

Abstract
Drug resistance is a growing concern with clinical use of tyrosine kinase inhibitors. Utilizing in vitro models of intrinsic drug resistance and stromal-mediated chemoresistance, as well as functional mouse models of progressive and residual disease, we attempted to develop a potential therapeutic approach designed to suppress leukemia recurrence following treatment with selective kinase inhibitors. The novel IAP inhibitor, LCL161, was observed to potentiate the effects of tyrosine kinase inhibition against leukemic disease both in the absence and presence of a stromal protected environment. LCL161 enhanced the proapoptotic effects of nilotinib and PKC412, respectively, against leukemic disease in vitro and potentiated the activity of both kinase inhibitors against leukemic disease in vivo. In addition, LCL161 synergized in vivo with nilotinib to reduce leukemia burden significantly below the baseline level suppression exhibited by a moderate-to-high dose of nilotinib. Finally, LCL161 displayed antiproliferative effects against cells characterized by intrinsic resistance to tyrosine kinase inhibitors as a result of expression of point mutations in the protein targets of drug inhibition. These results support the idea of using IAP inhibitors in conjunction with targeted tyrosine kinase inhibition to override drug resistance and suppress or eradicate residual disease.
Introduction
The development of resistance in leukemia patients to treatment with targeted tyrosine kinase inhibitors is a growing area of concern. For instance, the ABL inhibitor imatinib1,2 has proven to be a highly effective, front line therapy for chronic myeloid leukemia (CML), a hematopoietic malignancy caused by the product of a reciprocal t(9;22) chromosomal translocation, BCR-ABL.3 However, accelerated or blast crisis phase CML patients often relapse due to drug resistance resulting from the emergence of imatinib-resistant point mutations within the BCR-ABL tyrosine kinase domain4 or amplification of the target gene5. As an approach to overriding imatinib resistance, we developed the 2nd generation ABL inhibitor, nilotinib (AMN107, Tasigna™)6, which has been FDA approved for imatinib-resistant disease due to the development of BCR-ABL point mutations, as well as for newly diagnosed CML patients.
Approximately 30% of acute myeloid leukemia (AML) patients express a mutated form of the class III receptor tyrosine kinase, FLT37. We developed the FLT3 inhibitor, PKC4128, which was administered in sequential and simultaneous combinations with daunorubicin and cytarabine induction and high-dose cytarabine consolidation and yielded clinical responses with transient and/or reversible side effects9. Generally, though, FLT3 inhibitors tested thus far clinically induce only partial and transient responses in patients when used as single agents. This suggests a need for development of novel agents that can either be used effectively alone or combined with other agents to suppress disease progression and prolong lifespan.
In addition to identifying and developing potent kinase inhibitors representative of novel and unique structural classes with the ability to override drug resistance due to changes in the target protein, there is a push toward gaining a better understanding of the mechanisms underlying drug resistance in CML and AML as they relate to the leukemic cell microenvironment. Clinical trial data with tyrosine kinase inhibitors show that while the peripheral blasts are rapidly depleted, residual disease is apparent in patient bone marrow. Stromal cells have been implicated, as they provide viability signals to leukemic cells that protect them from inhibitor effects. Indeed, the quantity of leukemic stem cells that rely on stroma to survive is predictive of disease outcome.10
Thus, deregulated signaling molecules associated with viability/apoptotic signaling represent attractive targets for therapeutic intervention. Several strategies have emerged that may be effective in preventing drug resistance due to this, including the combination of targeted inhibitors with small molecule inhibitors of key components of major signaling pathways affecting the viability/expansion of leukemic cells. One potentially important therapeutic target is the viability signaling factor, second mitochondria-derived activator of caspase (Smac), which mediates apoptosis through the intrinsic pathway11 and binds to and inhibits the Inhibitors of Apoptosis (IAP) family of proteins.12,13
Recent work has indicated that IAP inhibitors can target multiple IAPs (i.e. XIAP, c-IAP) to enhance the activity of different pro-apoptotic signaling pathways.14 However, the role of XIAP in intrinsic versus extrinsic death pathways is unclear; recent studies suggest that it plays a more significant role in regulating death receptor-mediated apoptosis than intrinsic pathway-mediated cell death15.
We have developed effective inhibitors of the inhibitor of apoptosis (IAP) family of proteins12,13, such as LBW24216, and its structural analog, LCL161, which bind to and inhibit multiple IAPs (i.e. XIAP, c-IAP) to enhance the activity of different proapoptotic signaling pathways. LBW242 and LCL161 are of similar molecular weight and are related structurally. The C-terminal half of LCL161 differs from LBW242, but the N-terminal half is similar. LCL161 has been optimized for potency and PK, and is a candidate for clinical trial investigation for solid tumors.
We have previously demonstrated the ability of LBW242 to synergize with PKC412 in vivo against progressive mutant FLT3-positive leukemia16. Here, we show the ability of the LBW242 structural analog, LCL161, to kill both kinase inhibitor-sensitive and kinase inhibitor–resistant mutant FLT3- and BCR-ABL-positive cells. As observed with LBW242, LCL161 similarly synergizes- both in vitro and in vivo- with PKC412 against progressive mutant FLT3-positive leukemia. However, LCL161 also synergizes in vitro and in vivo with nilotinib against BCR-ABL-positive leukemia. In addition, the use of LCL161 in combination with nilotinib was demonstrated to significantly delay the onset of disease recurrence in an in vivo model of BCR-ABL-positive leukemia. These data underscore the potential clinical advantage to using a proapoptotic agent, such as an IAP inhibitor, in combination with kinase inhibition to potentially improve patient responsiveness to tyrosine kinase inhibitor treatment.
Materials and Methods
Cell lines and cell culture
Ba/F3.p210 cells were obtained by transfecting the IL-3-dependent marine hematopoietic Ba/F3 cell line with a pGD vector containing p210BCR-ABL (B2A2) cDNA.17,18,19 Murine hematopoietic 32D cells were transduced with retrovirus to express p210 Bcr-ABL (32D.p210 cells).20 Ba/F3 cells were stably transfected by electroporation with imatinib-resistant BCR-ABL constructs (pCI-neo Mammalian Expression Vector; Promega (#E1841) harboring the point mutations T315I, F317L, F486S, and M351T; transfectants were selected for neomycin resistance and IL-3-independent growth6.
The IL-3-dependent murine hematopoietic cell line Ba/F3 was transduced with WT-FLT3, FLT3-ITD- or FLT3-D835Y- containing MSCV retroviruses harboring a neomycin selectable marker, and selected for resistance to neomycin.21,22 Mutant FLT3-transduced cells were selected for growth in G418 (1mg/ml).
PKC412-resistant Ba/F3 cell lines expressing FLT3 harboring mutations in the ATP-binding pocket (Ba/F3-N676D, Ba/F3-G697R) were previously developed.23 The human AML-derived, FLT3-ITD-expressing cell line, MOLM-13 (DSMZ (German Resource Centre for BiologicalMaterial), was engineered to express luciferase fused to neomycin phosphotransferase (pMMP-LucNeo) by transduction with a VSVG-pseudotyped retrovirus as previously described.24
All cell lines were cultured with 5% CO2 at 37°C in RPMI (Mediatech, Inc., Herndon, VA) with 10% fetal calf serum (FCS) and supplemented with 1% L-glutamine. Parental Ba/F3 cells were similarly cultured with 15% WEHI-conditioned medium as a source of IL-3. Transfected cell lines were cultured in media supplemented with 1mg/ml G418.
Chemical compounds and biologic reagents
Nilotinib, imatinib, PKC412, and LCL161 were synthesized by Novartis Pharma AG, Basel, Switzerland. Compounds were initially dissolved in DMSO to make 10 mM stock solutions, and then were serially diluted to obtain final concentrations for in vitro experiments. Ara-c and doxorubicin were purchased from Sigma Chemical Co (St Louis, MO).
Normal bone marrow colony assays
Human bone marrow cells were obtained from normal donors after obtaining informed consent on an institutional IRB approved protocol. Mononuclear cells were isolated from normal bone marrow by density gradient centrifugation through Ficoll-Plaque Plus (Amersham Pharmacia Biotech AB, Uppsala, Sweden) at 2000 rpm for 30 minutes, followed by two washes in 1X PBS. Normal human bone marrow was analyzed in a colony assay: plates of 5×104 cells in “complete” methylcellulose medium containing recombinant cytokines (contents: fetal bovine serum, rh SCF, rh GM-CSF, rh IL-3, Bovine Serum Albumin, methylcellulose in Iscove’s MDM, 2-Mercaptoethanol, rh Erythropoietin, L-Glutamine) (MethoCult GFH4434, StemCell Technologies, Inc., Vancouver, BC) were prepared. The plates also contained LCL161 at the indicated concentrations. The plates were incubated at 37°C in 5% CO2 for > 1 week, and then myeloid and erythroid colonies (early progenitors with erythroid and myeloid components: CFU-GM, CFU-E, BFU-E, and CFU-GEMM) were counted on an inverted microscope.
Human bone marrow samples obtained from normal healthy donors were also investigated for responsiveness to LCL161 in liquid culture [Iscove’s MDM, supplemented with 20%FCS, L-glutamine and a cytokine cocktail containing rh Flt-3 ligand (100 ng/mL), rh SCF (100 ng/mL), rh IL-3 (20 ng/mL), rh IL-6 (20 ng/mL) and rh GM-CSF (10ng/mL)] in the presence of different concentrations of drug.
AML patient cells
Frozen vials of bone marrow from AML patients identified as harboring mutant FLT3 were thawed prior to processing using Ficoll-Plaque-Plus (Amersham Pharmacia Biotech AB, Uppsala, Sweden) for the isolation of mononuclear cells. Mononuclear cells were isolated from normal bone marrow by density gradient centrifugation through Ficoll-Plaque Plus at 2000 rpm for 30 minutes, followed by two washes in 1X PBS. Mononuclear cells were then tested in liquid culture (Iscove’s MDM, supplemented with 20% FCS) in the presence of different concentrations of HG-7-85-01. All bone marrow samples from AML patients were obtained under approval of the Dana Farber Cancer Institute Institutional Review Board.
Human AML peripheral blood and bone marrow samples were investigated for responsiveness to LCL161 in liquid culture [Iscove’s MDM, supplemented with 20%FCS and L-glutamine] in the presence of different concentrations of drug. Human AML samples were also analyzed via colony assay, as described above.
Proliferation studies, apoptosis assays, and cell cycle analysis
Cell counts for proliferation studies were obtained using the trypan blue exclusion assay, as previously described.8 Error bars represent the standard error of the mean for each data point. Programmed cell death of inhibitor-treated cells was determined using the Annexin-V-Fluos Staining Kit (Boehringer Mannheim, Indianapolis, IN), as previously described.8 Cell cycle analysis was performed as previously described.8
Drug combination studies
For drug combination studies, compounds were added simultaneously at fixed ratios to cells, and cell viability was determined by trypan blue exclusion and expressed as the function of growth affected (FA) drug-treated versus control cells. Synergy was assessed by Calcusyn software (Biosoft, Ferguson, MO and Cambridge, UK), using the Chou-Talalay method.25 The combination index=[D]1 [Dx]1 + [D]2/[Dx]2, where [D]1 and [D]2 are the concentrations required by each drug in combination to achieve the same effect as concentrations [Dx]1 and [Dx]2 of each drug alone. Generally, values less than one indicate synergy, whereas values greater than one indicate antagonism.
Bioluminescent mutant FLT3 model of AML
Ba/F3-FLT3-ITD cells were transduced with a VSVG-pseudotyped retrovirus comprised of the firefly luciferase coding region (from pGL3-basic; Promega, Madison, WI) cloned into PMSCV puro (Clonetech, Mountain View, CA). Cells were neomycin selected to produce the Ba/F3-FLT3-ITD (luc+) cell line.
Bioluminescence imaging was carried out as previously described6. Briefly, for administration to male NCR-nude mice (5–6 weeks of age; Taconic, NY), virus- and Mycoplasma-free cells were washed and resuspended in Hank’s Balanced Salt Solution (HBSS; Mediatech, Inc., VA) and administered via IV tail vein injection (800,000 cells/mouse). Anesthesized mice were imaged 1–3 days post IV-injection to generate a baseline used to establish treatment cohorts with matched tumor burden, and total body luminescence was measured as previously described.24
Bioluminescent Bcr-Abl model of CML
32D.p210 cells were transduced with a retrovirus encoding firefly luciferase (MSCV-Luc), and selected with G418 at a concentration of 1mg/ml to produce the 32D.p210-luciferase (luc+) cell line. 32D.p210-luc+ cells free of Mycoplasma and viral contamination were washed once with Hank’s Balanced Salt Solution (HBSS; Mediatech, Inc., VA), and resuspended in HBSS prior to administration to mice.
Bioluminescence imaging was carried out as previously described6. Briefly, for administration to male or female NCR-nude mice (5–6 weeks of age; Taconic, Hudson, NY), virus- and Mycoplasma-free cells were washed and resuspended in Hank’s Balanced Salt Solution (HBSS; Mediatech, Inc., VA) and administered via IV tail vein injection (650,000–1,000,000 cells/mouse). Anesthesized mice were imaged 1–2 days post IV-injection to generate a baseline used to establish treatment cohorts with matched tumor burden, and total body luminescence was measured as previously described.24
Drug formulations for in vivo studies
For the in vivo model of BCR-ABL-positive leukemia, solutions of nilotinib were prepared just prior to administration, by dissolving 100 mg in 1.0mL of NMP to give a clear solution and diluting with 9.0 mL PEG300. For the in vivo model of mutant FLT3-positive leukemia, 6% w/w PKC412 in Gelucire® 44/14 (Gattefosse, France) was diluted with 1X PBS and warmed in a 42°C water bath until liquid. The solution was then stored at 4°C until used for gavage treatment of mice. For both CML and AML in vivo models, LCL161 was prepared by first wetting 10mg powder stock with 30 μl water, and then dissolving in two equivalents (0.73μl per mg of compound) of 6.0 N HCl. The resulting (clear) solution was brought up to 1 ml in pH 4.6 acetate buffer, and the resulting stock was stored frozen at −20°C until used for gavage treatment of mice.
Results
Effects of IAP inhibitor treatment and FLT3 inhibition, alone and combined, on the growth, viability, and cell cycle progression of mutant FLT3-expressing cells in vitro
We were interested in investigating the ability of LCL161 to inhibit the growth of mutant FLT3-expressing cells, both as a single agent, as well as combined with PKC412. LCL161 modestly inhibits the growth of FLT3-ITD-expressing cells when administered alone, with an IC50 ranging from approximately 0.5μM (Ba/F3-FLT3-ITD cells, data not shown) to approximately 4 μM (MOLM13-luc+ cells (Figure 1 and Supplementary Figure 1). The potency of LCL161 against the D835Y mutant was observed to be considerably higher, with an IC50 of approximately 50 nM when tested against Ba/F3-D835Y cells (Supplementary Figure 2). Treatment of MOLM13-luc+ cells with a combination of LCL161 and PKC412 led to significantly more killing of cells than either agent alone, with Calcusyn combination indices suggestive of synergy (Figure 1A, Supplementary Figure 1, Table I).
Figure 1. Effects of IAP inhibitor treatment and FLT3 inhibition, alone and combined, on the growth and viability of mutant FLT3-expressing cells in vitro.

(A) Approximately 3-day treatment of MOLM13 cells with LCL161, PKC412, or a combination of LCL161+PKC412. Calcusyn-derived combination indices derived from data plotted in (A) are shown in Table 1. Data shown are representative of three independent studies. (B) Induction of apoptosis of MOLM13-luc+ cells by LCL161, PKC412, or a combination of the two agents following treatment for approximately 3 days. Data shown are representative of two independent studies.
Table I. Combination indices calculated for LCL161 combined with other agents against mutant FLT3-expressing cells.
Values 0.3–0.7 indicate synergy. Values 0.7–0.85 indicate moderate synergy. Values 0.85–0.90 indicate slight synergy. Values 0.9–1.1 indicate nearly additive effects. Values 1.10–1.20 indicate slight antagonism. Values 1.20–1.45 indicate moderate antagonism. Values 1.45–3.3 indicate antagonism.
| Combination Indices: | |||||
|---|---|---|---|---|---|
| Drug Combination | Cell Lines | ED25 | ED50 | ED75 | ED90 |
| LCL161+ PKC412 | Ba/F3-FLT3-ITD | 1.76736 | 1.06067 | 0.78854 | 0.69819 |
| LCL161+ Ara-c | Ba/F3-FLT3-ITD | 1.07626 | 0.63892 | 0.39867 | 0.26063 |
| LCL161+ doxorubicin | Ba/F3-FLT3-ITD | 4.63909 | 1.17180 | 0.59149 | 0.44495 |
| LCL161+ PKC412 | Ba/F3-D835Y | 2.39437 | 1.83940 | 1.43329 | 1.13536 |
| LCL161+ Ara-c | Ba/F3-D835Y | 0.92474 | 0.70927 | 0.55572 | 0.44349 |
| LCL161+ doxorubicin | Ba/F3-D835Y | 0.48173 | 0.62391 | 0.80902 | 1.05030 |
| LCL161+ PKC412 | MOLM13 | 0.58877 | 0.54114 | 0.49766 | 0.45794 |
| LCL161+ imatinib | Ba/F3.p210 | 0.73555 | 0.72434 | 0.76076 | 0.84933 |
PKC412 and LCL161, respectively, induced apoptosis of MOLM13-luc+ cells, as evidenced by an elevation in apoptotic fraction of drug-treated cells as compared to vehicle-control-treated cells (Figure 1B, Supplementary Figure 1). The combination of PKC412 and LCL161 led to a higher induction of apoptosis than either agent alone (Figure 1B).
We then investigated the effects of PKC412 and LCL161 on cell cycle progression of mutant FLT3-expressing cells. As shown in Figure 2, PKC412 induced G1 arrest in mutant FLT3-expressing cells, similar to what has been previously observed.8 In contrast, LCL161 had little effect on cell cycle progression as a single agent. G1 arrest was observed at highest concentrations of LCL161 and PKC412 combined (Figure 2).
Figure 2. Analysis of cell cycle progression of LCL161−, PKC412−, or combination-treated MOLM13-luc+ cells.

(A–C). 48 hour time point.
The ability of LCL161 to override stromal-mediated rescue of mutant FLT3-expressing cells through positive combination with PKC412
Having established the ability of LCL161 to positively combine with targeted kinase inhibitors against mutant FLT3-positive leukemia, we were interested in exploring the potential of LCL161 to enhance the activity of targeted therapy in the context of residual disease resulting from a cytoprotective microenvironment. We first investigated whether or not stromal-derived, secreted factor(s) could protect leukemia cells against the inhibitory effects of selective tyrosine kinase inhibition. We have previously demonstrated that partial stroma-mediated protection of BCR-ABL-positive leukemic cells from nilotinib treatment involved the cooperative interaction of members of a select panel of stromal-secreted viability factors, including IL-6 and GM-CSF.26 Similar results were observed with mutant FLT3-positive leukemia cells treated with PKC412 in the presence of HS-5 stromal-conditioned media (SCM) versus the same panel of stromal-secreted viability factors.27 Here, we validated these earlier results in the context of stromal-mediated cytoprotection of PKC412 mutant FLT3-expressing cells. Stromal-mediated rescue of PKC412-treated, mutant FLT3-ITD-expressing cells was demonstrated using SCM derived from the HS-5 stromal cell line, as well as primary mouse stroma flushed from mouse femurs (Figure 3A, B). SCM obtained from the flushed femurs of mice was also able to partially protect imatinib-treated 32D.p210-luc+ cells (Supplementary Figure 3). These results support the notion that stromal-mediated protection of kinase inhibitor-treated leukemic cells appears to involve viability signals in the form of secreted growth factors.
Figure 3. Effects of SCM on kinase inhibitor-treated leukemia cells in vitro.

(A) MOLM13-luc+ cells treated for approximately 3 days by PKC412 in the absence and presence of SCM. Media was collected from HS-5 stromal cells after 1 week from an already-established monolayer. HS-5 stromal cells were originally seeded 12 days prior to removal of conditioned media. Cells were then further cultured in fresh media for 7 days. Conditioned media was then collected for use in this study. (B) Approximately 3-day treatment of Ba/F3-FLT3-ITD cells in the presence and absence of SCM from primary murine stroma. Primary stromal cells were seeded following femur flush from C67BL/6J mice. SCM was collected and pooled from an already-established mouse primary stroma monolayer. Cells were cultured for 16 days in fresh media prior to media pool and collection. (C) Proliferation studies of MOLM13-luc+ cells treated for approximately 3 days by PKC412+/−LCL161 in the presence of HS-5 SCM. Cell counts were obtained via Trypan Blue exclusion. (D) Viability assays corresponding to study shown in C. Data points shown are the percentage of viable cells (as determined by Annexin-V-Fluos Staining) shown as a percent of untreated controls.
We investigated the ability of LCL161 to override stromal-mediated chemoresistance by studying its effects on MOLM13-luc+, cultured in the presence of SCM. Significantly, the combination of PKC412 and LCL161 leads to more killing of MOLM13-luc+ cells than either drug alone, both in the absence of SCM, as well as in its presence (Figure 3C, D, Supplementary Figure 3C). In the presence of SCM, LCL161 both enhanced the ability of PKC412 to inhibit proliferation of mutant FLT3-expressing cells (Figure 3C), as well as the ability of PKC412 to promote apoptosis of these cells (Figure 3D). It is important to note that while LCL161 enhances the ability of PKC412 to kill mutant FLT3-expressing cells in the presence of SCM, single agent LCL161-treated cells are protected by SCM to a similar extent as single agent PKC412-treated cells (Supplementary Figure 4).
Effects of IAP inhibitor treatment and FLT3 inhibition, alone and combined, on the growth of mutant FLT3-expressing cells in vivo
LCL161 significantly enhanced the ability of PKC412 to inhibit the growth of Ba/F3-FLT3-ITD-luc+ cells in vivo (Vehicle vs PKC412 (p=0.047); Vehicle vs PKC412+LCL161 (p=0.028)) (Figure 4). Differences between suppression of leukemia growth by PKC412 or LCL161 alone and the combination of PKC412+LCL161 were also significant (PKC412 vs combination (p=0.024); LCL161 vs combination (p=0.032)), and percent spleen weights were smaller for PKC412+LCL161 treated mice than for mice treated with vehicle or either agent alone (Figure 4).
Figure 4. Effects of IAP inhibitor treatment and FLT3 inhibition, alone and combined, on the growth of mutant FLT3-expressing cells in vivo.

(A–C) In vivo bioluminescence imaging study. Male NCr nude mice were administered, via tail vein injection, approximately 800,000 Ba/F3-FLT3-ITD-luc+ cells. Baseline imaging and randomization of mice were performed on day 1 post-IV injection of cells. Drug treatments were carried out for a total of 7 days, with final imaging performed on day 8 post-IV injection of cells. Results of Student t-test for statistical analysis of in vivo bioluminescence assay: Vehicle vs PKC412 (p=0.047); Vehicle vs LCL161 (p=0.142); Vehicle vs combination (p=0.028); PKC412 vs LCL161 (p=0.1505); PKC412 vs combination (p=0.024); LCL161 vs combination (p=0.032). (A) Mouse images obtained on day 8 post-IV injection of cells. (B) Plotted bioluminescence values for entire study. (C) Percent spleen weights obtained for mice sacrificed 9 days following the final imaging day. Vehicle mice: 2 viable at time of sacrifice. PKC412-tr mice: 3 viable at time of sacrifice. LCL161-tr mice: 2 viable at time of sacrifice. Combination-treated mice: 4 viable at time of sacrifice.
In addition to PKC412, LCL161 was also shown to positively combine with the standard chemotherapeutic agents, Ara-c and doxorubicin, against FLT3-ITD-expressing cells (Supplementary Figure 5) and against D835Y-expressing cells (Supplementary Figure 6). Combination indices generally suggested additive-to-synergistic effects for these combinations (Table I). In contrast, the combination of LCL161 and PKC412 against D835Y-expressing cells was antagonistic (Table I).
The clinical potential of LCL161 as a single agent was demonstrated by its ability to inhibit the growth of mutant FLT3-expressing cells derived from AML patients (Table II and Supplementary Figure 7). A normal bone marrow colony assay suggested a lack of drug effect at concentrations ≤2000 nM LCL161 (Supplementary Figure 7). One AML patient, “AML4,” responded to LCL161 at 100 nM with a loss of approximately 80% viability (Supplementary Figure 7).
Table II. Effect of LCL161 on human AML patient samples.
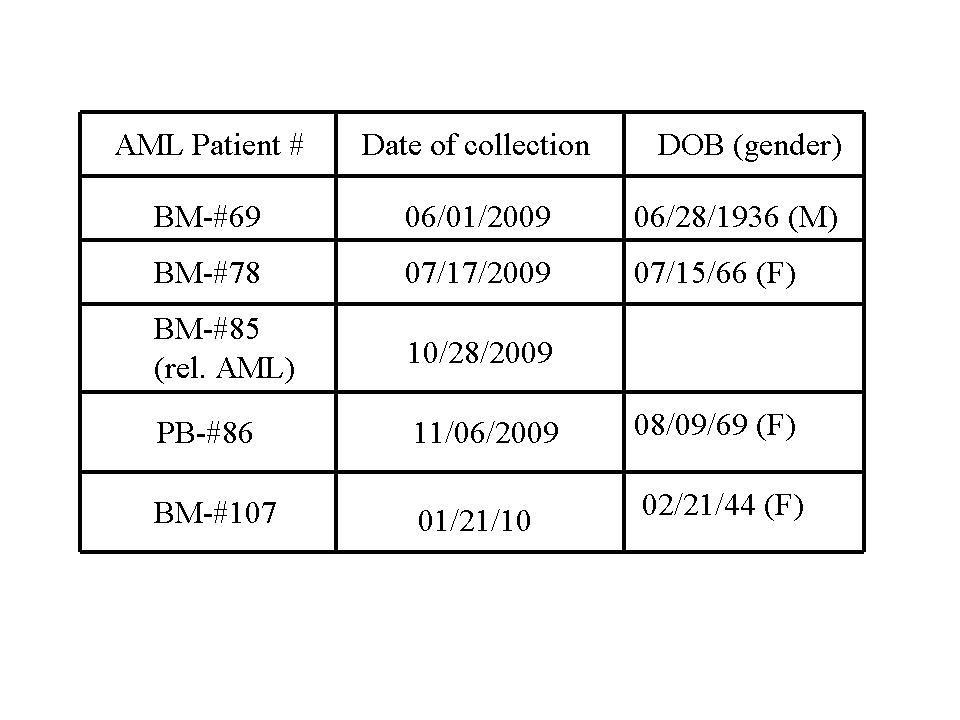
Colony assays performed on primary AML patient samples using Methocult without cytokines. Bone marrow (BM); peripheral blood (PB). Patient information is shown in Supplementary Table SI. Relapsed AML patient BM-#85 had 8% PB blasts and 15% BM blasts.
| AML Patient # | No. of colonies (LCL161-0 uM) | No. of colonies (LCL161-2 uM) | No. of colonies (LCL161-5 uM) |
|---|---|---|---|
| BM-#69 | 38 | 16 | 1 |
| BM-#78 | 7 | 0 | 0 |
| BM-#85 (rel. AML) | 15 | 1 | NA |
| PB-#86 | 10 | 2 | NA |
| BM-#107 | 39 | 7 | 3 |
Effects of IAP inhibitor treatment and Abl inhibition, alone and combined, on the growth of BCR-ABL-expressing cells in vitro and in vivo
LCL161 inhibited the growth of Ba/F3.p210 cells with an IC50 of approximately 100 nM (Figure 5A, Supplementary Figure 8). As with mutant FLT3-expressing cells, LCL161 induces apoptosis of BCR-ABL-expressing cells in a concentration-dependent manner, with no significant effect on cell cycle progression (Supplementary Figure 8). The combination of LCL161 and the Abl inhibitor, imatinib, was observed to be synergistic against BCR-ABL-expressing cells (Figure 5A, Table I). An in vivo study demonstrated a positive combination effect of 16-days of treatment with low-moderate dose (20 mg/kg) nilotinib and LCL161 against BCR-ABL-expressing cells (Figure 5B, C). Nilotinib and LCL161 both suppressed leukemia growth, and there was an additive effect achieved by combining both drugs, which was most apparent at the Day 17 imaging point (p=0.0002). After treatments were stopped on Day 17, there was rapid “catch-up growth” in the treated groups, and as such there was only marginal prolongation of survival comparing combo treated to single treatment groups. Survival was significantly prolonged in the drug-treated groups compared to the vehicle group (p < 0.0001) (Figure 5D).
Figure 5. Effects of IAP inhibitor treatment and Abl inhibition, alone and combined, on the growth of BCR-ABL-expressing cells in vitro and in vivo.

(A) Approximately 3-day treatment of Ba/F3.p210 cells with LCL161, imatinib, or a combination of LCL161+imatinib. Calcusyn-derived combination indices are shown in Table 1. (B–D) In vivo bioluminescence imaging study. Male NCr nude mice were administered, via tail vein injection, approximately 800,000 Ba/F3-FLT3-ITD-luc+ cells. 32 mice were injected with 32D-p210-LucNeo cells and imaged 3 days later to determine tumor burden. Following randomization, mice were divided into 4 treatment groups (n=8). Treatments were carried out via oral gavage 5X per week for a total of 3 weeks. Mice were imaged every 4–8 days and time-to-sac was recorded. Mice were administered vehicle, nilotinib at 20mg/kg 1X daily, LCL161 at 100mg/kg 1X daily, or a combination of nilotinib+LCL161. (B) Plotted bioluminescence values for entire study. (C) Plotted bioluminescence values on Day 17 of treatment (p=0.0002, ANOVA). (D) Survival curve (p < 0.0001, ANOVA).
Enhancement of in vivo effects of high-moderate doses of nilotinib by LCL161 on leukemia burden in mice
We were interested in expanding on the in vitro and in vivo data thus far generated to establish a therapeutic approach designed to suppress leukemia recurrence following treatment with selective kinase inhibitors. Specifically, we attempted to develop murine cell line-based models allowing noninvasive quantitative bioluminescent imaging as a measure of drug effects on both progressive and residual disease burden. For this purpose, we focused efforts on BCR-ABL-positive leukemia as our disease model, as tyrosine kinase inhibitors, such as imatinib, nilotinib, and dasatinib, are FDA-approved and in widespread clinical use for the treatment of CML. The determined ideal doses of nilotinib in these studies were sufficient to substantially reduce leukemia burden to visually undetectable levels, or levels comparable to starting, baseline, measurements. The effects of nilotinib were then tested in combination with LCL161, as a survival pathway blocking inhibitor, to determine if levels of baseline or “residual” leukemic disease could be suppressed further than what was achievable by nilotinib alone. Generally, the time of onset of disease recurrence following varying lengths of treatment with moderate-to-high dose nilotinib was compared between mice treated with nilotinib alone, mice treated with IAP inhibitor alone, and mice treated with a combination of the two agents.
An in vivo study was carried out that demonstrated a positive combination effect of 4-day, high dose (100mg/kg) nilotinib and LCL161 (40–100mg/kg) treatment against BCR-ABL-expressing cells. LCL161 showed transient activity as a single agent (Figure 6A, Supplementary Figure 9), and the addition of LCL161 to nilotinib delayed tumor regrowth by a short duration (Figure 6B, Supplementary Figure 9). In this study, high dose nilotinib treatment alone reduced leukemia burden initially to visually undetectable levels. This was followed by a steady rise in leukemia burden observed post-treatment, with nilotinib+LCL161 treatment groups showing significantly lower tumor burden as compared to nilotinib alone (Supplementary Figure 9).
Figure 6. Enhancement of in vivo effects of high-moderate doses of nilotinib by LCL161 on leukemia burden in mice.

(A–B) In vivo bioluminescence study investigating the effects of leukemia burden by short-term (4-day) treatment of BCR-ABL-harboring mice with high dose (100mg/kg) nilotinib, +/−LCL161 treatment. NCr nude mice (n=6 per treatment group) were injected via tail vein with 800,000 32D.p210-luc+ cells, and treated for a total of 4 days with vehicle, nilotinib (100mg/kg), LCL161 (40mg/kg), LCL161 (100mg/kg), a combination of nilotinib (100mg/kg)+LCL161 (40mg/kg), or a combination of nilotinib (100mg/kg)+LCL161 (100mg/kg). (A) Plotted bioluminescence values showing the effects of LCL161 on leukemia burden, as compared to vehicle control-treated mice, on Day 5 of treatment (p=0.0007, ANOVA). (B) Plotted bioluminescence values showing the effects of nilotinib alone, nilotinib+LCL161 (40mg/kg), or nilotinib+LCL161 (100mg/kg) on leukemia burden on Day 13 of treatment (p=0.002, ANOVA). (C) In vivo bioluminescence study investigating the effects on leukemia burden by long-term (several week) treatment of BCR-ABL-harboring mice with moderate-high dose (75mg/kg) nilotinib, +/−LCL161 treatment. NCr nude mice (n=9 for nilotinib and vehicle; n=10 for LCL161 and combination) were injected via tail vein with 800,000 32D.p210-luc+ cells, and treated for a total of 5 weeks with vehicle, nilotinib (75mg/kg 1X daily), LCL161 (100 mg/kg 1X daily), or a combination of nilotinib+LCL161. (C) Plotted bioluminescence values. ANOVA analysis of BLI comparing all groups at Day 8 of treatment (when all animals still alive) has p value <0.0001. Comparison of nilotinib to combo BLI on day 47 of treatment (last point before significant deaths in nilotinib group) p=0.04. (D) Survival curves. Survival comparison of all groups p<0.0001. Survival comparison of nilotinib and combo p=0.003.
In another in vivo study, a positive combination effect was demonstrated over 3 weeks of treatment with moderate-high dose (75mg/kg) nilotinib and LCL161 (100mg/kg) against BCR-ABL-expressing cells. Nilotinib alone initially suppressed leukemia growth, however whereas disease recurrence was observed with nilotinib as a single agent, leukemia levels continued to remain inhibited by nilotinib combined with LCL161 (Figure 6C).
Inhibition of proliferation of drug-resistant cell lines by LCL161
In addition to the ability of LCL161 to enhance the activity of targeted kinase inhibitor therapies, both in the absence and the presence of a stromal-protected microenvironment, LCL161 also has demonstrated activity against drug-resistant cells expressing point mutations in the target proteins. LCL161 at 1000 nM was able to mostly or completely kill Ba/F3-derived cell lines conferring resistance to PKC412, which express FLT3-ITD harboring point mutations in the ATP-binding pocket of FLT323 (Figure 7). LCL161 also showed activity at concentrations ranging from 100 nM to 1000 nM against Ba/F3 cells expressing various imatinib- and nilotinib-resistant BCR-ABL point mutations (Figure 7).
Figure 7. Inhibition of proliferation of drug-resistant cell lines by LCL161.

(A) Approximately 2-day LCL161 treatment of N676D-Ba/F3 cells, G697R-Ba/F3 cells, and wt FLT3-Ba/F3 cells. (B) Approximately 2-day LCL161 treatment of parental Ba/F3 cells, Ba/F3.p210, and Ba/F3 cells expressing the imatinib-resistant BCR-ABL mutants M351T, F317L, F486S, and T315I.. (C) Approximately 2-day imatinib treatment of Ba/F3, Ba/F3.p210, and Ba/F3 cells expressing imatinib-resistant BCR-ABL point mutants, shown as a control.
Discussion
A small pool of leukemic CD34+ cells can endure in the marrow of CML patients following years of treatment with imatinib. In similar fashion, clinical studies of advanced AML patients treated with FLT3 kinase inhibitors revealed a delayed or marginal decrease in levels of bone marrow blasts, in contrast to a sizable decrease in levels of blasts in patient peripheral blood. In fact, FLT3 inhibition in a defined “niche-like” in vitro environment was actually found to enhance, as opposed to inhibit, survival of CD34+CD38-CD123+ leukemic stem and progenitor cells.28 Taken together, these results suggest that this area of investigation is important for a better understanding of drug resistance mechanisms.
Highest tumor burden and residual disease were previously revealed in vivo in stroma-associated tissues in imatinib/nilotinib-treated mice, suggesting that significant reservoirs for tumor growth appear to be tissues that are able to support normal hematopoietic and malignant stem cell development.26 These studies, which showed a pattern of leukemia distribution consistent with what is observed in imatinib- and nilotinib-treated CML patients, were followed by a more in-depth analysis of stroma-leukemia cell interactions that lead to protection of leukemia cells from nilotinib-induced cytotoxicity. Partial stromal-mediated protection of BCR-ABL-positive leukemic cells from nilotinib treatment was found to involve the cooperative interaction of members of a select panel of stromal-secreted viability factors, including IL-6 and GM-CSF.26 Similar results were observed with mutant FLT3-positive leukemia cells treated with PKC412 in the presence of HS-5-stromal-conditioned media versus the same panel of stromal-secreted viability factors.27
We set out here to investigate whether pharmacological targeting of resistance mechanisms might synergize with existing, targeted therapies as a way to eventually improve patient outcomes by ablating, or delaying the onset of, residual disease. In our studies, we tested the combination of targeted therapies for CML and mutant FLT3+ AML, respectively, with a novel agent, LCL161, which inhibits signaling pathways critical for survival of leukemic cells. We confirmed the cytoprotective effects of stroma on kinase inhibitor-treated leukemia cells, and we utilized this assay system to explore and demonstrate the ability of the novel IAP inhibitor, LCL161, to enhance the efficacy of targeted therapies to override stromal-mediated chemoresistance.
We also developed an in vivo assay system that allows monitoring of the growth of progressive disease, as well as baseline level- or “residual” disease resulting from treatment with a moderate-to-high dose of kinase inhibitor. Importantly, we demonstrated the ability of LCL161 to delay the onset of recurring BCR-ABL-positive disease in mice carrying an extremely low tumor burden following treatment with moderate-to-high dose nilotinib.
The development of functional models of progressive and residual leukemic disease enable the potential identification and development of novel therapeutic strategies designed to prevent or delay the onset of CML and AML recurrence in patients following treatment with selective kinase inhibitors. Our studies point toward the potential clinical utility of IAP inhibitors, such as LCL161, as assets for combined therapy designed to override drug resistance and optimize the efficacy of existing targeted therapies for leukemia.
Supplementary Material
{kind=link}
Acknowledgments
Grant Support: J.D.G is supported by NIH grant CA66996, and a Specialized Center of Research Award from the Leukemia and Lymphoma Society. J.D.G. is also supported by NIH grants CA36167 and DK50654.
Footnotes
Conflict of Interest:
J.D.G., R.S., D.F. and A.L.K. have a financial interest with Novartis Pharma AG. Employees of Novartis include D.P., C.S., and L.Z. (formerly).
References
- 1.Druker BJ, Tamura S, Buchdunger E, Ohno S, Segal GM, Fanning S, et al. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat Med. 1996;2:561–566. doi: 10.1038/nm0596-561. [DOI] [PubMed] [Google Scholar]
- 2.Buchdunger E, Matter A, Druker BJ. Bcr-Abl inhibition as a modality of CML therapeutics. Biochim Biophys Acta. 2001;1551:M11–M18. doi: 10.1016/s0304-419x(01)00022-1. [DOI] [PubMed] [Google Scholar]
- 3.Deininger MW, Goldman JM, Melo JV. The molecular biology of chronic myeloid leukemia. Blood. 2000;96:3343–3356. [PubMed] [Google Scholar]
- 4.Gorre ME, Mohammed M, Ellwood K, Hsu N, Paquette R, Rao PN, et al. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science. 2001;293:876–880. doi: 10.1126/science.1062538. [DOI] [PubMed] [Google Scholar]
- 5.Weisberg E, Griffin JD. Mechanism of resistance to the ABL tyrosine kinase inhibitor STI571 in BCR/ABL-transformed hematopoietic cell lines. Blood. 2000;95:3498–505. [PubMed] [Google Scholar]
- 6.Weisberg E, Manley PW, Breitenstein W, Brüggen J, Cowan-Jacob SW, Ray A, et al. Characterization of AMN107, a selective inhibitor of native and mutant Bcr-Abl. Cancer Cell. 2005;7:129–141. doi: 10.1016/j.ccr.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 7.Stirewalt DL, Radich JP. The role of FLT3 in haematopoietic malignancies. Nat Rev Cancer. 2003;3:650–65. doi: 10.1038/nrc1169. [DOI] [PubMed] [Google Scholar]
- 8.Weisberg E, Boulton C, Kelly LM, Manley P, Fabbro D, Meyer T, et al. Inhibition of mutant FLT3 receptors in leukemia cells by the small molecule tyrosine kinase inhibitor PKC412. Cancer Cell. 2002;1:433–43. doi: 10.1016/s1535-6108(02)00069-7. [DOI] [PubMed] [Google Scholar]
- 9.Stone RM, De Angelo J, Galinsky I, Estey E, Klimek V, Grandin W, et al. PKC412 FLT3 inhibitor therapy in AML: results of a phase II trial. Ann Hematol. 2004;83(Suppl 1):S89–90. doi: 10.1007/s00277-004-0850-2. [DOI] [PubMed] [Google Scholar]
- 10.Kumagai M, Manabe A, Pui CH, Behm FG, Raimondi SC, Hancock ML, et al. Stroma-supported culture in childhood B-lineage acute lymphoblastic leukemia cells predicts treatment outcome. J Clin Invest. 1996;97:755–760. doi: 10.1172/JCI118474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Du C, Fang M, Li Y, Li L, Wang X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell. 2000;102:33–42. doi: 10.1016/s0092-8674(00)00008-8. [DOI] [PubMed] [Google Scholar]
- 12.Liu Z, Sun C, Olejniczak ET, Meadows RP, Betz SF, Oost T, et al. Structural basis for binding of Smac/DIABLO to the XIAP BIR3 domain. Nature. 2000;408:1004–8. doi: 10.1038/35050006. [DOI] [PubMed] [Google Scholar]
- 13.Wu G, Chai J, Suber TL, Wu JW, Du C, Wang X, et al. Structural basis of IAP recognition by Smac/DIABLO. Nature. 2000;408:1008–12. doi: 10.1038/35050012. [DOI] [PubMed] [Google Scholar]
- 14.Galban S, Hwang C, Rumble JM, Oetjen KA, Wright CW, Boudreault A, et al. Cytoprotective effects of IAPs revealed by a small molecule antagonist. Biochem J. 2009;417:765–71. doi: 10.1042/BJ20081677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sensintaffar J, Scott FL, Peach R, Hager JH. XIAP is not required for human tumor cell survival in the absence of an exogenous death signal. BMC Cancer. 2010;10:11. doi: 10.1186/1471-2407-10-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weisberg E, Kung AL, Wright RD, Moreno D, Catley L, Ray A, et al. Potentiation of antileukemic therapies by Smac mimetic, LBW242: effects on mutant FLT3-expressing cells. Mol Cancer Ther. 2007;6:1951–61. doi: 10.1158/1535-7163.MCT-06-0810. [DOI] [PubMed] [Google Scholar]
- 17.Daley GQ, Baltimore D. Transformation of an interleukin 3-dependent hematopoietic cell line by the chronic myeloid leukemia-specific p210 BCR-ABL protein. Proc Natl Acad Sci USA. 1988;85:9312–9316. doi: 10.1073/pnas.85.23.9312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Okuda K, Golub TR, Gilliland DG, Griffin JD. p210BCR-ABL, p190BCR-ABL, and TEL/ABL activate similar signal transduction pathways in hematopoietic cell lines. Oncogene. 1996;13:1147–52. [PubMed] [Google Scholar]
- 19.Sattler M, Salgia R, Okuda K, Uemura N, Durstin MA, Pisick E, et al. The proto-oncogene product p120CBL and the adaptor proteins CRKL and c-CRK link c-ABL, p190BCR-ABL and p210BCR-ABL to the phosphatidylinositol-3′ kinase pathway. Oncogene. 1996;12:839–46. [PubMed] [Google Scholar]
- 20.Matulonis U, Salgia R, Okuda K, Druker B, Griffin JD. IL-3 and p210 BCR-ABL activate both unique and overlapping pathways of signal transduction in a factor-dependent myeloid cell line. Exp Hematol. 1993;21:1460–1466. [PubMed] [Google Scholar]
- 21.Kelly LM, Liu Q, Kutok JL, Williams IR, Boulton CL, Gilliland DG. FLT3 internal tandem duplication mutations associated with human acute myeloid leukemias induce myeloproliferative disease in a murine bone marrow transplant model. Blood. 2002;99:310–318. doi: 10.1182/blood.v99.1.310. [DOI] [PubMed] [Google Scholar]
- 22.Jiang J, Paez JG, Lee JC, Bo R, Stone RM, DeAngelo DJ, et al. Identifying and characterizing a novel activating mutation of the FLT3 tyrosine kinase in AML. Blood. 2004;104:1855–1858. doi: 10.1182/blood-2004-02-0712. [DOI] [PubMed] [Google Scholar]
- 23.Cools J, Mentens N, Furet P, Fabbro D, Clark JJ, Griffin JD, et al. Prediction of resistance to small molecule FLT3 inhibitors: implications for molecularly targeted therapy of acute leukemia. Cancer Res. 2004;64:6385–6389. doi: 10.1158/0008-5472.CAN-04-2148. [DOI] [PubMed] [Google Scholar]
- 24.Armstrong SA, Kung AL, Mabon ME, Silverman LB, Stam RW, Den Boer ML, et al. Validation of a therapeutic target identified by gene expression based classification. Cancer Cell. 2003;3:173–83. doi: 10.1016/s1535-6108(03)00003-5. [DOI] [PubMed] [Google Scholar]
- 25.Chou T-C, Talalay P. Quantitative analysis of dose-effect relationships: The combined effects of multiple drugs or enzyme inhibitors. Adv Enz Regul. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 26.Weisberg E, Wright RD, McMillin DW, Mitsiades C, Ray A, Barrett R, et al. Stromal-mediated protection of tyrosine kinase inhibitor-treated BCR-ABL-expressing leukemia cells. Mol Cancer Ther. 2008;7:1121–9. doi: 10.1158/1535-7163.MCT-07-2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Weisberg E, Barrett R, Liu Q, Stone R, Gray N, Griffin JD. FLT3 inhibition and mechanisms of drug resistance in mutant FLT3-positive AML. Drug Resist Updat. 2009;12:81–9. doi: 10.1016/j.drup.2009.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mony U, Jawad M, Seedhouse C, Russell N, Pallis M. Resistance to FLT3 inhibition in an in vitro model of primary AML cells with a stem cell phenotype in a defined microenvironment. Leukemia. 2008;22:1395–1401. doi: 10.1038/leu.2008.125. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.