

Abstract
The BH3-only Bid protein is a critical sentinel of cellular stress in the liver and the hematopoietic system. Bid's initial `claim to fame' came from its ability—as a caspase-truncated product—to trigger the mitochondrial apoptotic program following death receptor activation. Today we know that Bid can response to multiple types of proteases, which are activated under different conditions such as T-cell activation, ischemical reperfusion injury and lysosomal injury. Activation of the mitochondrial apoptotic program by Bid—via its recently identified receptor mitochondrial carrier homolog 2—involves multiple mechanisms, including release of cytochrome c and second mitochondria-derived activator of caspase (Smac), alteration of mitochondrial cristae organization, generation of reactive oxygen species and engagement of the permeability transition pore. Bid is also emerging—in its full-length form—as a pivotal sentinel of DNA damage in the bone marrow regulated by the ataxia telangiectasia mutated (ATM)/ataxia telangiectasia and Rad3-related (ATR) kinases. The ATM/ATR-Bid pathway is critically involved in preserving the quiescence and survival of hematopoietic stem cells both in the absence and presence of external stress, and a large part of this review will be dedicated to recent advances in this area of research.
Keywords: Bid, apoptosis, mitochondria, DNA damage, ATM/ATR
ROLE OF BID IN CELL DEATH AND LIVER INJURY
The Bcl-2 family of proteins function as major regulators of the intrinsic apoptotic pathway.1 Following a death stimulus, the multi-domain pro-apoptotic family members, Bax and Bak form homo and hetero-oligomers to trigger mitochondrial outer-membrane permeabilization (MOMP), allowing the release of inter-membrane space proteins such as cytochrome c and activation of the downstream apoptotic pathway. The BH3-only family members serve as sensors of cellular damage. Post-translational modifications allow these proteins to translocate to the mitochondria, where they activate Bax and Bak. Although Bax and Bak are master regulators of apoptosis, and mice deficient in both of these proteins die in embryogenesis because of failure of essential developmental deaths, the BH3-only proteins have roles that are signal and tissue specific.
Bid is a BH3-only pro-death Bcl-2 family molecule. Bid was first identified in 1996 through expression cloning using Bcl-2 and Bax as baits.2 The ability to interact with the multi-domain pro-death molecules Bax or Bak is a distinguished feature of this molecule among the Bcl-2 family proteins. This feature constitutes the basis of how the BH3-only molecules may induce apoptosis by either inactivating the anti-death molecules and/or directly activating a multi-domain pro-death molecule.
Bid was re-cloned in 1998 during the screening for caspase substrates and confirmed to be cleaved by caspase-8 following Fas-mediated apoptosis3,4 (Figure 1). It was independently shown that tumor necrosis factor alpha (TNFα) stimulation could also trigger Bid cleavage by caspase-8.5 These studies further indicated that cleaved Bid could translocate from the cytosol to the mitochondria and induce the release of cytochrome c from the mitochondria. Together they demonstrated that Bid can bridge the death receptor apoptosis pathway to the mitochondrial apoptosis pathway.
Figure 1.
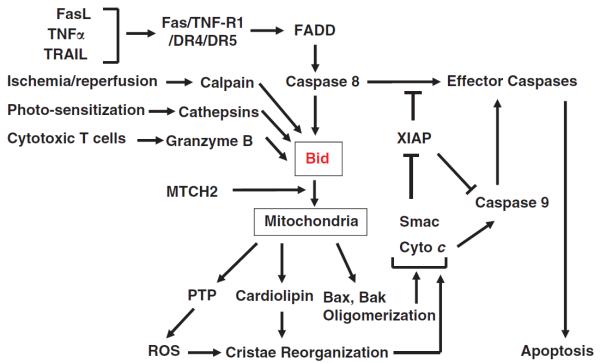
Bid can be activated by multiple proteases in response to a variety of pathophysiological signals. Cleaved Bid is recruited to the mitochondria with the participation of MTCH2, a mitochondrial outer membrane protein that interacts with Bid. Bid can interact with the multi-domain anti-death Bcl-2 family members, Bax and Bak, to induce their oligomerization, leading to the formation of a membrane pore and the release of molecules residing at the intermembrane space. Bid can also interact with cardiolipin, located at the contact site of the outer membranes, to promote cristae reorganization, which is also facilitated by ROS that could be resulted from the activation of the permeability transition pore under the influence of Bid. Cristae reorganization can cause the mobilization of cytochrome c residing at the inner membranes to maximize their release into the cytosol through the Bax/Bak pore. Although released cytochrome c is important for the activation of caspase-9 through the apoptosome, Smac is needed to reverse the suppression of caspase-9 and effector caspases by XIAP. This mitochondrial pathway is particularly critical in death receptor-initiated apoptosis in type II cells, such as hepatocytes, but would also have an important roles in amplifying apoptosis in other scenarios.
Subsequent studies indicated that Bid could actually be cleaved by many other proteases, such as granzyme B, calpains and cathepsins, in the same regulatory α2 loop where caspases act, generating the functional truncated Bid that can induce mitochondrial apoptosis (reviewed in Yin6). These proteases are activated under a variety of pathological conditions, suggesting that Bid is a sentinel of these types of insults and could contribute to the cellular injury in these scenarios. As such Bid serves a critical role in connecting these stimuli to the mitochondria, thus allowing the death process to be either advanced or amplified. In addition, these studies suggest that proteolytic cleavage of Bid may be the most common mechanism for Bid activation, although full-length Bid could trigger apoptosis as well.6,7
The functional role of Bid has been studied in many disease models.6 However, it is most extensively studied in murine models of liver injury triggered by the death receptor agonists, where the best mechanistic insight of how Bid contributes to apoptosis was obtained. Although engagement of the death receptors could trigger apoptosis without the participation of the mitochondria pathway in certain types of cells (that is, type I cells), the latter is required in other kinds of cells (that is, type II cells).8 Although this classification was initially defined in vitro with cancer cell lines, hepatocytes were the first type II cells defined in vivo when it was shown that bid-deficient mice are resistant to anti-Fas antibody-induced liver injury.9 More recently, pancreatic beta-cells were also defined as type II cells.10
In type I cells, engagement of the death receptors by the specific ligands or agonists could trigger caspase-8 activation, which can directly activate the effector caspases to launch a full scale apoptosis. In type II cells, effector caspases cannot be efficiently activated by caspase-8, leading to a halt in the progression of apoptosis. In bid-deficient mice stimulated by anti-Fas antibodies caspase-8 is appropriately activated, but there is no mitochondrial release of apoptogenic factors and the downstream effector caspase-3 is not activated.9,11 It was quickly realized that caspase-3 activation was arrested in bid-deficient hepatocytes in a pattern consistent with being suppressed by X-linked inhibitor of apoptosis protein (XIAP).11 XIAP belongs to the IAP family of anti-apoptosis molecules and can suppress caspase activation by direct binding.12 Such inhibition could be relieved by mitochondria-released second mitochondria-derived activator of caspase (Smac)/DIABLO through its competitive binding to XIAP.13 Indeed, one of the key functions of Bid in activating mitochondria is to trigger the release of mitochondrial Smac/DIABLO, in addition to cytochrome c and other factors.11 The resistance of bid-deficient hepatocytes to anti-Fas antibody-induced apoptosis could be biochemically attributed to the failure of de-repression of XIAP by Smac/DIABLO.11 The same explanation could account for the role of mitochondria in the Fas-activated type II lymphoid cells.14 The genetic and pharmacological evidence was provided by showing that deletion of XIAP or administration of Smac/DIABLO mimetics in bid-deficient mice reversed the sensitivity to Fas-induced apoptosis in both hepatocytes and pancreatic beta-cells.15
Notably, the dependence of Fas-induced hepatocyte apoptosis on Bid could also be bypassed by other means. Co-administration of a high dose of hepatocyte growth factor with anti-Fas antibodies triggered hepatocyte apoptosis in the absence of Bid.16 This is likely due to the binding of hepatocyte growth factor with its receptor, c-Met, which dissociates the interaction of c-Met with Fas. It has been shown that the availability of additional Fas receptors results in a stronger apoptotic response.17 Such a switch from type I to type II cells in response to Fas stimulation could be also observed in hepatocytes under different culture conditions that relate to extracellular matrix–Fas interactions.18 Conversely, stronger stimulation with a hexameric form of sFasL could also engage more Fas leading to a more potent activation of caspases, which kills hepatocytes without the need for Bid.19 Taken together, these studies indicate that Fas-mediated apoptosis in type II cells is subjected to strong inhibition mediated by XIAP, which could be overcome by Bid-mediated mitochondrial release of Smac, or by stronger stimulations at the receptor level via increased ligand–receptor interactions.
Bid is also important for TNFα- and TNF-related apoptosis-inducing ligand (TRAIL)-initiated apoptosis. Bid is cleaved and activated on TNFα/cycloheximide stimulation5 and it promotes TNFα-induced hepatocyte apoptosis in vivo and in vitro.20–22 Similarly, TRAIL-induced mitochondrial activation can also be mediated by Bid, which could be critical to the killing of cancer cells23 and to the synergistic effects of TRAIL and DNA-damaging agents.24 A notable difference seen in TNFα-induced liver injury is that deletion of Bid impedes, but does not prevent TNFα-induced killing.22,25,26 This is likely due to the fact that TNFα can activate several other death effector mechanisms, among which c-Jun N-terminal kinase and Bim, another BH3-only molecule that can be activated by c-Jun N-terminal kinase, turn out to be crucial players.25,27–30 Combined deletion of Bid and Bim could protect mice from lipopolysaccharide/D-galactosamine (LPS/GalN)-induced liver injury, which is mediated by TNFα.27
Under physiological conditions without exogenous stimulation, a small but significant amount of cleaved Bid is found in wild-type mouse livers, which can cause cytochrome c release.31 However, such a condition does not lead to noticeable liver injury unless an anti-apoptotic molecule such as Bcl-xL is deleted, or suppressed by the pharmacological inhibitor ABT-737. Further studies showed that deletion of Mcl-1, another anti-death Bcl-2 family molecule constitutively expressed in the liver, can also result in spontaneous hepatocyte apoptosis.32 Co-deletion of Bid suppressed the spontaneous apoptosis.31 Taken together, these studies indicated that Bid is important to hepatocyte homeostasis under both physiological and pathological conditions.
The impact of Bid on the mitochondria is significant, causing MOMP resulting in the release of many inter-membrane space proteins, including cytochrome c and Smac.3–5,11 In addition, the mitochondrial permeability transition pore is activated21 and reactive oxygen species (ROS) production is enhanced.20 Morphologically, mitochondria could present a significant level of cristae reorganization that could be related to ROS and the interaction of Bid with cardiolipin.20,33–37 All of these events could contribute to the progression of apoptosis.
These effects of Bid are mediated by a series of interactions between Bid and other mitochondrial proteins and between Bid and mitochondrial membrane lipids. The interaction of Bid with the multi-domain pro-death molecules Bax or Bak seems to be the most important for its pro-death effect, which can be antagonized by its interaction with the anti-death Bcl-2 molecules.2,38–40 The BH3 domain of Bid is required for these interactions, and Bax or Bak can become oligomerized to trigger MOMP. Indeed, mice deficient in both Bax and Bak are much like Bid-deficient mice with respect to resistance to anti-Fas antibody-induced hepatocyte apoptosis in vivo.40 Among the other Bid interactors is mitochondrial carrier homolog 2 (MTCH2),41 which facilitates the recruitment of truncated Bid to the mitochondria. Importantly, mice deficient in liver MTCH2 are much like Bid-deficient mice with respect to resistance to anti-Fas antibody-induced hepatocyte apoptosis in vivo.42
As mentioned above, Bid has also been shown to bind to cardiolipin, a phospholipid present only in the mitochondria membranes.33,35,36,43 Overall, Bid–cardiolipin interaction is independent of Bid's BH3 domain,33 but dependent on its α4–α6 helices,44 which are required for Bid membrane insertion.33,43 Such interactions seem to be relevant to Bid-induced cristae reorganization that mobilizes the cytochrome c pool.20,33,34,37 Thus, Bid–cardiolipin interactions seem to be coordinated with the Bid-Bax/Bak interactions that lead to MOMP.
One of the non-pro-death functions of Bid is that it can promote cell proliferation.45,46 Studies done on other Bcl-2 family proteins reveal that pro-death molecules, such as Bax, Bad and Bid, also possess a pro-proliferation function, whereas anti-death molecules, such as Bcl-2 and Bcl-xL, possess an anti-proliferation function.6,47,48 Bcl-2 family members seem to regulate cell proliferation at the G0/G1 cell cycle entry point. For example in hepatocytes, Bid was found to be required for endoplasmic reticulum (ER) calcium regulation of cyclin D1 expression following mitogen stimulation.46 Notably, Bax and Bak are required to control T-cell proliferation through a similar mechanism of ER calcium regulation,48 which was first characterized in an apoptosis scenario.49 On the other hand, Bcl-2 and Bcl-xL have been shown to regulate ER calcium storage in the opposite way,50,51 which would result in an opposite phenotype of delayed cell cycle entry. It has been pointed out that the regulation of ER calcium would result in an impact on either cell death or cell growth depending on whether the triggering signal is apoptogenic or mitogenic. The ability of Bid to regulate proliferation can affect tumor growth as shown in a mouse model of liver cancer initiated by the chemical carcinogen diethylnitrosamine. Under these conditions, deletion of Bid renders a retarded growth of tumors.45 This phenotype is similar to the one seen in mice overexpressing Bcl-2, which would exert an anti-proliferative phenotype.52 The lesson learnt from these studies is that the Bcl-2 family of proteins can affect tumor growth at both the cell death and cell proliferation levels, which could result in different phenotypes depending on the actual context of tissue types and mechanisms of tumorigenesis.
BID INTEGRATES STRESS SIGNALS IN HEMATOPOIESIS
Bid is highly expressed in the hematopoietic system, and the hematopoietic system as well as the liver, represent the best-characterized organs in terms of Bid's role. Hematopoietic homeostasis requires stringent regulation and coordination of cell proliferation, self-renewal and differentiation. Hematopoietic stem cells (HSCs) are responsible for maintaining the blood system over the life of an organism. HSCs exist predominantly in the quiescent state. This quiescent state protects HSCs from genotoxic damage acquired during replication, preserving genomic integrity in the hematopoietic system.
In addition, quiescence maintains HSC function. Quiescent cells are less vulnerable to DNA damage from exogenous toxins as well as the chromatin packed DNA is protected from modification by ROS generated by normal cellular metabolism. In addition, many signals that stimulate entry into the cell cycle, such as mitogen-activated protein kinases, also induce differentiation, thus limiting long-term HSC function.53
Following hematopoietic stress from trauma or toxins, HSCs enter the cell cycle (mobilize) and undergo both proliferation as well as differentiation. It is the committed progenitor populations that then rapidly proliferate to reconstitute the hematopoietic system. These progenitor cells are thus particularly vulnerable to genotoxic stress during replication. Control of cell death in these myeloid progenitor cells (MPCs) is critical: too much cell death of MPCs leads to increased cycling of HSCs. This excessive HSC cycling results in eventual exhaustion and bone marrow failure. Expression of proteins involved in cell cycle checkpoints as well as DNA repair are increased in progenitor cells,54 and increased DNA repair ability is found in these cycling progenitors. Loss of checkpoint control in these progenitor cells leads to bone marrow failure because of increased cycling of stem cells, leading to exhaustion.55,56
Loss of Bid in mice results in abnormal myeloid homeostasis and tumorigenesis.57,58 Initially, Bid −/− mice maintain hematopoietic homeostasis with normal blood counts in all lineages. After 1 year of age, Bid −/− mice display a decrease in blood counts, with anemia and thrombocytopenia. This progresses to an increase in cells of the myeloid lineage, culminating in a disorder that closely resembles chronic myelomonocytic leukemia with evidence of significant chromosomal instability.
Two groups subsequently showed that Bid is a substrate of the DNA damage kinases, ATM and ATR. In addition, Bid has a role in S phase checkpoint regulation following DNA damage.59,60 This cell cycle checkpoint role requires phosphorylation at serine 78, but does not require the BH3 or death domain of Bid.60Bid −/− MPCs display decreased viability in culture.57,58 Cultured Bid −/− activated T cells and MPCs are hypersensitive to hydroxyurea (HU) but not ionizing radiation treatment in vitro.60Bid −/− bone marrow is hypersensitive to intraperitoneal injection of HU but not to ionizing radiation in vivo,57 consistent with an impaired DNA damage response (DDR) to replicative stress. Indeed, Bid −/− MPCs and Bid-deficient U2OS cells demonstrate limited ATR function following HU treatment.57 Furthermore, Bid associates with the ATR/Atrip/replication protein A (RPA) complex,57 and the association between ATR/Atrip and RPA is significantly diminished in Bid-deficient cells following replicative stress,57 suggesting that Bid has a role to maintain the DNA damage sensor complex. Bid is thus positioned to have a role in the DDR to replicative stress in the hematopoietic system.
During hematopoietic stress, it is the progenitor populations that proliferate to expand and to refill the bone marrow. These populations are thus vulnerable to the effects of replicative stress. Bid expression is increased in myeloid progenitor populations, especially in the granulocyte/macrophage progenitors population61 (Figure 2). Given the role of Bid in the ATR-mediated DDR, we investigated the in vivo response of wild-type and Bid −/− LSK cells and committed progenitors to replicative stress. HU depletes the nucleotide pool, stalling the DNA polymerase, and inducing replicative stress and an ATR-mediated DDR. Interestingly, in wild-type mice under conditions of repeated replicative stress, the LSK and MPC cell populations are expanded, and bone marrow function is maintained as demonstrated by the ability to compete with unchallenged bone marrow to adequately repopulate lethally irradiated recipient mice.62Bid −/− MPCs are more sensitive to replicative stress, resulting in depletion of MPCs following HU. HSCs respond to this loss of progenitor cells by mobilizing to enter the cell cycle. Notably, Bid −/− HSCs demonstrate increased cycling and bromodeoxyuridine incorporation. Eventually, Bid −/− HSCs are exhausted, as demonstrated by decreased competitive repopulating ability in serial bone marrow transplant assays.
Figure 2.
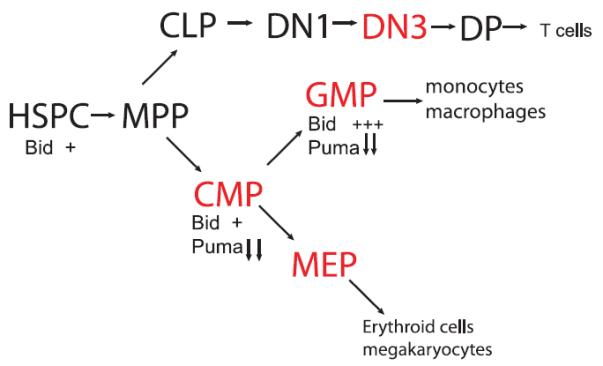
Bid has a role to monitor the DDR in vulnerable hematopoietic cell populations.
Two groups have published competitive bone marrow transplants of Bid −/− bone marrow.58,63 The number of bone marrow cells transplanted was markedly different in the two studies, complicating direct comparison. Nonetheless, both studies demonstrated increased competitive reconstitution of Bid-deficient myeloid cells, and decreased competitive reconstitution of T lymphoid cells. Bid thus appears to have a pro-apoptotic role in mature myeloid cells, and perhaps a survival function in lymphoid cells.
Interestingly, two groups have reported a role for Bid in early thymic development. Bid is downregulated by pre-T cell receptor survival signals.64 Bid is activated by p53 in DN3 cells during the DDR elicited by T cell receptor recombination. Loss of Bid in addition to the pre-T cell receptor, prevents death of DN3 cells. In thymic reconstitution studies, Bid −/− bone marrow reconstitution results in a decreased spleen and thymus size, with a disproportionate percentage of DN3 T cells.63 These results are consistent with decreased death of DN3 T cells. In addition, these studies demonstrate decreased T cells beyond the DN3 stage in the absence of Bid. Although these results are consistent with a defect in T-cell differentiation in Bid −/− thymocytes, the studies do not directly demonstrate decreased differentiative capacity. One alternative possibility to explain these results is that Bid−/− DN3 cells do not activate the S phase checkpoint that is activated by the DDR in wild-type DN3 cells, and therefore expand because of increased proliferation. The expanded Bid−/− DN3 population might not give rise to more mature cells because of death of later T-cell stages from accumulation of increased DNA damage. Further experiments will be necessary to sort out these possibilities.
Bid's role in hematopoietic stem and early progenitor function was evaluated both by immunophenotype as well as long-term reconstitution. Under homeostatic conditions, there is no difference in the number of HSC and progenitor cells.58,63,65Bid mRNA levels are increased in the granulocyte monocyte progenitor population relative to the HSC or common myeloid progenitor populations. Two studies evaluated competitive reconstitution of Bid−/− bone marrow, using very different conditions. With high numbers of transplanted cells (2 million wild-type and 2 million Bid−/− bone marrow cells transplanted), there is a decreased competitive repopulating ability of Bid−/− lymphocytes, and an advantage for Bid−/− myeloid cells.63 When 8000 Bid−/− or Bid+/+ cells were reconstituted at a 1:20 ratio with competing congenic wild-type cells, there was no significant difference in repopulating ability between wild-type and Bid−/− bone marrow for lymphocytes, but a competitive advantage that increased over time for myeloid cells.58
In summary, Bid has an important role in hematopoietic homeostasis, to monitor the DDR to stress. In MPCs that are subject to replicative stress following bone marrow insult, Bid facilitates the DDR to replicative stress mediated by ATR.57,62,66 Loss of Bid results in increased death of MPCs, increased HSC mobilization and eventual exhaustion of bone marrow function. In addition, the surviving bone marrow progenitors display increased propensity to transformation, and the mice develop a malignant, clonal disorder closely resembling chronic myelomonocytic leukemia.58 Bid further monitors the DDR in DN3 T cells, the population that undergoes pre-TCR rearrangement. Loss of Bid leads to accumulation of these DN3 cells, but relative depletion of more mature forms. Further studies will be necessary to determine the effect of loss of Bid on T-cell transformation.
BID IS A CRITICAL `GATE-KEEPER' OF HSC MOBILIZATION
The ATM kinase was previously demonstrated to have a role in regulating the self-renewal and quiescence of HSCs.67 This novel and unexpected role of ATM awaited identification of an ATM effector to provide further support for the importance of this pathway. Previous data demonstrated that Bid is a nuclear ATM effector in the DDR phosphorylated on serines 61 and 78.59 Using BidS61A/S78A (BidAA) knock in mice it was revealed that ATM-mediated Bid phosphorylation has an unexpected role in maintaining the quiescence of HSCs in the absence of hematopoietic stress from trauma or toxins.65 Loss of Bid phosphorylation leads to escape from quiescence of HSCs, resulting in exhaustion of the HSC pool and a marked reduction of HSC repopulating potential in vivo. Bid phosphorylation also has a role in protecting HSCs from irradiation, and regulating both quiescence and survival of HSCs depends on Bid's ability to regulate oxidative stress at the mitochondria.
Mammalian HSCs are kept quiescent in the endosteal niche, a hypoxic zone of the bone marrow, and redox signaling has emerged as an important regulator of HSC self-renewal and mobilization.68–71 Previously it was demonstrated that ATM-deficient HSCs accumulate elevated levels of ROS, and treating the null mice with the anti-oxidant N-acetyl-l-cystein resulted in complete reversal of the phenotype.67,72 Importantly, treatment of BidAA mice with N-acetyl-l-cystein restored HSC quiescence and the number of HSCs to wild-type levels.65 Thus, the elevated ROS present in BidAA HSCs is the primary cause of the defect in quiescence. Taken together, these studies are consistent with the idea that the ATM–Bid pathway regulates the self-renewal/quiescence of HSCs via regulation of oxidative stress.
Identifying Bid as an ATM effector in both the DDR and the self-renewal/quiescence of HSCs suggests that these two pathways are related. A hallmark defect in the ATM pathway is increased radiation sensitivity,73–75 and this hypersensitivity is believed to be due to defects in the DDR (defects in cell cycle arrest, DNA repair, and so on). The findings demonstrating that N-acetyl-l-cystein injection both rescues BidAA HSC quiescence and partially protects BidAA mice from total body irradiation65 are consistent with the idea that defects in HSC quiescence contribute to hypersensitivity to radiation. For example, a defect in self-renewal/quiescence of HSCs may result in a DDR defect as HSCs in cycle are more sensitive to stress.76 In support of this hypothesis, ATM−/− mice treated with N-acetyl-l-cystein are also partially protected from radiation.77
How does phosphorylated Bid regulate ROS levels and the cell cycle status of HSCs? Loss of Bid phosphorylation or ATM knockout leads to an increase in mitochondrial Bid, which correlates with an increase in mitochondrial ROS (mitoROS) in vivo.65 Importantly, loss of Bid phosphorylation also triggers an oxidative stress-dependent upregulation of genes involved in the transition from early to late G1 (R point; for example, cyclin D1 and CDK4), and genes involved in the G1/S transition (for example, cyclin E1). Quiescent HSCs presumably re-enter the cell cycle in the early G1 phase,53 and thus keeping in check the proteins promoting R point transition is likely to be important for maintaining HSC quiescence. Thus, Bid accumulation at the mitochondria, which is negatively regulated by ATM, triggers a metabolic change in mitochondria that includes an increase in ROS and perhaps changes in other metabolites that signal back to the nucleus to regulate gene transcription leading to cell cycle progression (Figure 3).
Figure 3.
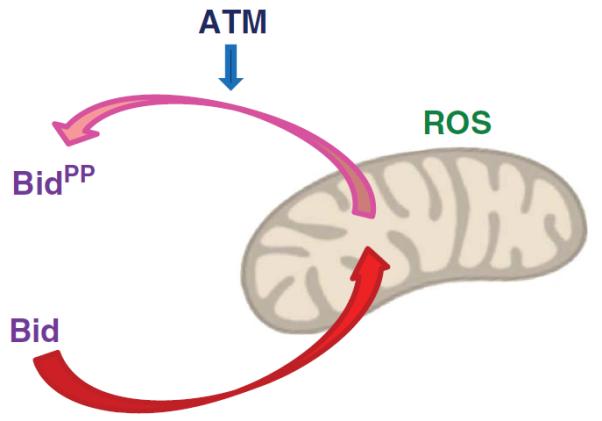
Bid accumulation at the mitochondria, which is negatively regulated by ATM, triggers a metabolic change in mitochondria that includes an increase in ROS.
Bid accumulation at the mitochondria also leads to activation of the mitochondrial cell death program through activation of Bax/Bak and MOMP. In addition, Bid is upregulated in granulocyte/macrophage progenitor cells.61 Thus, it is also possible that Bid's apoptotic role is activated in hematopoietic progenitor cells of BidAA mice. This would lead to increased cell death of progenitor cells and compensatory increased mobilization of HSCs, and would contribute to the HSC exhaustion phenotype. As granulocyte/macrophage progenitor cells have an important role in maintaining viability following irradiation,54 increased death of granulocyte/macrophage progenitors, mediated by increased production of ROS in the BidAA mice may also contribute to the decreased viability of the mice.
Interestingly, exposing wild-type mice to irradiation also triggers an increase in mitochondrial Bid (which includes phosphorylated Bid) and an increase in mitoROS,65 suggesting that the ATM–Bid complex responds as a metabolic rheostat to changes in DNA damage levels. Taken together, these results are consistent with the idea that at low levels of DNA damage ATM phosphorylates Bid to keep it away from the mitochondria resulting in low levels of ROS. Increasing the levels of DNA damage proportionally increases the levels of mitochondrial Bid, which leads to a proportional increase in mitoROS. This increase in mitoROS might be the signal for HSCs to exit quiescence and enter the cell cycle to replenish the blood system.
Surprisingly, Bid−/− mice display similar phenotypes to Bid+/+ and not to the BidAA mice.65 In Bid−/− mice, Bid would not translocate to the mitochondria to induce ROS and thus these mice are expected not to have the same phenotypes as the BidAA mice. The differential sensitivity between these mice in response to DNA damage supports the idea that the increased baseline ROS level per se rather than Bid itself dictates the sensitivity to DNA damage.
It is well established that ATM is a nuclear kinase and it was previously demonstrated that part of the Bid protein pool is localized to the nucleus.59,60 Thus, it is most likely that ATM phosphorylates Bid in the nucleus and that this phosphorylation inhibits Bid nuclear exit and thereby its accumulation in mitochondria. However, most recently, it was demonstrated that ATM is also localized to mitochondria.78 Thus, an alternative possibility is that ATM phosphorylates Bid at the mitochondria leading to its retrotranslocation into the cytosol, as previously shown for the Bcl-xL protein that retrotranslocates pro-apoptotic Bax from the mitochondria into the cytosol.79 As in the case of Bcl-xL and Bax where loss of survival signals leads to inhibition of Bax removal into the cytosol and to its accumulation in mitochondria, increasing the levels of DNA damage may inhibit the removal of phosphorylated Bid into the cytosol and to its accumulation in mitochondria.
How does the ATM–Bid complex regulate the metabolic status/ROS of mitochondria? As mentioned above, MTCH2 serves as a mitochondrial receptor for Bid important for Fas-induced liver apoptosis.42 Interestingly, MTCH2 was previously identified in a genome-wide association study as one of six new gene loci associated with body mass index (BMI) in humans.80 Body mass index is the most commonly used quantitative measure of adiposity, and adults with high values of body mass index are termed obese. Thus, MTCH2 may also be involved in fatty acid/glucose metabolism in mitochondria. It remains an open and exciting avenue for future research how MTCH2's metabolic activity is related to Bid's role in regulating the quiescence of HSCs. Finally, ATM-deficiency worsens features of the metabolic syndrome in ApoE−/−, Ob/Ob and Db/Db mice,81 and cells from ATM−/− mice and from ataxia telangiectasia patients show increased mitoROS.78 Thus, ATM may regulate the metabolic status of mitochondria via the Bid–MTCH2 complex.
ACKNOWLEDGEMENTS
AG is supported by the Israel Science Foundation, USA-Israel Binational Science Foundation and the German-Israel Foundation, and is the incumbent of the Armour Family Career Development Chair of Cancer Research.
Footnotes
CONFLICT OF INTEREST The authors declare no conflict of interest.
REFERENCES
- 1.Danial NN, Korsmeyer SJ. Cell death: critical control points. Cell. 2004;116:205–219. doi: 10.1016/s0092-8674(04)00046-7. [DOI] [PubMed] [Google Scholar]
- 2.Wang K, Yin XM, Chao DT, Milliman CL, Korsmeyer SJ. BID: a novel BH3 domain-only death agonist. Genes Dev. 1996;10:2859–2869. doi: 10.1101/gad.10.22.2859. [DOI] [PubMed] [Google Scholar]
- 3.Li H, Zhu H, Xu CJ, Yuan J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell. 1998;94:491–501. doi: 10.1016/s0092-8674(00)81590-1. [DOI] [PubMed] [Google Scholar]
- 4.Luo X, Budihardjo I, Zou H, Slaughter C, Wang X. Bid a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell. 1998;94:481–490. doi: 10.1016/s0092-8674(00)81589-5. [DOI] [PubMed] [Google Scholar]
- 5.Gross A, Yin XM, Wang K, Wei MC, Jockel J, Milliman C, et al. Caspase cleaved BID targets mitochondria and is required for cytochrome c release, while BCL-XL prevents this release but not tumor necrosis factor-R1/Fas death. J Biol Chem. 1999;274:1156–1163. doi: 10.1074/jbc.274.2.1156. [DOI] [PubMed] [Google Scholar]
- 6.Yin XM. Bid, a BH3-only multi-functional molecule, is at the cross road of life and death. Gene. 2006;369:7–19. doi: 10.1016/j.gene.2005.10.038. [DOI] [PubMed] [Google Scholar]
- 7.Sarig R, Zaltsman Y, Marcellus RC, Flavell R, Mak TW, Gross A. BID-D59A is a potent inducer of apoptosis in primary embryonic fibroblasts. J Biol Chem. 2003;278:10707–10715. doi: 10.1074/jbc.M210296200. [DOI] [PubMed] [Google Scholar]
- 8.Scaffidi C, Fulda S, Srinivasan A, Friesen C, Li F, Tomaselli KJ, et al. Two CD95 (APO-1/Fas) signaling pathways. Embo J. 1998;17:1675–1687. doi: 10.1093/emboj/17.6.1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yin XM, Wang K, Gross A, Zhao Y, Zinkel S, Klocke B, et al. Bid-deficient mice are resistant to Fas-induced hepatocellular apoptosis. Nature. 1999;400:886–891. doi: 10.1038/23730. [DOI] [PubMed] [Google Scholar]
- 10.McKenzie MD, Carrington EM, Kaufmann T, Strasser A, Huang DC, Kay TW, et al. Proapoptotic BH3-only protein Bid is essential for death receptor-induced apoptosis of pancreatic beta-cells. Diabetes. 2008;57:1284–1292. doi: 10.2337/db07-1692. [DOI] [PubMed] [Google Scholar]
- 11.Li S, Zhao Y, He X, Kim T-H, Kuharsky DK, Rabinowich H, et al. Relief of extrinsic pathway inhibition by the Bid-dependent mitochondrial release of Smac in Fas-mediated hepatocyte apoptosis. J Biol Chem. 2002;277:26912–26920. doi: 10.1074/jbc.M200726200. [DOI] [PubMed] [Google Scholar]
- 12.Eckelman BP, Salvesen GS, Scott FL. Human inhibitor of apoptosis proteins: why XIAP is the black sheep of the family. EMBO Rep. 2006;7:988–994. doi: 10.1038/sj.embor.7400795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shi Y. A conserved tetrapeptide motif: potentiating apoptosis through IAP-binding. Cell Death Differ. 2002;9:93–95. doi: 10.1038/sj.cdd.4400957. [DOI] [PubMed] [Google Scholar]
- 14.Sun XM, Bratton SB, Butterworth M, MacFarlane M, Cohen GM. Bcl-2 and Bcl-xL inhibit CD95-mediated apoptosis by preventing mitochondrial release of Smac/DIABLO and subsequent inactivation of X-linked inhibitor-of-apoptosis protein. J Biol Chem. 2002;277:11345–11351. doi: 10.1074/jbc.M109893200. [DOI] [PubMed] [Google Scholar]
- 15.Jost PJ, Grabow S, Gray D, McKenzie MD, Nachbur U, Huang DC, et al. XIAP discriminates between type I and type II FAS-induced apoptosis. Nature. 2009;460:1035–1039. doi: 10.1038/nature08229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhao Y, Difrancesca D, Wang X, Zarnegar R, Michalopoulos GK, Yin XM. Promotion of Fas-mediated apoptosis in type II cells by high doses of hepatocyte growth factor bypasses the mitochondrial requirement. J Cell Physiol. 2007;213:556–563. doi: 10.1002/jcp.21136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang X, DeFrances MC, Dai Y, Pediaditakis P, Johnson C, Bell A, et al. A mechanism of cell survival: sequestration of Fas by the HGF receptor Met. Mol Cell. 2002;9:411–421. doi: 10.1016/s1097-2765(02)00439-2. [DOI] [PubMed] [Google Scholar]
- 18.Walter D, Schmich K, Vogel S, Pick R, Kaufmann T, Hochmuth FC, et al. Switch from type II to I Fas/CD95 death signaling on in vitro culturing of primary hepatocytes. Hepatology. 2008;48:1942–1953. doi: 10.1002/hep.22541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schungel S, Buitrago-Molina LE, Nalapareddy P, Lebofsky M, Manns MP, Jaeschke H, et al. The strength of the Fas ligand signal determines whether hepatocytes act as type 1 or type 2 cells in murine livers. Hepatology. 2009;50:1558–1566. doi: 10.1002/hep.23176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ding WX, Ni HM, DiFrancesca D, Stolz DB, Yin XM. Bid-dependent generation of oxygen radicals promotes death receptor activation-induced apoptosis in murine hepatocytes. Hepatology. 2004;40:403–413. doi: 10.1002/hep.20310. [DOI] [PubMed] [Google Scholar]
- 21.Zhao Y, Ding WX, Qian T, Watkins S, Lemasters JJ, Yin XM. Bid activates multiple mitochondrial apoptotic mechanisms in primary hepatocytes after death receptor engagement. Gastroenterology. 2003;125:854–867. doi: 10.1016/s0016-5085(03)01066-7. [DOI] [PubMed] [Google Scholar]
- 22.Zhao Y, Li S, Childs EE, Kuharsky DK, Yin X-M. Activation of pro-death Bcl-2 family proteins and mitochondria apoptosis pathway in tumor necrosis factor-alpha-induced liver injury. J Biol Chem. 2001;276:27432–27440. doi: 10.1074/jbc.M102465200. [DOI] [PubMed] [Google Scholar]
- 23.Deng Y, Lin Y, Wu X. TRAIL-induced apoptosis requires Bax-dependent mitochondrial release of Smac/DIABLO. Genes Dev. 2002;16:33–45. doi: 10.1101/gad.949602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Broaddus VC, Dansen TB, Abayasiriwardana KS, Wilson SM, Finch AJ, Swigart LB, et al. Bid mediates apoptotic synergy between tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) and DNA damage. J Biol Chem. 2005;280:12486–12493. doi: 10.1074/jbc.M408190200. [DOI] [PubMed] [Google Scholar]
- 25.Chen X, Ding WX, Ni HM, Gao W, Shi YH, Gambotto AA, et al. Bid-independent mitochondrial activation in tumor necrosis factor alpha-induced apoptosis and liver injury. Mol Cell Biol. 2007;27:541–553. doi: 10.1128/MCB.01166-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kaufmann T, Tai L, Ekert PG, Huang DC, Norris F, Lindemann RK, et al. The BH3-only protein bid is dispensable for DNA damage- and replicative stress-induced apoptosis or cell-cycle arrest. Cell. 2007;129:423–433. doi: 10.1016/j.cell.2007.03.017. [DOI] [PubMed] [Google Scholar]
- 27.Kaufmann T, Jost PJ, Pellegrini M, Puthalakath H, Gugasyan R, Gerondakis S, et al. Fatal hepatitis mediated by tumor necrosis factor TNFalpha requires caspase-8 and involves the BH3-only proteins Bid and Bim. Immunity. 2009;30:56–66. doi: 10.1016/j.immuni.2008.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ni HM, Chen X, Ding WX, Schuchmann M, Yin XM. Differential roles of JNK in ConA/GalN and ConA-induced liver injury in mice. Am J Pathol. 2008;173:962–972. doi: 10.2353/ajpath.2008.080358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ni HM, Chen X, Shi YH, Liao Y, Beg AA, Fan J, et al. Genetic delineation of the pathways mediated by bid and JNK in tumor necrosis factor-alpha-induced liver injury in adult and embryonic mice. J Biol Chem. 2009;284:4373–4382. doi: 10.1074/jbc.M807259200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schmich K, Schlatter R, Corazza N, Sa Ferreira K, Ederer M, Brunner T, et al. Tumor necrosis factor alpha sensitizes primary murine hepatocytes to Fas/CD95-induced apoptosis in a Bim- and Bid-dependent manner. Hepatology. 2011;53:282–292. doi: 10.1002/hep.23987. [DOI] [PubMed] [Google Scholar]
- 31.Hikita H, Takehara T, Kodama T, Shimizu S, Hosui A, Miyagi T, et al. BH3-only protein bid participates in the Bcl-2 network in healthy liver cells. Hepatology. 2009;50:1972–1980. doi: 10.1002/hep.23207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hikita H, Takehara T, Shimizu S, Kodama T, Li W, Miyagi T, et al. Mcl-1 and Bcl-xL cooperatively maintain integrity of hepatocytes in developing and adult murine liver. Hepatology. 2009;50:1217–1226. doi: 10.1002/hep.23126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim TH, Zhao Y, Ding WX, Shin JN, He X, Seo YW, et al. Bid-cardiolipin interaction at mitochondrial contact site contributes to mitochondrial cristae reorganization and cytochrome C release. Mol Biol Cell. 2004;15:3061–3072. doi: 10.1091/mbc.E03-12-0864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Scorrano L, Ashiya M, Buttle K, Weiler S, Oakes SA, Mannella CA, et al. A distinct pathway remodels mitochondrial cristae and mobilizes cytochrome c during apoptosis. Dev Cell. 2002;2:55–67. doi: 10.1016/s1534-5807(01)00116-2. [DOI] [PubMed] [Google Scholar]
- 35.Kuwana T, Mackey MR, Perkins G, Ellisman MH, Latterich M, Schneiter R, et al. Bid, Bax, and lipids cooperate to form supramolecular openings in the outer mitochondrial membrane. Cell. 2002;111:331–342. doi: 10.1016/s0092-8674(02)01036-x. [DOI] [PubMed] [Google Scholar]
- 36.Ott M, Zhivotovsky B, Orrenius S. Role of cardiolipin in cytochrome c release from mitochondria. Cell Death Differ. 2007;14:1243–1247. doi: 10.1038/sj.cdd.4402135. [DOI] [PubMed] [Google Scholar]
- 37.Yamaguchi R, Lartigue L, Perkins G, Scott RT, Dixit A, Kushnareva Y, et al. Opa1-mediated cristae opening is Bax/Bak and BH3 dependent, required for apoptosis, and independent of Bak oligomerization. Mol Cell. 2008;31:557–569. doi: 10.1016/j.molcel.2008.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cheng EH, Wei MC, Weiler S, Flavell RA, Mak TW, Lindsten T, et al. BCL-2, BCL-X(L) sequester BH3 domain-only molecules preventing BAX- and BAK-mediated mitochondrial apoptosis. Mol Cell. 2001;8:705–711. doi: 10.1016/s1097-2765(01)00320-3. [DOI] [PubMed] [Google Scholar]
- 39.Desagher S, Osen-Sand A, Nichols A, Eskes R, Montessuit S, Lauper S, et al. Bid-induced conformational change of Bax is responsible for mitochondrial cytochrome c release during apoptosis. J Cell Biol. 1999;144:891–901. doi: 10.1083/jcb.144.5.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wei MC, Lindsten T, Mootha VK, Weiler S, Gross A, Ashiya M, et al. tBID, a membrane-targeted death ligand, oligomerizes BAK to release cytochrome c. Genes Dev. 2000;14:2060–2071. [PMC free article] [PubMed] [Google Scholar]
- 41.Grinberg M, Schwarz M, Zaltsman Y, Eini T, Niv H, Pietrokovski S, et al. Mitochondrial carrier homolog 2 is a target of tBID in cells signaled to die by tumor necrosis factor alpha. Mol Cell Biol. 2005;25:4579–4590. doi: 10.1128/MCB.25.11.4579-4590.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zaltsman Y, Shachnai L, Yivgi-Ohana N, Schwarz M, Maryanovich M, Houtkooper RH, et al. MTCH2/MIMP is a major facilitator of tBID recruitment to mitochondria. Nat Cell Biol. 2010;12:553–562. doi: 10.1038/ncb2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lutter M, Fang M, Luo X, Nishijima M, Xie X, Wang X. Cardiolipin provides specificity for targeting of tBid to mitochondria. Nat Cell Biol. 2000;2:754–761. doi: 10.1038/35036395. [DOI] [PubMed] [Google Scholar]
- 44.Liu J, Weiss A, Durrant D, Chi NW, Lee RM. The cardiolipin-binding domain of Bid affects mitochondrial respiration and enhances cytochrome c release. Apoptosis. 2004;9:533–541. doi: 10.1023/B:APPT.0000038034.16230.ea. [DOI] [PubMed] [Google Scholar]
- 45.Bai L, Ni HM, Chen X, DiFrancesca D, Yin XM. Deletion of Bid impedes cell proliferation and hepatic carcinogenesis. Am J Pathol. 2005;166:1523–1532. doi: 10.1016/S0002-9440(10)62368-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ni HM, Baty CJ, Li N, Ding WX, Gao W, Li M, et al. Bid agonist regulates murine hepatocyte proliferation by controlling endoplasmic reticulum calcium homeostasis. Hepatology. 2010;52:338–348. doi: 10.1002/hep.23672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cory S, Huang DC, Adams JM. The Bcl-2 family: roles in cell survival and oncogenesis. Oncogene. 2003;22:8590–8607. doi: 10.1038/sj.onc.1207102. [DOI] [PubMed] [Google Scholar]
- 48.Jones RG, Bui T, White C, Madesh M, Krawczyk CM, Lindsten T, et al. The proapoptotic factors Bax and Bak regulate T cell proliferation through control of endoplasmic reticulum Ca(2+) homeostasis. Immunity. 2007;27:268–280. doi: 10.1016/j.immuni.2007.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Scorrano L, Oakes SA, Opferman JT, Cheng EH, Sorcinelli MD, Pozzan T, et al. BAX and BAK regulation of endoplasmic reticulum Ca2+: a control point for apoptosis. Science. 2003;300:135–139. doi: 10.1126/science.1081208. [DOI] [PubMed] [Google Scholar]
- 50.Oakes SA, Scorrano L, Opferman JT, Bassik MC, Nishino M, Pozzan T, et al. Proapoptotic BAX and BAK regulate the type 1 inositol trisphosphate receptor and calcium leak from the endoplasmic reticulum. Proc Natl Acad Sci USA. 2005;102:105–110. doi: 10.1073/pnas.0408352102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.White C, Li C, Yang J, Petrenko NB, Madesh M, Thompson CB, et al. The endoplasmic reticulum gateway to apoptosis by Bcl-X(L) modulation of the InsP3R. Nat Cell Biol. 2005;7:1021–1028. doi: 10.1038/ncb1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vail ME, Chaisson ML, Thompson J, Fausto N. Bcl-2 expression delays hepatocyte cell cycle progression during liver regeneration. Oncogene. 2002;21:1548–1555. doi: 10.1038/sj.onc.1205212. [DOI] [PubMed] [Google Scholar]
- 53.Orford KW, Scadden DT. Deconstructing stem cell self-renewal: genetic insights into cell-cycle regulation. Nat Rev Genet. 2008;9:115–128. doi: 10.1038/nrg2269. [DOI] [PubMed] [Google Scholar]
- 54.Akashi K, He X, Chen J, Iwasaki H, Niu C, Steenhard B, et al. Transcriptional accessibility for genes of multiple tissues and hematopoietic lineages is hierarchically controlled during early hematopoiesis. Blood. 2003;101:383–389. doi: 10.1182/blood-2002-06-1780. [DOI] [PubMed] [Google Scholar]
- 55.Cheng T, Rodrigues N, Shen H, Yang Y, Dombkowski D, Sykes M, et al. Hematopoietic stem cell quiescence maintained by p21cip1/waf1. Science. 2000;287:1804–1808. doi: 10.1126/science.287.5459.1804. [DOI] [PubMed] [Google Scholar]
- 56.Hock H, Hamblen MJ, Rooke HM, Schindler JW, Saleque S, Fujiwara Y, et al. Gfi-1 restricts proliferation and preserves functional integrity of haematopoietic stem cells. Nature. 2004;431:1002–1007. doi: 10.1038/nature02994. [DOI] [PubMed] [Google Scholar]
- 57.Liu Y, Bertram CC, Shi Q, Zinkel SS. Proapoptotic Bid mediates the Atr-directed DNA damage response to replicative stress. Cell Death Differ. 2011;18:841–852. doi: 10.1038/cdd.2010.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zinkel SS, Ong CC, Ferguson DO, Iwasaki H, Akashi K, Bronson RT, et al. Proapoptotic BID is required for myeloid homeostasis and tumor suppression. Genes Dev. 2003;17:229–239. doi: 10.1101/gad.1045603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kamer I, Sarig R, Zaltsman Y, Niv H, Oberkovitz G, Regev L, et al. Proapoptotic BID is an ATM effector in the DNA-damage response. Cell. 2005;122:593–603. doi: 10.1016/j.cell.2005.06.014. [DOI] [PubMed] [Google Scholar]
- 60.Zinkel SS, Hurov KE, Ong C, Abtahi FM, Gross A, Korsmeyer SJ. A role for proapoptotic BID in the DNA-damage response. Cell. 2005;122:579–591. doi: 10.1016/j.cell.2005.06.022. [DOI] [PubMed] [Google Scholar]
- 61.Mohrin M, Bourke E, Alexander D, Warr MR, Barry-Holson K, Le Beau MM, et al. Hematopoietic stem cell quiescence promotes error-prone DNA repair and mutagenesis. Cell Stem Cell. 2010;7:174–185. doi: 10.1016/j.stem.2010.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liu Y, Aiello A, Zinkel SS. Bid protects the mouse hematopoietic system following hydroxyurea-induced replicative stress. Cell Death Differ. 2012;19:1602–1612. doi: 10.1038/cdd.2012.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shen H, Yu H, Liang PH, Xufeng R, Song Y, Hu X, et al. Bid is a positive regulator for donor-derived lymphoid cell regeneration in gamma-irradiated recipients. Exp Hematol. 2011;39:947–57. e1. doi: 10.1016/j.exphem.2011.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mandal M, Crusio KM, Meng F, Liu S, Kinsella M, Clark MR, et al. Regulation of lymphocyte progenitor survival by the proapoptotic activities of Bim and Bid. Proc Natl Acad Sci USA. 2008;105:20840–20845. doi: 10.1073/pnas.0807557106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Maryanovich M, Oberkovitz G, Niv H, Vorobiyov L, Zaltsman Y, Brenner O, et al. The ATM-BID pathway regulates the quiescence and survival of haematopoietic stem cells. Nat Cell Biol. 2012;14:535–541. doi: 10.1038/ncb2468. [DOI] [PubMed] [Google Scholar]
- 66.Liu Y, Vaithiyalingam S, Shi Q, Chazin WJ, Zinkel SS. BID binds to replication protein A and stimulates ATR function following replicative stress. Mol Cell Biol. 2011;31:4298–4309. doi: 10.1128/MCB.05737-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ito K, Hirao A, Arai F, Matsuoka S, Takubo K, Hamaguchi I, et al. Regulation of oxidative stress by ATM is required for self-renewal of haematopoietic stem cells. Nature. 2004;431:997–1002. doi: 10.1038/nature02989. [DOI] [PubMed] [Google Scholar]
- 68.Jang YY, Sharkis SJ. A low level of reactive oxygen species selects for primitive hematopoietic stem cells that may reside in the low-oxygenic niche. Blood. 2007;110:3056–3063. doi: 10.1182/blood-2007-05-087759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Parmar K, Mauch P, Vergilio JA, Sackstein R, Down JD. Distribution of hematopoietic stem cells in the bone marrow according to regional hypoxia. Proc Natl Acad Sci USA. 2007;104:5431–5436. doi: 10.1073/pnas.0701152104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tesio M, Golan K, Corso S, Giordano S, Schajnovitz A, Vagima Y, et al. Enhanced c-Met activity promotes G-CSF-induced mobilization of hematopoietic progenitor cells via ROS signaling. Blood. 2011;117:419–428. doi: 10.1182/blood-2009-06-230359. [DOI] [PubMed] [Google Scholar]
- 71.Juntilla MM, Patil VD, Calamito M, Joshi RP, Birnbaum MJ, Koretzky GA. AKT1 and AKT2 maintain hematopoietic stem cell function by regulating reactive oxygen species. Blood. 2010;115:4030–4038. doi: 10.1182/blood-2009-09-241000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ito K, Hirao A, Arai F, Takubo K, Matsuoka S, Miyamoto K, et al. Reactive oxygen species act through p38 MAPK to limit the lifespan of hematopoietic stem cells. Nat Med. 2006;12:446–451. doi: 10.1038/nm1388. [DOI] [PubMed] [Google Scholar]
- 73.Barlow C, Hirotsune S, Paylor R, Liyanage M, Eckhaus M, Collins F, et al. Atm-deficient mice: a paradigm of ataxia telangiectasia. Cell. 1996;86:159–171. doi: 10.1016/s0092-8674(00)80086-0. [DOI] [PubMed] [Google Scholar]
- 74.Celeste A, Petersen S, Romanienko PJ, Fernandez-Capetillo O, Chen HT, Sedelnikova OA, et al. Genomic instability in mice lacking histone H2AX. Science. 2002;296:922–927. doi: 10.1126/science.1069398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ward IM, Minn K, van Deursen J, Chen J. p53 Binding protein 53BP1 is required for DNA damage responses and tumor suppression in mice. Mol Cell Biol. 2003;23:2556–2563. doi: 10.1128/MCB.23.7.2556-2563.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sugiyama T, Kohara H, Noda M, Nagasawa T. Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity. 2006;25:977–988. doi: 10.1016/j.immuni.2006.10.016. [DOI] [PubMed] [Google Scholar]
- 77.Ito K, Takubo K, Arai F, Satoh H, Matsuoka S, Ohmura M, et al. Regulation of reactive oxygen species by Atm is essential for proper response to DNA double-strand breaks in lymphocytes. J Immunol. 2007;178:103–110. doi: 10.4049/jimmunol.178.1.103. [DOI] [PubMed] [Google Scholar]
- 78.Valentin-Vega YA, Maclean KH, Tait-Mulder J, Milasta S, Steeves M, Dorsey FC, et al. Mitochondrial dysfunction in ataxia telangiectasia. Blood. 2012;119:1490–1500. doi: 10.1182/blood-2011-08-373639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Edlich F, Banerjee S, Suzuki M, Cleland MM, Arnoult D, Wang C, et al. Bcl-x(L) retrotranslocates Bax from the mitochondria into the cytosol. Cell. 2011;145:104–116. doi: 10.1016/j.cell.2011.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Willer CJ, Speliotes EK, Loos RJ, Li S, Lindgren CM, Heid IM, et al. Six new loci associated with body mass index highlight a neuronal influence on body weight regulation. Nat Genet. 2009;41:25–34. doi: 10.1038/ng.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Schneider JG, Finck BN, Ren J, Standley KN, Takagi M, Maclean KH, et al. ATM-dependent suppression of stress signaling reduces vascular disease in metabolic syndrome. Cell Metab. 2006;4:377–389. doi: 10.1016/j.cmet.2006.10.002. [DOI] [PubMed] [Google Scholar]