

Abstract
A new measure for gene prediction in eukaryotes is presented. The measure is based on the Discrete Fourier Transform (DFT) phase at a frequency of 1/3, computed for the four binary sequences for A, T, C, and G. Analysis of all the experimental genes of S. cerevisiae revealed distribution of the phase in a bell-like curve around a central value, in all four nucleotides, whereas the distribution of the phase in the noncoding regions was found to be close to uniform. Similar findings were obtained for other organisms. Several measures based on the phase property are proposed. The measures are computed by clockwise rotation of the vectors, obtained by DFT for each analysis frame, by an angle equal to the corresponding central value. In protein coding regions, this rotation is assumed to closely align all vectors in the complex plane, thereby amplifying the magnitude of the vector sum. In noncoding regions, this operation does not significantly change this magnitude. Computing the measures with one chromosome and applying them on sequences of others reveals improved performance compared with other algorithms that use the 1/3 frequency feature, especially in short exons. The phase property is also used to find the reading frame of the sequence.
Gene prediction analysis, and specifically, the computational methods for finding the location of protein-coding regions in uncharacterized genomic DNA sequences, is one of the central issues in bioinformatics (Fickett 1996; Salzberg et al. 1998). For a given DNA sequence of an organism, in which the genes and other functional structures are not already known, it is very important to have an accurate and reliable tool for automatic annotation of the sequence: the number and location of genes, the location of exons and introns (in eukaryotes), and their exact boundaries (Claverie 1997). Therefore, along with standard molecular methods, many new methods for finding distinctive features of protein-coding regions have been proposed in the past two decades (see reviews by Fickett 1996; Claverie 1997; Mathé et al. 2002). These methods are based on different measures for discriminating between protein-coding regions and noncoding regions. Some of the measures are based on statistical regularities in genes or exons, which are not present in introns and intergenic sections, such as, for example, differences in codon usage (Staden and McLachlan 1982), hexamer counts (Claverie and Bouguelerat 1986; Farber et al. 1992; Fickett and Tung 1992), codon position asymmetry (Fickett 1982), different periodicities (Fickett 1982; Silverman and Linsker 1986; Chechetkin and Turygin 1995; Tiwari et al. 1997; Herzel et al. 1999; Trifonov 1998; Anastassiou 2000), autocorrelations, nucleotide frequencies (Shulman et al. 1981; Fickett 1982; Borodovsky et al. 1986, 1994), entropy measures (Almagor 1985), and many others. Other measures are based on signals of the gene expression machinery (reviewed in Mathé et al. 2002). Sophisticated algorithms for gene prediction based on both types of measures have been proposed. These algorithms use, for instance, artificial neural networks (Lapedes et al. 1990; Uberbacher and Mural 1991; Farber et al. 1992; Xu et al. 1994; Snyder and Stormo 1995), Hidden Markov Models (Krogh et al. 1994; Baldi and Brunak 2001), and linguistic methods (Searls 1992; Dong and Searls 1994; Mantegna et al. 1994).
Despite the extensive research in the area of gene prediction, current predictors do not provide a complete solution to the problem of gene identification. Short exons are difficult to locate, because discriminative statistical characteristics are less likely to appear in short strands. Furthermore, some genes do not possess the characteristic features that identify most genes, and hence it is not possible to track them using gene predictors that rely on these features.
In this paper a new discriminating feature for gene prediction is proposed. This measure is based on the arguments of the Discrete Fourier Transform (DFT), and is shown to be a potential candidate for locating short genes and exons. The paper is organized as follows: In the Methods section, the first part deals with the frequency analysis of DNA sequence; the second part details the Fourier analysis at a frequency of 1/3, and discusses the relationship between spectral arguments and the position frequencies. The first part of the Results section introduces the distribution of the arguments of the Fourier spectra; the last two parts describe the applications for gene prediction.
METHODS
Periodicities in DNA Sequences and DFT Analysis
The importance of measuring different periodicities for a given DNA in order to determine the locations of protein-coding regions has already been addressed by Fickett (1992), and these periodicities have been used as discriminant features in several studies of gene prediction (Silverman and Linsker 1986; Fickett and Tung 1992; Chechetkin and Turygin 1995; Tiwari et al. 1997; Herzel et al. 1999; Anastassiou 2000). The Discrete Fourier Transform (DFT) is a powerful tool for studying periodicities.
The DFT of a given numeric sequence x(n) of length N is defined by
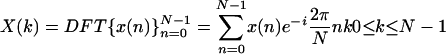 |
2.1 |
(Oppenheim and Schafer 1999), where n is the sequence index, and k corresponds to a period of N/k samples, or discrete frequency of (2π/N)k.
Because the DNA sequence is a character string, numerical values must be assigned to each character: A, T, C, and G. One possible way of performing this conversion is to assign a binary sequence to each of the four bases (Voss 1992). This binary sequence will take a value of 1 or 0 at location n of the sequence, depending on the existence or absence of that base. Thus we have four binary sequences, one for each base, denoted by uA(n), uT(n), uC(n), and uG(n), respectively (Voss 1992; Anastassiou 2000). Applying the DFT to each of these sequences produces four spectral representations, denoted as UA(k), UT(k), UC(k), and UG(k), respectively. That is, for a base b (b = A,T,C, or G), the DFT of the binary sequence ub(n) of length N is
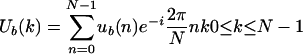 |
2.2 |
The total frequency spectrum of the given DNA character string is defined as:
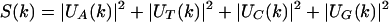 |
2.3 |
(Silverman and Linsker 1986; Tiwari et al. 1997).
A distinctive feature of protein-coding regions in DNA is the existence of short-range correlations in the nucleotide arrangement, especially a 1/3-periodicity (Fickett 1982), arising from the fact that coding DNA consists of triplets (codons). As a consequence, the total Fourier spectrum of protein coding DNA (equation 2.3) typically has a peak at the frequency k = N/3, whereas the total Fourier spectrum of noncoding DNA generally does not have any significant peaks (Tsonis et al. 1991; Voss 1992; Chechetkin and Turygin 1995). Tiwari et al. (1997) used the measure in equation 2.3 with k = N/3, known as the Spectral Content measure, to construct a gene predictor. It can be shown that this measure is the same (up to a 3/2 multiplicative factor) as the sum of the four Position Asymmetry measures (Fickett and Tung 1992), namely
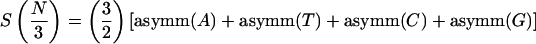 |
2.4 |
Where, for a base b (b = A,T,C, or G) asymm 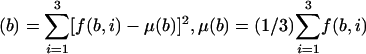 , and f (b,i) is the frequency of b in the codon position i, i = 1,2,3.
, and f (b,i) is the frequency of b in the codon position i, i = 1,2,3.
Anastassiou (2000) introduced the Optimized Spectral Content measure:
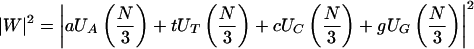 |
2.5 |
In this measure, the coefficients a, t, c, and g are calculated using an optimization technique applied to the known genes of a given organism. The measure in equation 2.5 shows significant improvement over the measure presented by Tiwari et al. (1997) in predicting genes in S. cerevisiae (Anastassiou 2000).
In the following sections we show how signal processing measures for gene prediction can be improved by considering a new feature of protein-coding DNA regions, which can be measured by DFT, namely, the arguments of the Fourier spectra at N/3. We show that the arguments of the Fourier spectra in coding regions are narrowly distributed around corresponding central values.
Relationship Between Spectral Arguments and Position Frequencies
As evident from equation 2.4, the peak of the total Fourier spectrum at k = N/3 in protein-coding DNA sequences is directly related to the asymmetric distribution of each of the four bases among the three codon positions. This asymmetry is strongly related to the codon usage of the particular organism. For any given organism, most genes have similar codon usage; therefore, in most protein-coding regions, the ratios between each pair of the counters 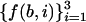 for each base b can be expected to be close to some constant values. These ratios determine the value of arg [Ub(N/3)]. To demonstrate, let s be a DNA sequence. The (N/3)th element of the DFT of the binary sequence ub(n) of length N associated with the base b (b = A, T, C, or G) is obtained by substituting k = N/3 in equation 2.2:
for each base b can be expected to be close to some constant values. These ratios determine the value of arg [Ub(N/3)]. To demonstrate, let s be a DNA sequence. The (N/3)th element of the DFT of the binary sequence ub(n) of length N associated with the base b (b = A, T, C, or G) is obtained by substituting k = N/3 in equation 2.2:
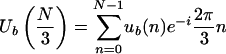 |
3.1 |
Since ub(n) = 0 or 1, there are three distinct possible nonzero terms in the sum in equation 3.1, namely 1, e-i(2π/3), and ei(2π/3), and equation 3.1 takes the form:
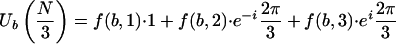 |
3.2 |
Since, 1 + e-i(2π/3) + ei(2π/3) = 0 equation 3.2 can be expressed as follows
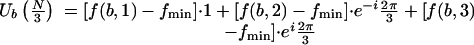 |
3.3 |
where 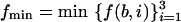 . If all f(b,i), i = 1,2,3, are equal, then Ub(N/3) = 0. Figure 1 illustrates the case where fmin = f(b,1). A simple trigonometric computation yields:
. If all f(b,i), i = 1,2,3, are equal, then Ub(N/3) = 0. Figure 1 illustrates the case where fmin = f(b,1). A simple trigonometric computation yields:
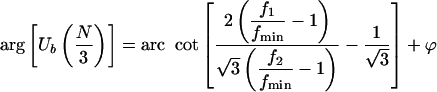 |
3.4 |
where f1 and f2 are the other two counters, numbered in counter-clockwise direction from the vector corresponding to fmin, and ϕ is the argument of the vector corresponding to f1(0, 2π/3, or -2π/3).
Figure 1.

Computing Ub(N/3) in the case fminf(b,1).
It can be seen that arg[Ub(N/3)] will shift by -2π/3 or 2π/3 for reading frames 2 and 3, respectively.
Since it was assumed above that the ratios between each pair of the counters 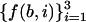 in most coding regions are close to some constant values, the same holds for Ub (N/3), where b is one of the bases A, T, C, or G.
in most coding regions are close to some constant values, the same holds for Ub (N/3), where b is one of the bases A, T, C, or G.
RESULTS
The Distribution of the Arguments of the Fourier Spectra
Let s be a DNA strand, and for each base b, denote b(s) = Ub(N/3). We calculated the values of arg(A[s]), arg(T[s]), arg(C[s]), and arg(G[s]) in coding and noncoding regions, for different organisms. The histograms describing these distributions for all experimental genes in the 16 chromosomes of S. cerevisiae (multiple-exon genes were concatenated to single strands; GenBank acc. nos. NC001133–NC001148, at http://www.ncbi.nlm.nih.gov) are shown in Figure 2. (Because the arguments are originally in principal values [between -π and π], a 2π shift was applied to part of the data so that the histograms are plotted around the angular mean.) As the figure reveals, in all four nucleotides the distributions of the arguments taper around a central value, with the distributions of arg(G[s]) and arg(T[s]) being much narrower than the other two. Similar results, in both shape and statistics, were obtained for each of the 16 chromosomes of S. cerevisiae.
Figure 2.

Argument distributions for all experimental genes in all chromosomes in S. cerevisiae.
The corresponding histograms for noncoding regions (intergenic spacers and introns in all genes [experimental and not experimental]) in the 16 chromosomes appear in Figure 3. The distributions for noncoding regions seem to be close to uniform, and very different from the distributions that were obtained for coding regions. A similar pattern was observed for each separate chromosome.
Figure 3.

Argument distribution for noncoding regions in all chromosomes in S. cerevisiae.
To make sure that the former results are not unique for S. cerevisiae, the same analysis was performed on other organisms. The resulting histograms (Fig. 4A,B,C) show the argument distributions for S. cerevisiae, S. pombe, (chromosomes 2 and 3; acc. nos. NC003423 and NC003421, respectively), and Guillardia theta (chromosome I; acc. no. AF165818), respectively. It is readily evident that the three histograms greatly resemble each other, although the exact statistical values differ somewhat. In particular, the central value of arg(G), in all three organisms, is located somewhere in the vicinity of 0. This means that the base G appears in the first codon position much more often than in the second and third positions. This is consistent with the findings of Trifonov (1987).
Figure 4.

(A) Argument distribution for all experimental genes in all chromosomes of S. cerevisiae. (B) Argument distribution for all genes in chromosomes 2 and 3 of S. pombe. (C) Argument distribution for all genes in chromosome 1 of Guillardia theta.
In the following section we show how the difference between coding and noncoding regions in terms of argument distribution can be applied to gene prediction.
Rotational Measures for Gene Prediction
Several measures were constructed using the argument distribution described above. These measures are based on the notion of spectral rotation and alignment.
Assume we have an organism for which arg(A[s]), arg(T[s]), arg(C[s]), and arg(G[s]) are distributed in a similar manner to that observed in the organisms described above (i.e., bell-shaped in coding regions, and close to uniform in noncoding regions). Let μA, μT, μC, and μG, be the approximated average values, in coding regions, of arg(A[s]), arg(T[s]), arg(C[s]), and arg(G[s]), respectively. Since for a typical coding sequence s it is expected that arg(A[s]) ≈ μA, arg(T[s]) ≈ μT, arg(C[s]) ≈ μC, and arg(G[s]) ≈ μG, rotating the vectors A(s), T(s), C(s), and G(s) clockwise, each by the corresponding argument μA, μT, μC, and μG (multiplication by e-iμA, e-iμT, e-iμC, and e-iμG respectively) will yield four vectors pointing roughly in the same direction: towards the positive real axis for reading frame 1, and at -2π/3 and 2π/3 for reading frames 2 and 3, respectively. Hence the vector sum
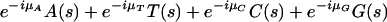 |
5.1 |
will be of large magnitude compared to the case where the vectors point in different directions, as is most likely the case for a noncoding sequence. Figure 5A,B illustrates this idea for the sum of two vectors e-iμTT(s) + e-iμGG(s).
Figure 5.


(A) Rotation and alignment of the vectors G(s) and T(s), when arg(T[s]) ≈ μT and arg(G[s]) ≈ μG. (B) Rotation and alignment of the vectors G(s) and T(s), when arg(T[s]) and arg(G[s]) are any random values.
Dividing each term in equation 5.1 by the corresponding angular deviation (σA, σT, σC, and σG of A[s], T[s], C[s], and G[s], and respectively) will give more weight to narrower distributions, yielding the measure
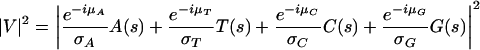 |
5.2 |
which we call a Spectral Rotation (or SR) measure. This resembles the Optimized Spectral Content measure of Anastassiou (2000) given in equation 2.5.
Table 1 compares the performance of four measures: two introduced here, namely the SR measure and the G Rotation measure (described below), and two known measures based on Fourier spectrum: the Spectral Content measure (Tiwari et al. 1997), and Optimized Spectral Content measure (Anastassiou 2000). All measures were tested on all experimental exons, and noncoding strands (intergenic spacers and introns) from the first 15 chromosomes of S. cerevisiae (because the measures were calculated using chromosome 16 of S. cerevisiae, their performance was tested on the remaining 15 chromosomes) of a length greater than 50 bp. The results were obtained by sliding windows of sizes 90, 120, 180, 240, 300, and 351 bp (an analysis frame of 351 bp is used by Anastassiou [2000] and by Tiwari et. al. [1997]. The choice of this size of frame is explained in the latter), with gaps of size 30, 40, 60, 60, 75, and 99 bp, respectively. The threshold in each case was chosen so that the percentage of introns falsely detected as exons (false positives) is 10%. As Table 1 indicates, the SR measure shows better performance, especially in smaller analysis frames.
Table 1.
Performance of Fourier Spectrum Measures on All Experimental Exons and All Noncoding Strands of Length Greater Than 50 bp in S. cerevisiae, Using Different Window Sizes
|
% of exons detected for 10% false positive
|
||||||
|---|---|---|---|---|---|---|
| Measure | 90 bp | 120 bp | 180 bp | 240 bp | 300 bp | 351 bp |
| Spectral Rotation | 84.5 | 88.0 | 90.8 | 91.9 | 92.7 | 93.0 |
| Optimized spectral content (Anastassiou 2000) | 82.1 | 86.0 | 89.3 | 91.0 | 91.7 | 92.2 |
| Spectral content (Tiwari et al. 1997) | 76.0 | 79.4 | 86.3 | 89.4 | 90.0 | 90.4 |
| G Rotation | 83.3 | 86.2 | 89.6 | 90.4 | 90.8 | 90.7 |
Figure 6 compares the probability density functions of the Spectral Content measure and the SR measure. The values of the Spectral Content measure were scaled so that intersection points of the two curves of each color are vertically aligned. The better performance of the SR measure is illustrated by the fact that the distance between the bold curves is greater than the distance between the fine curves.
Figure 6.

Probability density functions for Spectral Rotation (bold) and Spectral Content (fine) measures (solid lines represent exons and dashed lines represent noncoding regions).
Detection of short exons may be rendered more effective by using one statistical parameter that is narrowly distributed when calculated over short strands. As shown in Figure 7, the distribution of arg(G[s]) in the genes of S. cerevisiae is narrow when calculated over coding strands of length 120 bp.
Figure 7.

Argument distribution of coding DNA strands of length 120 bp in S. cerevisiae.
The following measure uses only arg(G[s]). Since only the vector G(s) is rotated in this measure, we need a fixed reference vector to maximize the vector sum. Suppose R is a real number. If s belongs to a coding region, and is in reading frame 1, the vector G(s), rotated clockwise by the approximated average argument μG will most likely be directed towards the positive real axis, that is, in the same direction as the vector R. On the other hand, if s does not belong to a coding region, the rotated vector may point in any direction. The vector sum of R and the rotated G(s) will discriminate best between coding and noncoding regions, when R is of the same order of magnitude as |G(s)|. Thus, we choose |G(s)| as the reference vector. In order to identify genes in all three reading frames, we define the G Rotation measure as
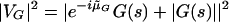 |
5.3 |
where ˜μG is chosen from the set {μG, μG + (2π/3), μG - (2π/3)}, so that the value of the measure in equation 5.3 is maximal. Table 1 compares the performance of the G Rotation measure with that of other measures, on the experimental genes and exons of S. cerevisiae.
When using a measure calculated from data of one organism to predict genes in another organism, it may be preferable to use a subset of the vectors A(s), T(s), C(s), and G(s). For example, the vectors T(s) and G(s), which have narrowly distributed arguments, can be aligned to yield the TG-Rotation measure |VTG|2 = |(e-iμT/σT)T(s) + (e-iμG/σG)G(s)|2. This measure is determined by |μG - μT|. Hence, where this value happens to be similar in two organisms (e.g., S. cerevisiae and S. pombe; see Fig. 4A,B), it is possible to predict genes in one organism by using the parameters of another.
Figure 8A shows the curve of the TG-Rotation measure, constructed from data in chromosome 16 of S. cerevisiae, on a typical split gene of S. pombe (gene SPBC582.08 in chromosome 2). For comparison, Figure 8B shows the graph of the Codon Usage measure on the same gene. The horizontal lines represent the actual location of the three exons. Note the short intron between the second and third exons. In general, one should be wary about using data from one organism to predict genes in another organism, because the respective central argument values in different organisms may not be similar. For example, note that in Figure 4A,B,C the central values of arg(A) and arg(C) are very different in the three organisms.
Figure 8.

Graphs of gene prediction applied on the gene SPBC582.08 in chromosome 2 of S. pombe, using a sliding window of 180 bp: (A) TG-Rotation measure; (B) Codon Usage measure. The horizontal segments represent the actual location of the three exons. To get the actual base location in the chromosome, add 300,000 bp to the numbers on the horizontal axis.
Table 2 summarizes the data on gene SPBC582.08.
Table 2.
Gene SPBC582.08 in Chromosome 2 of S. pombe
| Exon | Start base | End base | Length |
|---|---|---|---|
| 1 | 352249 | 352711 | 463 |
| 2 | 352903 | 353702 | 800 |
| 3 | 353756 | 354010 | 255 |
Complementary Sequences and Reading Frame Identification
Genes on the complementary strand can be detected using the following transformation from Anastassiou (2000):
If V = aA(s) + tT(s) + cC(s) + gG(s), then the predictor for the complementary strand is Ṽ = ãA(s) + t̃T(s) + c̃C(s) + g̃G(s), where ã = e-i(2π/3)t′, t̃ = e-i(2π/3)a′, c̃ = e-i(2π/3)g′, and g̃ = e-i(2π/3)c′, and a′, t′, c′, and g′, are the complex conjugates of a, t, c, and g, respectively.
An nonannotated strand is examined using both measures |V|2 and |Ṽ|2. A detected gene will be considered complementary if |V|2 > |Ṽ|2.
As mentioned in the Methods section, arg[Ub(N/3)] will shift by -2π/3 or 2π/3, relative to its value for reading frame 1, if the actual reading frames are 2 and 3, respectively (see Anastassiou 2000). As explained in the previous section, the rotational measures identify exons, regardless of their reading frame. To identify the reading frame, for the SR measure, we look at arg(V). For a coding sequence in reading frame 1, the rotated vectors will be aligned close to the positive real axis, and thus arg(V) should be close to zero. For reading frames 2 and 3, arg(V) will be in the vicinity of -2π/3 and 2π/3, respectively. This is illustrated by the following two examples.
Figure 9A depicts the graph of the SR measure on the gene SPBC1685.08 in chromosome 2 of S. pombe (acc. no. NC003423). The measure's parameters were calculated from the genes in the smaller chromosome 3 (acc. no. NC003421). Figure 9B depicts the graph of arg(V). The graphs were obtained by calculating the measure with a sliding window of 351 bp, using a step of 3 bp. The gene has three exons, in reading frames 1, 3, and 2 respectively. Table 3 summarizes the data on the gene. Note the short intron between the second and third exons.
Figure 9.

Graphs of the SR measure on the gene SPBC1685.08 in chromosome 2 of S. pombe, using a sliding window of 351 bp. (A) The measure; (B) arg(V). The horizontal segments represent the actual location of the exons. To get the actual base location in the chromosome, add 400,000 bp to the numbers on the horizontal axis.
Table 3.
Gene SPBC1685.08 in Chromosome 2 of S. pombe
| Exon | Start base | End base | Length | Reading frame |
|---|---|---|---|---|
| 1 | 433915 | 434252 | 338 | 1 |
| 2 | 434423 | 434626 | 204 | 3 |
| 3 | 434671 | 435403 | 733 | 2 |
Figure 9 illustrates how the curve of arg(V) can be used to identify the exact boundaries of an exon. It is expected that along an exon the value of arg(V) will remain in the vicinity of one of the values 0, -2π/3, or 2π/3, while outside the exon, the value of arg(V) will change to some “random” value.
Figure 10A depicts the graphs of the SR measure on the gene SPBC1709.08 in chromosome 2 of S. pombe (acc. no. NC003423). The measure's parameters were calculated as in the previous example. Figure 10B depicts the graph of arg(V). In this example, the gene has one exon, between nucleotides 1033037 and 1037362, in reading frame 2. The fact that the curve of arg(V) is constant at around the value of -2π/3 along the whole gene indicates that the gene consists of one exon, and not of multiple exons, as might be incorrectly deduced by looking only at the curve of Figure 10A. This procedure can therefore assist in differentiating between multiple exons and single exons.
Figure 10.

Graphs of the SR measure on the gene SPBC1709.08 in chromosome 2 of S. pombe, using a sliding window of 351 bp. (A) The measure; (B) arg(V). The horizontal segment represents the actual location of the gene. To get the actual base location in the chromosome, add 1,000,000 bp to the numbers on the horizontal axis.
DISCUSSION
In this paper a new method for gene prediction is proposed, based on several measures of protein coding regions. The measures are derived from a regularity of the spectral phase within coding regions. In this study we found that the phase of the DFT at a frequency of 1/3 is distributed with a bell-shaped curve around a central value in coding regions, whereas in noncoding regions, the distribution was close to uniform. This behavior was shown to exist in all chromosomes of S. cerevisiae, and also in two other organisms, S. pombe and Guillardia theta. This regularity was used for the construction of measures for discriminating between coding and noncoding regions in a given nonannotated DNA sequence. The measures are constructed by clockwise rotation of the vectors, which are the values obtained by DFT analysis for the four binary sequences of each nucleotide, with the corresponding central values. After such rotation, the four vectors in coding regions tend to be aligned close to each other, whereas the arrangement of vectors in noncoding regions is random. Earlier studies described proposed measures for gene prediction based on Fourier transform at a frequency of 1/3 or at other frequencies (Trifonov and Sussman 1980; Fickett 1982; Silverman and Linsker 1986; Fickett and Tung 1992; Tiwari et al. 1997; Anastassiou 2000). In most of these studies, the information was derived from the magnitude of the DFT, whereas the information of the phase component was not explicitly used. Tiwari et al. (1997) used the magnitude to construct the so-called Spectral Content measure (see equation 2.3). Anastassiou (2000) improved on the former measure by proposing the Optimized Spectral Content measure (see equation 2.5), which is based on an optimization technique. In this measure, the Fourier component for each nucleotide was multiplied by a coefficient in order to maximize an optimization criterion for discrimination between coding regions and random DNA sequence. However, this technique was not justified analytically in order to explain why it yields better performance than the measure of Tiwari et al. (1997), and its optimization criterion, which discerns between introns (and intergenic spacers) and exons, is based on random DNA. Because introns and intergenic spacers might reveal nonrandom characteristics, it is assumed that better results could be achieved if introns or sequences from intergenic spacers were used in the optimization. However, in the construction of the measures proposed in the present work, there is no need for random DNA or for introns, because the rotation parameters are the central values of the spectral phases in coding regions.
The attempt to use parameters derived from one organism to recognize genes in another organism is based on an implicit assumption of the universality of genes, at least with regard to the structure that elicits the above spectral features. However, as this study (and also previous ones) show, the peak at a frequency of 1/3 is attributed to position asymmetry of the nucleotide within the three possible locations in the codon. This asymmetry was shown to be the result of codon usage (Tsonis et al. 1991) and codon bias. Because different organisms exhibit different codon usage, it is expected that such prediction will not be optimal for use in organisms with different codon usage. Using the Spectral Rotation measure presented here, better performance was achieved than in both studies mentioned above (Tiwari et al. 1997 and Anastassiou 2000; see Table 1). As described, the measures proposed in the current study yielded improved results even in short analysis frames (120 bp and even 90 bp). This was notably true for the measure based only on G. Assuming a narrow distribution of arg(G), as is the case for the organisms studied, the relative simplicity of computing the DFT for only one nucleotide makes the G Rotation measure a fitting candidate to serve for identification of short genes and exons. Indeed, in this work it was shown that this measure outperforms other known measures (Table 1). For other organisms, if the existing gene data enable identification of the base b (b = A, T, C, or G) for which arg(b) is most narrowly distributed, it is possible to construct a b Rotation measure accordingly.
Considering the argument distributions obtained in this study, it was predicted that wherever an analysis frame slides within a protein-coding region, the value of arg(V) (the vector sum of the rotated spectra) will be close to one of three possible values (0, -2π/3, or 2π/3, according to the reading frame), and random in introns or between genes. Furthermore, the slope of the curve will be close to zero in sections corresponding to protein-coding regions, and will have a noisy unpredicted appearance elsewhere. Therefore the plot of arg(V) can be a tool for finding the reading frame. Moreover, as shown in the third part of the Results, plotting the graph of the SR measure, along with arg(V) can help to distinguish between one long exon and multiple exons spaced by short introns. Whereas the angle's slope will tend to be close to zero in the former case, it will have a noisy structure in the intron sections in the latter. This feature was also shown to help in the exact demarcation of the exon-intron boundaries.
Last, a comment about the length of the analysis frame. A short analysis frame (less than 180 bp) may detect short exons and short introns, whereas frames of over 300 bp may miss them. However, there is a tradeoff, because the use of shorter analysis frames causes more statistical flunctuations, resulting in more false negatives and false positives. Hence, it is important to have a measure that still performs reasonably with short frames.
In summary, we suggest that considering the arguments of the Fourier spectra at k=N/3 yields more information about a DNA sequence than the corresponding magnitudes alone. However, it should be noted that these two values (namely, the magnitude and the argument) are not independent. A large magnitude of a Fourier spectrum at k=N/3 is a result of a sharp position asymmetry in the corresponding base. If a sharp position asymmetry is characteristic of the coding regions of an organism, then the value of arg[Ub(N/3)] will be more stable; that is, its distribution over the genes of the organism will have low variance. However, as shown in this work, incorporating data about the distribution of the arguments of the Fourier spectra at k=N/3, along with their magnitudes, into a measure, yields a measure that is more sensitive to exon-intron transition than a measure that uses the magnitudes alone.
Acknowledgments
We thank Mr. Efim Yakir for preparing part of the figures and for technical support. We also thank Dr. Dorit Shweiki for reading the manuscript and for valuable discussions.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.1261703.
Footnotes
Article published online before print in July 2003.
References
- Almagor, H. 1985. Nucleotide distribution and the recognition of coding regions in DNA sequences: An information theory approach. J. Theor. Biol. 117: 127-136. [DOI] [PubMed] [Google Scholar]
- Anastassiou, D. 2000. Frequency-domain analysis of biomolecular sequences. Bioinformatics 16: 1073-1082. [DOI] [PubMed] [Google Scholar]
- Baldi, P. and Brunak S. 2001. Bioinformatics: The machine learning approach 2nd ed., chapter 7. MIT Press, Cambridge, MA.
- Borodovsky, M.Y., Sprizhitsky, Y.A., Golovanov, E.I., and Alexandrov, A.A. 1986. Statistical patterns in the primary structure of the functional regions of the Escherichia coli genome. II. Nonuniform Markov models. Mol. Biol. 20: 833-840. [Google Scholar]
- Borodovsky, M.Y., Koonin, E.V., and Rudd, K.E. 1994. New genes in old sequence: A strategy for finding genes in the bacterial genome. Trends Biochem. Sci. 19: 309-313. [DOI] [PubMed] [Google Scholar]
- Chechetkin, V.R. and Turygin, A.Y. 1995. Size-dependence of three-periodicity and long-range correlations in DNA sequences. Phys. Lett. A. 199: 75-80. [Google Scholar]
- Claverie, J.-M. 1997. Computational methods for the identification of genes in vertebrate genomic sequences. Hum. Mol. Genet. 6: 1735-1744. [DOI] [PubMed] [Google Scholar]
- Claverie, J.M. and Bougueleret, L. 1986. Heuristic informational analysis of sequences. Nucleic. Acids Res. 14: 179-196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong, S. and Searls, D.B. 1994. Gene structure prediction by linguistic methods. Genomics 23: 540-551. [DOI] [PubMed] [Google Scholar]
- Farber, R.B., Lapedes, A.S., and Sirotkin, K.M. 1992. Determination of eukaryotic protein coding regions using neural networks and information theory. J. Mol. Biol. 226: 471-479. [DOI] [PubMed] [Google Scholar]
- Fickett, J.W. 1982. Recognition of protein coding regions in DNA sequences. Nucleic Acids Res. 10: 5303-5318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fickett, J.W. 1996. The gene identification problem: An overview for developers. Comput. Chem. 20: 103-118. [DOI] [PubMed] [Google Scholar]
- Fickett, J.W. and Tung, C.S. 1992. Assessment of protein coding measures. Nucleic Acids Res. 20: 6441-6450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzel, H., Weiss, O., and Trifonov, E.N. 1999. 10–11 bp periodicities in complete genomes reflect protein structure and protein folding. Bioinformatics 15: 187-193. [DOI] [PubMed] [Google Scholar]
- Krogh, A., Mian, I.S., and Haussler, D. 1994. A hidden Markov model that finds genes in E. coli DNA. Nucleic Acids Res. 22: 4768-4778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapedes, A.S., Barnes, C., Burks, C., Farber, R.M., and Sirotkin, K.M. 1990. Application of neural networks and other machine learning algorithms to DNA sequence analysis. In Computers and DNA (eds. G. Bell. and T. Marr), pp. 157-182. Addison-Wesley, Redwood City, CA.
- Mantegna, R.N., Buldyrev, S.V., Goldberger, A.L., Havlin, S., Peng, C.-K., Simmons, M., and Stanley, H.E. 1994. Linguistic features of noncoding DNA sequences. Phys. Rev. Lett. 73: 3169-3172. [DOI] [PubMed] [Google Scholar]
- Mathé, C., Sagot, M-F., Schiex, T., and Rouzé, P. 2002. Current methods of gene prediction, their strength and weaknesses. Nucleic Acids Res. 30: 4103-4117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oppenheim, A.V. and Schafer, R.W. 1999. Discrete-time signal processing, chapter 8 Prentice Hall, Upper Saddle River, NJ.
- Salzberg, S.L., Searls, D.B., and Kasif, S., eds. 1998. Computational methods in molecular biology, chapter 1. Elsevier, Amsterdam.
- Searls, D.B. 1992. The Linguistics of DNA. Amer. Scientist 80: 579-591. [Google Scholar]
- Shulman, M.J., Steiberg, C.M., and Westmoreland, B. 1981. The coding function of nucleotide sequences can be discerned by statistical analysis J. Theor. Biol. 88: 409-420. [DOI] [PubMed] [Google Scholar]
- Silverman, B.D. and Linsker, R. 1986. A measure of DNA periodicity. J. Theor. Biol. 118: 295-300. [DOI] [PubMed] [Google Scholar]
- Snyder, E.E. and Stormo, G.D. 1995. Identification of protein coding regions in genomic DNA. J. Mol. Biol. 258: 1-18. [DOI] [PubMed] [Google Scholar]
- Staden, R. and McLachlan, A.D. 1982. Codon preference and its use in identifying protein coding regions in long DNA sequences. Nucleic Acids Res. 10: 141-156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiwari, S., Ramachandran, S., Bhattacharya., A., Bhattacharya, S., and Ramaswamy, R. 1997. Prediction of probable genes by Fourier analysis of genomic sequences. Comput. Appl. Biosci. 113: 263-270. [DOI] [PubMed] [Google Scholar]
- Trifonov, E.N. 1987. Translation framing code and framing-monitoring mechanism as suggested by the analysis of mRNA and 16S rRNA nucleotide sequences. J. Mol. Biol. 194: 643-652. [DOI] [PubMed] [Google Scholar]
- Trifonov, E.N. 1998. 3-, 10.5-, 200- and 400-base periodicities in genome sequences. Phys. A. 249: 511-516. [Google Scholar]
- Trifonov, E.N. and Sussman, J.L. 1980. The pitch of chromatin DNA is reflected in its nucleotide sequence. Proc. Natl. Acad. Sci. 77: 3816-3820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsonis, A.A., Elsner, J.B., and Tsonis, P.A., 1991: Periodicity in DNA coding sequences: Implications in gene evolution. J. Theor. Biol. 151: 323-331. [DOI] [PubMed] [Google Scholar]
- Uberbacher, E.C. and Mural, R.J. 1991. Locating protein-coding regions in human DNA sequences by a multiple sensor-neural network approach. Proc. Natl. Acad. Sci. 88: 11261-11265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voss, R. 1992. Evolution of long-range fractal correlations and 1/f noise in DNA base sequences. Phys. Rev. Lett. 68: 3805-3808. [DOI] [PubMed] [Google Scholar]
- Xu, Y., Mural, R.J., and Uberbacher, E.C. 1994. Constructing gene models from accurately predicted exons: An application of dynamic programming. Comput. Appl. Biosci. 10: 613-623. [DOI] [PubMed] [Google Scholar]
WEB SITE REFERENCES
- http://www.ncbi.nlm.nih.gov/GenBank; National Center for Biotechnology Information.