

Abstract

Silylene transfer to allylic sulfides results in a formal 1,2-sulfide migration. The rearrangement yields substituted silacyclobutanes, not the expected silacyclopropanes. The silacyclobutanes were elaborated by insertions of carbonyl compounds selectively into one carbon–silicon bond. A mechanism for the 1,2-sulfide migration is proposed involving an episulfonium ion intermediate.
Silacyclobutanes are unique strained-ring compounds1 that have been used in a variety of chemical applications, including ring-opening polymerization.2 These silanes are synthesized conventionally by intramolecular Wurtz-type coupling3-6 or [2+2] cycloaddition reactions,7,8 making additional synthetic routes desirable. Unlike three-membered ring silanes, silacyclobutanes are often air stable, which facilitates their handling and manipulation.9 Carbonyl insertions with strained silacyclobutanes provide a variety of synthetically useful transformations.6,10-13
In the course of exploring silylene transfer reactions, we discovered that our silylene transfer conditions14-16 provided a synthesis of four-membered ring silanes when applied to allylic sulfides (Table 1). In contrast to metal-catalyzed silylene insertion reactions with allylic ethers,17,18 no carbon–sulfur bond insertion products were observed. Instead, the products resulted from formal 1,2-sulfur migration.19 A variety of metal salts catalyzed the silylene transfer reaction at ambient temperature in moderate yields. AgO2CCF3 was found to be the optimal catalyst (entry 1). Silylene transfer with silacyclobutane formation occurred in the absence of catalyst, but elevated temperatures were required (entry 8).
Table 1. Metal Catalysts Utilized for Silylene Transfer to Allylic Sulfide 1a.
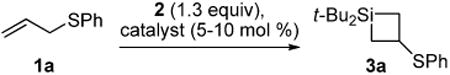
| |||
|---|---|---|---|
|
| |||
| entry | catalyst | T (°C) | yield (%)a |
| 1 | AgO2CCF3 | 22 | 72 |
| 2 | AgOTf | 22 | 61 |
| 3 | Ag3PO4 | 22 | 70 |
| 4 | (CuOTf)·2PhMe | 22 | 42 |
| 5 | CuBr | 22 | 63 |
| 6 | CuI | 22 | 35 |
| 7 | AuBr3 | 22 | 58 |
| 8 | -- | 70 | 43 |
As determined by 1H NMR spectroscopic analysis relative to an internal standard (PhSiMe3).
The reaction was general for a variety of substituted allylic sulfides, although yields were lower (Table 2). Silylene transfer to α-methyl substituted allylic sulfide 1b afforded silacyclobutane trans-3b with high diastereoselectivity (entry 1). An increase in steric hindrance at the α position was observed to increase diastereoselectivity in the reaction, but resulted in low yield of the desired silacyclobutane (entry 2). Silylene transfer to crotyl sulfide 1d gave silacyclobutane trans-3d with lower diastereoselectivity (entry 3). When the reaction was attempted with α-methyl substituted crotyl sulfide 1e, product 3e was obtained in low yield and with low diastereoselectivity (entry 4).
Table 2. Silylene Transfer to Substituted Allylic Sulfides.

| ||||||
|---|---|---|---|---|---|---|
|
| ||||||
| entry | substrate | R1 | R2 | product | dra,b | yield (%)a |
| 1 | 1b | H | Me | 3b | 92:8 | 41 |
| 2 | 1c | H | i -Pr | 3c | 100:0 | 17 |
| 3 | 1d | Me | H | 3d | 86:14 | 55 |
| 4 | 1e | Me | Me | 3e | 66:34 | 19c |
As determined by 1H NMR spectroscopic analysis relative to an internal standard (PhSiMe3).
Relative stereochemistry was determined by NOE analysis of the products. Details are provided as Supporting Information.
Five equivalents of cyclohexene silacyclopropane 2 were required for this reaction to proceed to completion.20
To assess if other functional groups could undergo 1,2-migration,21-23 the silylene transfer conditions were applied to allylic silane 4 and allyl bromide. Silylene transfer to allylic silane 4 did not provide 1,2-silyl migration and afforded silacyclopropane 5, which was subjected to the two-step, one-flask carbonyl insertion reaction24 to afford a mixture of products 6a-6c (Scheme 1). Allylic silane 6a can result from hydrogen-atom transfer processes,25,26 and dioxasilacyclopentane 6c is the product of silylene transfer to two equivalents of benzaldehyde.27,28 The same conditions with allyl bromide resulted in decomposition of the starting materials.
Scheme 1. Silylene Transfer to an Allylic Silane.

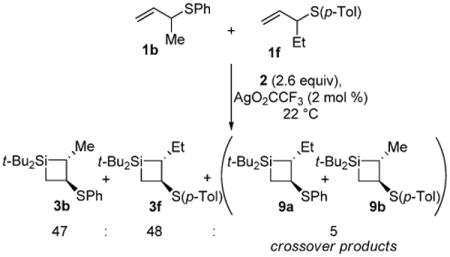
A crossover experiment was performed with allylic sulfides 1b and 1f to provide insight into the mechanism for the allylic sulfide rearrangement (eq 1). Only trace amounts of crossover products were observed, indicating that the 1,2-sulfide migration occurs through a stepwise mechanism in which fragments largely combine intramolecularly. The likely mechanism involves episulfonium ion formation29-33 with the electrophilic silylenoid15,34,35 to give intermediate 10. The silver-bound silyl species could open the episulfonium ion concurrent to four-membered ring closure to afford silacyclobutane 3 (Scheme 2). The trace crossover products could arise from loss and recombination of the sulfide group on intermediate 10.36
Scheme 2. Episulfonium Ion Formation in the 1,2-Sulfide Migration Mechanism.

 |
(1) |
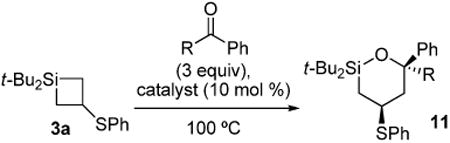
Carbonyl insertion reactions were performed with silacyclobutane 3a to afford oxasilacyclohexanes as single diastereomers (Table 3).6,11-13 Various metal catalysts were employed in the insertion reaction with silacyclobutane 3a, and zinc catalysts were observed to be optimal (entry 2). No reaction was observed in the absence of catalyst. Benzaldehyde and acetophenone were competent in the insertion reaction, but both linear and branched aliphatic aldehydes and ketones were not.25 The observed diastereoselectivity could occur to minimize the unfavorable 1,3-diaxial interaction that would arise between the phenyl group of the carbonyl and a tert-butyl group on silicon.
Table 3. Carbonyl Insertion Reactions with Silacyclobutane 3a.
| |||||
|---|---|---|---|---|---|
|
| |||||
| entry | R | catalyst | product | yield (%)a | dra |
| 1 | H | (CuOTf)·2PhMe | 11a | 68 | 100:0 |
| 2 | H | ZnI2 | 11a | 70 | 100:0 |
| 3 | H | Zn(OTf)2 | 11a | 70 | 100:0 |
| 4 | Me | ZnI2 | 11b | 63 | 96:4 |
As determined by 1H NMR spectroscopic analysis relative to an internal standard (PhSiMe3).
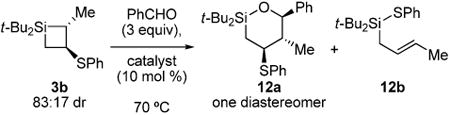
The increased ring substitution on silacyclobutane 3b decreased the efficiency of the carbonyl insertion reaction (Table 4). The most competent catalyst in the benzaldehyde insertion reaction was ZnI2. The yield was moderate, however, and isolation of the product proved difficult due to the significant number of unidentifiable decomposition products. Carbonyl insertion occurred into the more substituted carbon–silicon bond in moderate yield to afford oxasilacyclohexane 12a as a single diastereomer. Allyl silane 12b was observed as a minor product resulting from metal-mediated rearrangement of the starting material.37 The regiochemistry is in contrast to that observed for analogous zinc-catalyzed carbonyl insertions into silacyclopropanes.38 The formation of allylic silane 14 from silacyclobutane 3a in the absence of aldehyde supports the existence of transmetalation intermediate 13 (eq 2). The regioselectivity of this reaction is analogous to the copper-mediated transmetalation observed for silacyclopropanes.39,40
Table 4. Carbonyl Insertion Reactions with Silacyclobutane 3b.
| ||
|---|---|---|
|
| ||
| entry | catalyst | yield 12a (%)a |
| 1 | (CuOTf)·2PhMe | 25 |
| 2 | ZnI2 | 35 |
| 3 | Zn(OTf)2 | 29 |
As determined by 1H NMR spectroscopic analysis relative to an internal standard (PhSiMe3).
 |
(2) |
To increase the general utility of the 1,1-di-tert-butyl-3-thiophenyl-1-silacyclobutanes, lithiation reactions were performed to see if the sulfide moiety could be functionalized selectively. Silacyclobutane 3a underwent lithium–sulfide exchange41-45 followed by trapping with chlorotrimethylsilane to afford silacyclobutane 15 (eq 3). Silacyclobutane 15 did not undergo carbonyl insertion when subjected to the zinc-catalyzed conditions.46
 |
(3) |
In conclusion, metal-catalyzed silylene transfer conditions were utilized in a rearrangement reaction with allylic sulfides to afford silacyclobutanes. The resultant four-membered ring compounds were subjected to carbonyl insertion reaction conditions to afford substituted oxasilacyclohexanes. A mechanism for the 1,2-sulfide migration was proposed utilizing a sulfonium ion intermediate.
Experimental Section
Silylene Transfer to Allylic Sulfides (Procedure A)
Silacyclobutane 3a
To a solution of allylic sulfide 1a (5.00 g, 33.3 mmol) in toluene (140 mL) was added cyclohexene silacyclopropane 215,47 (8.96 g, 39.9 mmol) and AgO2CCF3 (0.073 g, 0.33 mmol). The reaction mixture was stirred overnight, filtered through a pad of SiO2 with hexanes to remove residual catalyst, and concentrated in vacuo. Purification by column chromatography (hexanes) afforded silacyclobutane 3a as a white solid (6.2 g, 64%): mp 36–38 °C; 1H NMR (400 MHz, C6D6) δ 7.40 (d, J = 7.2, 2H), 7.10 (t, J = 7.6, 2H), 6.98 (t, J = 7.3, 1H), 3.88 (quint, J = 9.2, 1H), 1.57–1.50 (m, 2H), 1.32–1.25 (m, 2H), 0.93 (s, 9H), 0.92 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 137.5, 130.0, 128.7, 125.8, 37.8, 28.0, 27.8, 19.3, 18.9, 18.7; 29Si NMR (99.3 MHz, CDCl3) δ 18.9; IR (thin film) 3082, 3059, 2935, 2854, 1583, 1464 cm-1; HRMS (GC-MS) m / z calcd for C17H29SSi (M + H)+ 293.1759, found 293.1759. Anal. Calcd for C17H28SSi: C, 69.79; H, 9.65. Found: C, 69.89; H, 9.65.
Silacyclobutane 3b
Procedure A was employed using allylic sulfide 1b (4.01 g, 24.4 mmol), AgO2CCF3 (0.53 g, 2.4 mmol), and cyclohexene silacyclopropane 215,47 (16.4 g, 73.1 mmol). Purification by column chromatography (hexanes) afforded a mixture of silacyclobutanes trans-3b and cis-3b (83:17 dr) as a pale yellow oil (1.0 g, 15%). Silacyclobutane trans-3b: 1H NMR (400 MHz, C6D6) δ 7.53 (d, J = 7.1, 2H), 7.08 (t, J = 7.4, 2H), 7.00 (t, J = 7.3, 1H), 3.51 (dt, J = 10.6, 9.8, 1H), 1.66 (dq, J = 11.3, 7.3, 1H), 1.54 (dd, J = 14.6, 8.9, 2H), 1.26 (d, J = 7.4, 3H), 0.98 (s, 9H), 0.85 (s, 9H); 13C NMR (125 MHz, C6D6) δ 136.4, 132.57, 128.6, 126.4, 48.4, 29.3, 28.4, 28.1, 19.8, 19.5, 18.2, 14.5; 29Si NMR (119.2 MHz, C6D6) 17.1; IR (neat) 3059, 2931, 2858, 1583, 1471, 1265 cm-1; HRMS (GC-MS) m / z calcd for C18H31SSi (M + H)+ 307.1916, found 307.1912. Silacyclobutane cis-3b: 1H NMR (400 MHz, C6D6, distinctive peaks) δ 7.34 (d, J = 7.3, 2H), 4.19 (q, J = 9.7, 1H), 2.14–2.05 (m, 1H), 1.37 (d, J = 8.2, 3H), 1.01 (s, 9H), 0.96 (s, 9H); 13C NMR (125 MHz, C6D6, distinctive peaks) δ 135.3, 132.56, 128.7, 124.7, 42.9, 28.7, 28.0, 17.2, 12.9.
Silacyclobutane 3c
Procedure A was employed using allylic sulfide 1c (0.55 mL, 0.24 M in C6D6, 0.13 mmol), AgO2CCF3 (0.001 g, 0.005 mmol), and cyclohexene silacyclopropane 215,47 (0.085 g, 0.38 mmol). After 24 h, the reaction had afforded silacyclobutane 3c as a single diastereomer in 17% yield as determined by 1H NMR spectroscopy relative to an internal standard (PhSiMe3) using a single scan: 1H NMR (400 MHz, C6D6, distinctive peaks) δ 3.57 (q, J = 9.5, 1H), 0.94 (s, 9H), 0.88 (s, 9H); LRMS (GCMS) Rt = 19.3 min; m / z calcd for C20H34SSi (M)+ 334.22, found 334.17; m / z calcd for C16H25SSi (M – C4H9)+ 277.14, found 277.19.
Silacyclobutane 3d (same as compound 3b)
Procedure A was employed using crotyl sulfide 1d (0.62 g, 3.7 mmol), cyclohexene silacyclopropane 215,47 (2.53 g, 11.3 mmol), and AgO2CCF3 (0.038 mg, 0.19 mmol). Purification by column chromatography (hexanes – 1:99 EtOAc/hexanes) afforded silacyclobutanes trans-3d and cis-3d (80:20 dr) as a colorless oil (0.28 g, 24%). Full characterization data was reported for silacyclobutane 3b, vide supra.
Silacyclobutane 3e
Procedure A was employed using crotyl sulfide 1e (0.372 g, 2.09 mmol), cyclohexene silacyclopropane 215,47 (2.36 g, 10.5 mmol), and AgO2CCF3 (0.023 g, 0.10 mmol). The crude mixture contained a mixture of silacyclobutanes trans-3e/cis-3e (66:34 dr). Purification by column chromatography (hexanes – 1:99 EtOAc/hexanes) afforded an impure yellow oil containing silacyclobutane cis-3e (0.06 g, 10%) in addition to an impure mixture containing silacyclobutanes trans-3e/cis-3e (55:45 dr) as a yellow oil (0.22 g, 32%). Silacyclobutane trans-3e: 1H NMR (400 MHz, C6D6, distinctive peaks) δ 7.58 (dd, J = 8.0, 2.0, 2H), 3.07 (t, J = 11.5, 1H). Silacyclobutane cis-3e: 1H NMR (400 MHz, C6D6, distinctive peaks) δ 7.37 (d, J = 7.3, 2H), 7.07 (t, J = 7.7, 2H), 3.96 (dd, J = 12.6, 9.3, 1H), 2.06 (quint, J = 8.8, 1H), 1.87–1.78 (m, 1H), 1.36 (d, J = 8.2, 3H), 1.29 (d, J = 5.7, 3H),48 1.03 (s, 9H), 0.98 (s, 9H); 29Si NMR (119.2 MHz, C6D6) δ 14.1; IR (neat) 3059, 2935, 2860, 1581, 1473, 1363 cm-1; HRMS (GC-MS) m / z calcd for C19H31SSi (M – H)+ 319.1916, found 319.1920.
Carbonyl Insertion into Silacyclobutanes (Procedure B)
Oxasilacyclohexane 11a
To a solution of silacyclobutane 3a (1.4 g, 4.8 mmol) in toluene (22 mL) was added benzaldehyde (1.5 mL, 15 mmol) and ZnI2 (0.16 g, 0.50 mmol). The reaction mixture was heated to 100 °C. After 30 h, the reaction mixture was filtered through a pad of SiO2/Celite (1:9) with hexanes to remove residual catalyst and concentrated in vacuo. Benzaldehyde was removed in vacuo at 100 °C (0.4 mm Hg). Purification by column chromatography (1:99 EtOAc/hexanes) afforded oxasilacyclohexane 11a as a white solid (0.69 g, 36%): mp 56–58 °C; 1H NMR (500 MHz, CDCl3) δ 7.42 (d, J = 8.1, 2H), 7.35–7.32 (m, 6H), 7.28–7.24 (m, 2H), 4.99 (appar d, J = 11.2, 1H), 3.73 (tdd, J = 12.7, 4.2, 2.3, 1H), 2.17 (appar dd, J = 13.9, 1.7, 1H), 1.60 (dt, J = 13.7, 11.7, 1H), 1.34 (ddd, J = 14.4, 4.5, 2.1, 1H), 1.09 (s, 9H), 1.07 (s, 9H), 0.89 (appar t, J = 13.8, 1H); 13C NMR (125 MHz, CDCl3) δ 145.0, 134.6, 132.4, 129.0, 128.3, 127.2, 127.1, 125.2, 77.3, 45.0, 44.7, 28.2, 27.6, 22.2, 20.2, 13.8; IR (thin film) 3061, 2931, 1601, 1583, 1471, 1092 cm-1; HRMS (GC-MS) m / z calcd for C24H35OSSi (M + H)+ 399.2178, found 399.2180. Anal. Calcd for C24H34OSSi: C, 72.30; H, 8.60. Found: C, 72.04; H, 8.59.
Oxasilacyclohexane 11b
Procedure B was employed using silacyclobutane 3a (0.29 g, 1.0 mmol), acetophenone (0.35 mL, 3.0 mmol), and ZnI2 (0.035 g, 0.11 mmol). Acetophenone was removed in vacuo at 100 °C (0.4 mm Hg). Purification by column chromatography (1:99 EtOAc/hexanes) afforded oxasilacyclohexane 11b as a colorless oil (0.22 g, 54%): 1H NMR (500 MHz, CDCl3) δ 7.44 (d, J = 7.2, 2H), 7.41 (d, J = 7.3, 2H), 7.36 (t, J = 7.6, 2H), 7.31–7.26 (m, 3H), 7.20 (t, J = 7.3, 1H), 3.78 (appar td, J = 12.7, 3.5, 1H), 2.45 (appar d, J = 13.7, 1H), 1.85 (appar t, J = 12.9, 1H), 1.46 (s, 3H), 1.30 (ddd, J = 14.2, 4.2, 2.2, 1H), 1.10 (s, 9H), 0.87 (s, 9H), 0.78 (appar t, J = 13.8, 1H); 13C NMR (125 MHz, CDCl3) δ 150.4, 135.2, 131.5, 129.0, 127.9, 126.9, 126.3, 124.2, 76.4, 45.2, 40.6, 32.0, 27.6, 27.4, 20.9, 20.6, 14.6; IR (neat) 3058, 2967, 1601, 1583, 1471, 1011 cm-1; HRMS (GC-MS) m / z calcd for C25H37OSSi (M + H)+ 413.2334, found 413.2328. Anal. Calcd for C25H36OSSi: C, 72.76; H, 8.79. Found: C, 72.55; H, 8.85.
Oxasilacyclohexane 12a and Allyl Silane 12b
Procedure B was employed using silacyclobutane 3b (0.319 g, 1.04 mmol, 83:17 dr), benzaldehyde (0.32 mL, 3.1 mmol), and ZnI2 (0.032 g, 0.10 mmol). The reaction mixture was heated to 70 °C. Benzaldehyde was removed in vacuo at 100 °C (0.4 mm Hg). Purification by column chromatography (1:199 EtOAc/hexanes) afforded oxasilacyclohexane 12a as a white solid (0.072 g, 16%) and allyl silane 12b as a colorless oil (0.019 g, 6%). Oxasilacyclohexane 12a: mp 80–82 °C; 1H NMR (500 MHz, CDCl3) δ 7.49 (d, J = 7.7, 2H), 7.40–7.34 (m, 8H), 4.61 (d, J = 9.5, 1H), 3.58 (ddd, J = 11.9, 10.5, 4.4, 1H), 1.88 (tq, J = 10.3, 6.7, 1H), 1.40 (dd, J = 14.9, 4.5, 1H), 1.15 (dd, J = 15.1, 11.9, 1H), 1.08 (s, 9H), 1.04 (s, 9H), 0.90 (d, J = 6.7, 3H); 13C NMR (125 MHz, CDCl3) δ 144.6, 135.2, 132.8, 128.9, 128.2, 127.6, 127.2, 127.0, 84.4, 53.0, 45.7, 28.4, 27.6, 22.2, 20.1, 17.6, 16.2; IR (thin film) 3070, 2929, 2856, 1581, 1471, 1387 cm-1; HRMS (GC-MS) m / z calcd for C25H37OSSi (M + H)+ 413.2334, found 413.2320. Anal. Calcd for C25H36OSSi: C, 72.76; H, 8.79. Found: C, 72.48; H, 8.91. Allyl Silane 12b: 1H NMR (500 MHz, CDCl3) δ 7.60–7.28 (m, 5H), 5.48 (dt, J = 15.1, 7.5, 1H), 5.37 (dq, J = 15.0, 6.3, 1H), 1.80 (d, J = 7.6, 2H), 1.67 (d, J = 6.3, 3H), 1.13 (s, 18H); 13C NMR (125 MHz, CDCl3) δ 136.2, 136.1, 128.7, 126.93, 126.87, 125.1, 28.9, 18.3, 18.1; IR (neat) 3072, 2933, 1768, 1583, 1473, 1389 cm-1; HRMS (GC-MS) m / z calcd for C18H34NSSi (M + NH4)+ 324.2181, found 324.2171.
Lithiation of the Sulfide Functionality
Silacyclobutane 15
A procedure reported by Cohen42 was adapted to prepare silacyclobutane 15. A cooled (−78 °C) solution of Li pellets (0.030 g, 4.3 mmol) in THF (1.0 mL) was prepared under an atmosphere of argon. A solution of naphthalene (0.014 g, 0.11 mmol) in THF (1.0 mL) was added, followed by a solution of silacyclobutane 3a (0.24 g, 0.80 mmol) in THF (1.5 mL). After 2 h at −78 °C, Me3SiCl (0.15 mL, 1.2 mmol) was added to the reaction mixture. After 2 h, the reaction mixture was warmed to 22 °C and diluted with pentane (10 mL). An aqueous solution of saturated NH4Cl was added and the layers were separated. The organic layer was washed with H2O and brine, dried over Na2SO4, and concentrated in vacuo. Purification by column chromatography (hexanes) afforded silacyclobutane 15 as a pale yellow oil (0.15 g, 70%): 1H NMR (500 MHz, CDCl3) δ 1.44 (quint, J = 10.8, 1H), 1.10 (s, 9H), 0.99 (s, 9H), 0.89 (d, J = 10.7, 4H), −0.04 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 28.0, 27.7, 20.3, 19.2, 17.3, 8.5, −3.6; IR (neat) 2951, 2858, 1470, 1248, 1115, 829 cm-1; HRMS (GC-MS) m / z calcd for C14H36NSi2 (M + NH4)+ 274.2386, found 274.2388.
Supplementary Material
Acknowledgments
This research was supported by the National Institute of General Medical Sciences of the National Institute of Health (GM-54909). K.M.B. and L.E.B. thank the Department of Education (GAANN) for pre-doctoral fellowships. B.J.A. thanks the National Science Foundation (Chem-SURF) for undergraduate research support. K.A.W. thanks Amgen and Lilly for awards to support research. We thank Dr. P. Dennison (UCI) for assistance with NMR spectroscopy and Dr. J. Greaves and Ms. S. Sorooshian (UCI) for mass spectroscopy.
Footnotes
Supporting Information Available: General experimental information and additional experimental procedures, spectroscopic, and analytical data for the products. This material is available free of charge via the Internet at http://pubs.acs.org.
References and Notes
- 1.Gordon MS, Boatz JA, Walsh RJ. J Phys Chem. 1989;93:1584–1585. [Google Scholar]
- 2.Matsumoto K, Shimazu H, Deguchi M, Yamaoka H. J Polym Sci, Part A: Polym Chem. 1997;35:3207–3216. [Google Scholar]
- 3.Laane J. J Am Chem Soc. 1967;89:1538–1540. [Google Scholar]
- 4.Damrauer R, Davis RA, Burke MT, Karn RA, Goodman GT. J Organomet Chem. 1972;43:117–120. [Google Scholar]
- 5.Damrauer R, Simon R, Laporterie A, Manuel G, Park YT, Weber WP. J Organomet Chem. 1990;391:7–12. [Google Scholar]
- 6.Okada K, Matsumoto K, Oshima K, Utimoto K. Tetrahedron Lett. 1995;36:8067–8070. [Google Scholar]
- 7.Sewald N, Ziche W, Wolff A, Auner N. Organometallics. 1993;12:4123–4134. [Google Scholar]
- 8.Auner N, Heikenwaelder CR, Herrschaft B. Organometallics. 2000;19:2470–2476. [Google Scholar]
- 9.Seyferth D, Haas CK, Annarelli DC. J Organomet Chem. 1973;56:C7–C10. [Google Scholar]
- 10.Tanaka Y, Yamashita H, Tanaka M. Organometallics. 1996;15:1524–1526. [Google Scholar]
- 11.Takeyama Y, Oshima K, Utimoto K. Tetrahedron Lett. 1990;31:6059–6062. [Google Scholar]
- 12.Hirano K, Yorimitsu H, Oshima K. Org Lett. 2008;10:2199–2201. doi: 10.1021/ol800603z. [DOI] [PubMed] [Google Scholar]
- 13.Hirano K, Yorimitsu H, Oshima K. Org Lett. 2006;8:483–485. doi: 10.1021/ol0527577. [DOI] [PubMed] [Google Scholar]
- 14.Ćiraković J, Driver TG, Woerpel KA. J Org Chem. 2004;69:4007–4012. doi: 10.1021/jo0355505. [DOI] [PubMed] [Google Scholar]
- 15.Driver TG, Woerpel KA. J Am Chem Soc. 2003;125:10659–10663. doi: 10.1021/ja0301370. [DOI] [PubMed] [Google Scholar]
- 16.Clark TB, Woerpel KA. J Am Chem Soc. 2004;126:9522–9523. doi: 10.1021/ja047498f. [DOI] [PubMed] [Google Scholar]
- 17.Bourque LE, Cleary PA, Woerpel KA. J Am Chem Soc. 2007;129:12602–12603. doi: 10.1021/ja073758s. [DOI] [PubMed] [Google Scholar]
- 18.Bourque LE, Haile PA, Loy JMN, Woerpel KA. Tetrahedron. 2009;65:5608–5613. doi: 10.1016/j.tet.2009.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sromek AW, Gevorgyan V. Top Curr Chem. 2007;274:77–124. [Google Scholar]
- 20.Silacyclobutanes trans-3e and cis-3e were the only identifiable products of this reaction. Details are provided as Supporting Information.
- 21.Suginome M, Takama A, Ho Y. J Am Chem Soc. 1998;120:1930–1931. and references cited therein. [Google Scholar]
- 22.Ishikawa M, Nakagawa KI, Kumada M. J Organomet Chem. 1981;214:277–288. [Google Scholar]
- 23.Miyake H, Yamamura K. Bull Chem Soc Jpn. 1988;61:3752–3754. [Google Scholar]
- 24.Buchner KM, Clark TB, Loy JMN, Nguyen TX, Woerpel KA. Org Lett. 2009;11:2173–2175. doi: 10.1021/ol900456v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Seyferth D, Duncan DP, Shannon ML. Organometallics. 1984;3:579–583. [Google Scholar]
- 26.Bodnar PM, Palmer WS, Ridgway BH, Shaw JT, Smitrovich JH, Woerpel KA. J Org Chem. 1997;62:4737–4745. [Google Scholar]
- 27.Bourque LE, Woerpel KA. Org Lett. 2008;10:5257–5260. doi: 10.1021/ol8018538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lim YM, Park CH, Yoon SJ, Cho HM, Lee ME, Baeck KK. Organometallics. 2010;29:1355–1361. [Google Scholar]
- 29.Bland JM, Stammer CH. J Org Chem. 1983;48:4393–4394. [Google Scholar]
- 30.Auvray P, Knochel P, Normant JF. Tetrahedron. 1988;44:4495–4508. [Google Scholar]
- 31.Hirabayashi K, Sato H, Kuriyama Y, Matsuo Ji, Sato S, Shimizu T, Kamigata N. Chem Lett. 2007;36:826–827. [Google Scholar]
- 32.Kim JT, Kel'in AV, Gevorgyan V. Angew Chem, Int Ed. 2003;42:98–101. doi: 10.1002/anie.200390064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Peng L, Zhang X, Zhang S, Wang J. J Org Chem. 2007;4:1192–1197. doi: 10.1021/jo0618674. [DOI] [PubMed] [Google Scholar]
- 34.Driver TG, Woerpel KA. J Am Chem Soc. 2004;126:9993–10002. doi: 10.1021/ja0306563. [DOI] [PubMed] [Google Scholar]
- 35.Mayoral JA, Rodríguez-Rodríguez S, Salvatella L. Eur J Org Chem. 2010:1231–1234. [Google Scholar]
- 36.Schmid GH, Fitzgerald PH. J Am Chem Soc. 1971;93:2547–2548. [Google Scholar]
- 37.Allyl silane 12b was isolated in 6% yield when the reaction was performed on preparative scale, although it was present in such small amounts that it was not observed by 1H NMR spectroscopic analysis of the unpurified reaction mixture.
- 38.Franz AK, Woerpel KA. Angew Chem, Int Ed. 2000;39:4295–4299. doi: 10.1002/1521-3773(20001201)39:23<4295::AID-ANIE4295>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 39.Franz AK, Woerpel KA. J Am Chem Soc. 1999;121:949–957. [Google Scholar]
- 40.The thiophenyl silane analogue of compound 14 was observed as a minor product due to rearrangement of the starting material. Details are provided as Supporting Information.
- 41.Screttas CG, Micha-Screttas M. J Org Chem. 1979;44:713–719. [Google Scholar]
- 42.Zhu S, Cohen T. Tetrahedron. 1997;53:17607–17624. [Google Scholar]
- 43.Yus M, Herrera RP, Guijarro A. Chem Eur J. 2002;8:2574–2584. doi: 10.1002/1521-3765(20020603)8:11<2574::AID-CHEM2574>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 44.Screttas CG, Heropoulos GA, Micha-Screttas M, Steele BR, Catsoulacos DP. Tetrahedron Lett. 2003;44:5633–5635. [Google Scholar]
- 45.Streiff S, Ribiero N, Desaubry L. J Org Chem. 2004;69:7592–7598. doi: 10.1021/jo049237u. [DOI] [PubMed] [Google Scholar]
- 46.The lithiation reaction with silacyclobutane 3a suffered from irreproducibility. Attempts to trap the alkyllithium intermediate with a carbon electrophile were unsuccessful, as were attempts to lithiate the sulfide functionality on oxasilacyclohexane 11a.
- 47.Boudjouk P, Samaraweera U, Sooriyakumaran R, Chrusciel J, Anderson KR. Angew Chem, Int Ed Engl. 1988;27:1355–1356. [Google Scholar]
- 48.An NOE enhancement was applied to resolve the splitting of this peak.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.