. 2004;172:1825–1832. doi: 10.4049/jimmunol.172.3.1825. [DOI] [PubMed] [Google Scholar]
- 28.Zhou YQ, Chen YQ, Fisher JH, Wang MH. Activation of the Ron receptor tyrosine kinase by macrophage-stimulating protein inhibits inducible cyclo-oxygenase-2 expression in murine macrophages. J Biol Chem. 2002;277:38104–38110. doi: 10.1074/jbc.M206167200. [DOI] [PubMed] [Google Scholar]
- 29.Deryckere F, Gannon F. A one-hour minipreparation technique for extraction of DNA-binding proteins from animal tissues. Biotechniques. 1994;16:405. [PubMed] [Google Scholar]
- 30.Schierwagen C, Bylund-Fellenius AC, Lundberg C. Improved method for quantification of tissue PMN accumulation measured by myeloperoxidase activity. J Pharmacol Methods. 1990;23:179–186. doi: 10.1016/0160-5402(90)90061-o. [DOI] [PubMed] [Google Scholar]
- 31.Lentsch AB, Czermak BJ, Bless NM, Ward PA. NF-kappaB activation during IgG immune complex-induced lung injury: requirements for TNF-alpha and IL-1beta but not complement. Am J Pathol. 1998;152:1327–1336. [PMC free article] [PubMed] [Google Scholar]
- 32.Nathens AB, Bitar R, Davreux C, Bujard M, Marshall JC, Dackiw AP, Watson RW, Rotstein OD. Pyrrolidine dithiocarbamate attenuates endotoxin-induced acute lung injury. Am J Respir Cell Mol Biol. 1997;17:608–616. doi: 10.1165/ajrcmb.17.5.2661. [DOI] [PubMed] [Google Scholar]
- 33.Kristof AS, Goldberg P, Laubach V, Hussain SN. Role of inducible nitric oxide synthase in endotoxin-induced acute lung injury. Am J Respir Crit Care Med. 1998;158:1883–1889. doi: 10.1164/ajrccm.158.6.9802100. [DOI] [PubMed] [Google Scholar]
- 34.Numata M, Suzuki S, Miyazawa N, Miyashita A, Nagashima Y, Inoue S, Kaneko T, Okubo T. Inhibition of inducible nitric oxide synthase prevents LPS-induced acute lung injury in dogs. J Immunol. 1998;160:3031–3037. [PubMed] [Google Scholar]
- 35.Shanley TP, Zhao B, Macariola DR, Denenberg A, Salzman AL, Ward PA. Role of nitric oxide in acute lung inflammation: lessons learned from the inducible nitric oxide synthase knockout mouse. Crit Care Med. 2002;30:1960–1968. doi: 10.1097/00003246-200209000-00003. [DOI] [PubMed] [Google Scholar]
- 36.Sherman MP, Aeberhard EE, Wong VZ, Griscavage JM, Ignarro LJ. Pyrrolidine dithiocarbamate inhibits induction of nitric oxide synthase activity in rat alveolar macrophages. Biochem Biophys Res Commun. 1993;191:1301–1308. doi: 10.1006/bbrc.1993.1359. [DOI] [PubMed] [Google Scholar]
- 37.Ulich TR, Watson LR, Yin SM, Guo KZ, Wang P, Thang H, del Castillo J. The intratracheal administration of endotoxin and cytokines. I. Characterization of LPS-induced IL-1 and TNF mRNA expression and the LPS-, IL-1–, and TNF-induced inflammatory infiltrate. Am J Pathol. 1991;138:1485–1496. [PMC free article] [PubMed] [Google Scholar]
- 38.Follenzi A, Bakovic S, Gual P, Stella MC, Longati P, Comoglio PM. Crosstalk between the proto-oncogenes Met and Ron. Oncogene. 2000;19:3041–3049. doi: 10.1038/sj.onc.1203620. [DOI] [PubMed] [Google Scholar]
- 39.Mera A, Suga M, Ando M, Suda T, Yamaguchi N. Induction of cell shape changes through activation of the interleukin-3 common beta chain receptor by the Ron receptor–type tyrosine kinase. J Biol Chem. 1999;274:15766–15774. doi: 10.1074/jbc.274.22.15766. [DOI] [PubMed] [Google Scholar]
- 40.Peace BE, Hill KJ, Degen SJ, Waltz SE. Cross-talk between the receptor tyrosine kinases Ron and epidermal growth factor receptor. Exp Cell Res. 2003;289:317–325. doi: 10.1016/s0014-4827(03)00280-5. [DOI] [PubMed] [Google Scholar]
- 41.Warner RL, Paine R, 3rd, Christensen PJ, Marletta MA, Richards MK, Wilcoxen SE, Ward PA. Lung sources and cytokine requirements for in vivo expression of inducible nitric oxide synthase. Am J Respir Cell Mol Biol. 1995;12:649–661. doi: 10.1165/ajrcmb.12.6.7539274. [DOI] [PubMed] [Google Scholar]
- 42.Bless NM, Huber-Lang M, Guo RF, Warner RL, Schmal H, Czermak BJ, Shanley TP, Crouch LD, Lentsch AB, Sarma V, et al. Role of CC chemokines (macrophage inflammatory protein-1 beta, monocyte chemoattractant protein-1, RANTES) in acute lung injury in rats. J Immunol. 2000;164:2650–2659. doi: 10.4049/jimmunol.164.5.2650. [DOI] [PubMed] [Google Scholar]
- 43.Shanley TP, Foreback JL, Remick DG, Ulich TR, Kunkel SL, Ward PA. Regulatory effects of interleukin-6 in immunoglobulin G immune-complex-induced lung injury. Am J Pathol. 1997;151:193–203. [PMC free article] [PubMed] [Google Scholar]
- 44.Shanley TP, Schmal H, Warner RL, Schmid E, Friedl HP, Ward PA. Requirement for C-X-C chemokines (macrophage inflammatory protein-2 and cytokine-induced neutrophil chemoattractant) in IgG immune complex-induced lung injury. J Immunol. 1997;158:3439–3448. [PubMed] [Google Scholar]