

Metal catalyzed reductive couplings of π-unsaturated reactants bypass use of premetallated C-nucleophiles in a range of carbonyl and imine additions.[1] Despite significant advances, catalytic intermolecular reductive couplings of simple alkenes or alkynes to ketones (activated or unactivated) remain uncommon.[2,3,4,5,6,7] In connection with efforts aimed at the development of hydrogen-mediated reductive couplings beyond hydroformylation,[1f,g,h] rhodium or iridium catalyzed hydrogenations of α-ketoesters in the presence of conjugated or nonconjugated alkynes, respectively, were found to promote formation of α-hydroxyesters (Figure 1).[6b,c,7] More recently, under the conditions of ruthenium(0) catalyzed transfer hydro-genation, it was found that activated secondary alcohols engage in C-C coupling to unsaturated reactants to form products of hydrohydroxyalkylation.[8,9,10,11] Specifically, the catalytic C-C coupling of α-hydroxy carbonyl compounds[8] or 1,2-diols[8d,f] with conjugated dienes,[8a,b,d,e] terminal olefins[8c] and α,β-unsaturated esters[8f] was achieved. The feasibility of coupling secondary alcohols to alkynes[6,7,9,12] under the conditions of ruthenium(0) catalyzed transfer hydrogenation was unclear, as ruthenium(0) complexes are efficient catalysts for alkyne [2+2+2]cycloaddition.[13,14] Here, we report that the ruthenium(0) catalyst formed in situ from Ru3(CO)12 and tricyclohexylphosphine, PCy3, promotes regio- and stereoselective alkyne-diol hydrohydroxyalkylation to form α-hydroxy-β,γ-unsaturated ketones in good to excellent yields (Figure 1). The reaction products engage in acid catalyzed cyclodehydration to form tetrasubstituted furans.[15]
Figure 1.
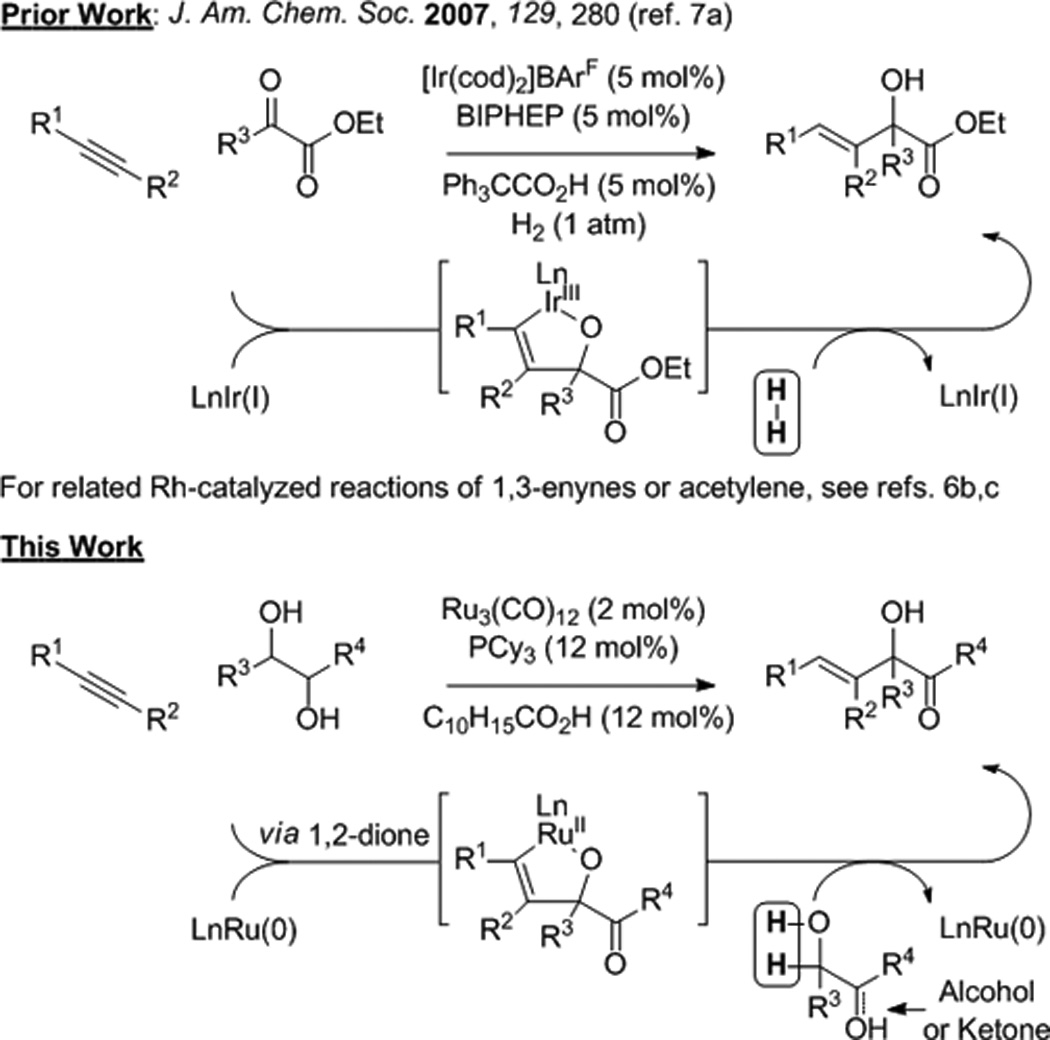
Hydrogen-mediated reductive coupling of alkynes to activated ketones and related hydrohydroxyalkylations.
In a preliminary experiment, racemic trans-1,2-cyclohexane diol 1b was exposed to 1-phenyl-1-propyne 2a under conditions established for the C-C coupling of dienes to α-hydroxy esters or α-hydroxy amides.[8a,b] The anticipated product of hydrohydroxyalkylation 3b was formed, but in only 14% yield (eq. 1). In related rhodium or iridium catalyzed alkyne-α-ketoester reductive couplings employing hydrogen as terminal reductant,[6b,c,7] carboxylic acid co-catalysts were found to increase rate and conversion. As supported by computational studies,[16] such acid co-catalysts bypass highly energetic 4-centered transition structures for direct hydrogenolysis of oxametallacyclic intermediates via σ-bond metathesis, instead protonating the metal-oxygen bond to form metal carboxylates, which then react with elemental hydrogen by way of lower energy 6-centered transition structures. This acid co-catalyst effect also is operative in ruthenium(0) catalyzed hydrohydroxyalkylations[8c,f] that proceed through oxametallacyclic intermediates cleaved through transfer hydrogenolysis. As borne out experimentally, use of 1-adamantanecarboxylic acid (12 mol%) under the aforesaid conditions led to a 95% isolated yield of adduct 3b as a single olefin stereoisomer (eq. 1).
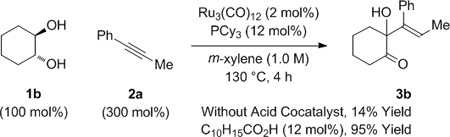 |
(eq. 1) |
Under these conditions, diverse diols 1a-1l react with 1-phenyl-1-propyne 2a to form the corresponding α-hydroxy ketones 3a-3l (Table 1). Diol stereochemistry is unimportant, as both cis- and trans-diastereomers engage in efficient C-C coupling. Both cyclic diols 1a-1d, 1i-1l and acyclic diols 1e-1h, deliver couplings products in good to excellent yields. In all but two cases, adducts 3c and 3d, complete levels of olefin stereocontrol are observed. For adducts 3c and 3d, longer reaction times result in a greater loss of E/Z stereoselectivity, suggesting these products initially form with high levels of E/Z stereoselectivity but isomerize under the reaction conditions. As will be discussed, the coupling of diol 1l proceeds with inversion of regioselectivity, as corroborated by single crystal X-ray diffraction. Using this specific catalyst system, terminal vicinal diols that incorporate primary alcohols provide complex stereoisomeric mixtures of mono- and bis-C-C coupling products. In the absence of diol, alkyne 2a is converted to products of [2+2+2]cycloaddition.[13,14]
Table 1.
Ruthenium(0) catalyzed hydrohydroxyalkylation of 1-phenyl-1-propyne 2a with diols 1a-1l.a
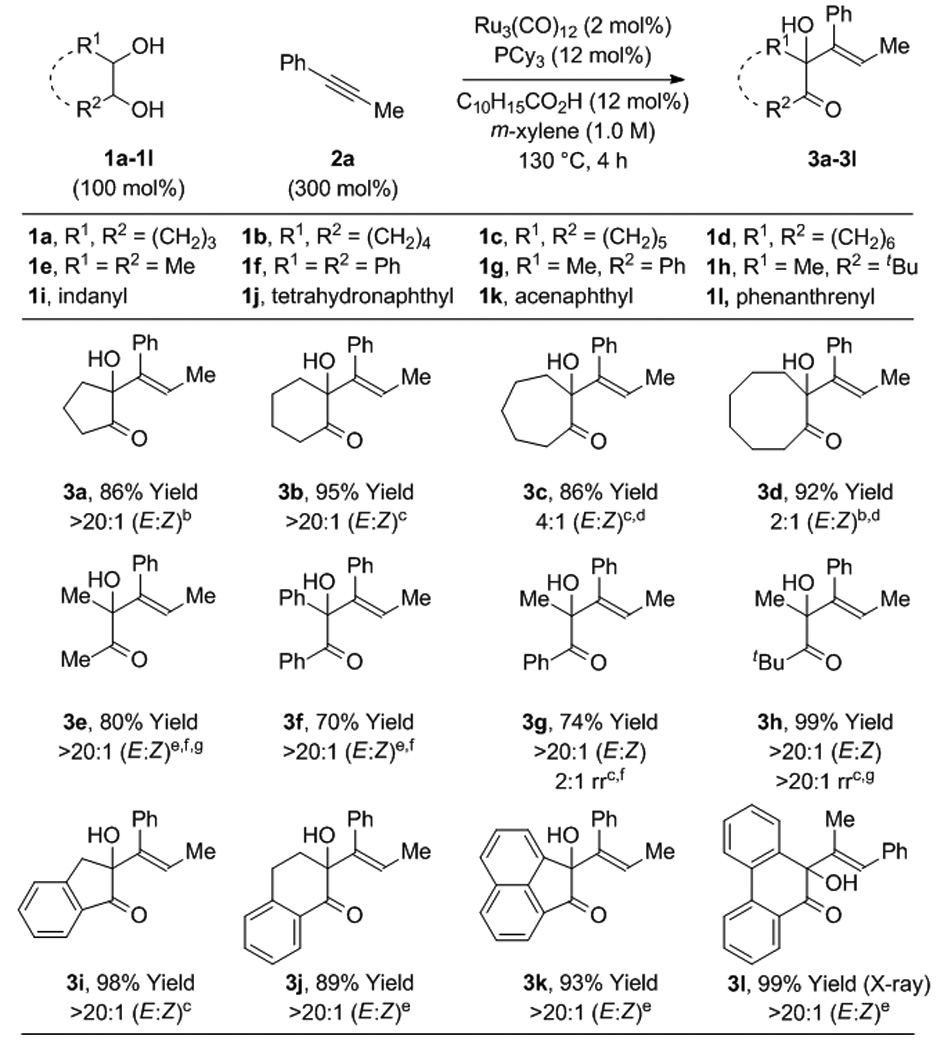 |
Yields are of material isolated by silica gel chromatography. C10H15CO2H refers to 1-adamantanecarboxylic acid. For acyclic 1,2-diols, the terminology “cis” and “trans” relates to the indicated reactant conformation. See Supporting Information for further details.
The cis-1,2-diol was employed.
The trans-1,2-diol was employed.
2 h.
A mixture of cis- and trans-1,2-diols was employed
DPPF (6 mol%) was used instead PCy3 (12 mol%).
20 h.
To further evaluate the scope of this process, the coupling of trans-1,2-cyclohexane diol 1b to alkynes 2a-2h was explored (Table 2). In all cases, good to excellent isolated yields of the corresponding coupling products 3b, 3n-3t were obtained. With the exception of adduct 3p, complete levels of olefin (E:Z)-stereoselectivity were observed, and for all nonsymmetric alkynes (2a, 2b, 2d-2h), complete levels of regioselectivity were observed. Terminal alkynes and dialkyl substituted alkynes do not engage in efficient coupling under these conditions. As illustrated in the reaction of racemic ethyl mandelate 1m to 1-phenyl-1-propyne 2a to form adduct 3m, other vicinally deoxygenated reactants participate in C-C bond formation under these conditions and require an acid cocatalyst (eq. 2). Finally, in a preliminary evaluation of the utility of the coupling products, adducts 3b, 3e, 3j, 3o, 3q, and 3t were exposed to catalytic p-toluenesulfonic acid in m-xylene at 80 °C. Cyclodehydration to form the corresponding substituted furans 4a-4f occurred in good yields (Table 3).[15]
Table 2.
Ruthenium(0) catalyzed hydrohydroxyalkylation of alkynes 2a-2h with cyclohexane diol 1b.a
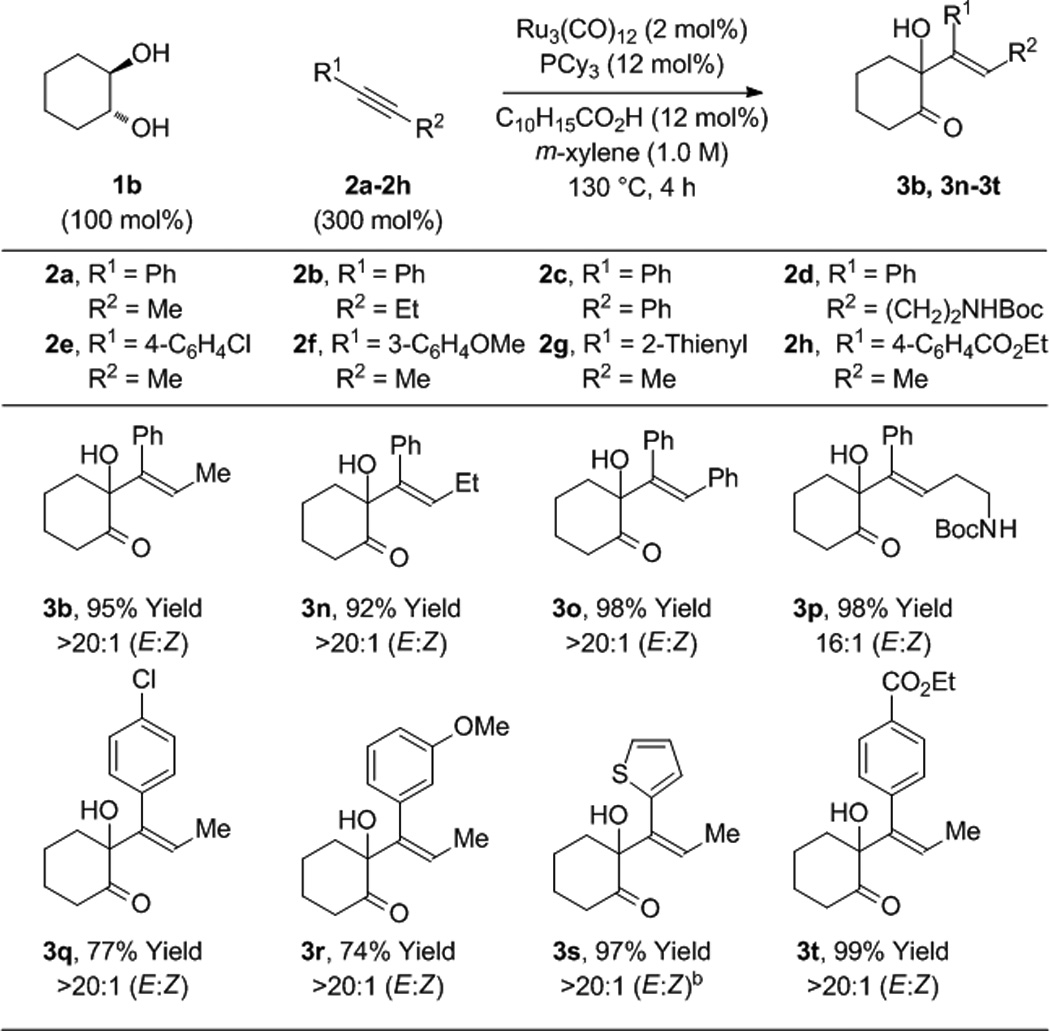 |
Yields are of material isolated by silica gel chromatography. C10H15CO2H refers to 1-adamantanecarboxylic acid. See Supporting Information for further details.
20 h.
Table 3.
Acid catalyzed cyclodehydration of adducts 3b, 3e, 3j, 3o, 3q, and 3t to form furans 4a-4f.a
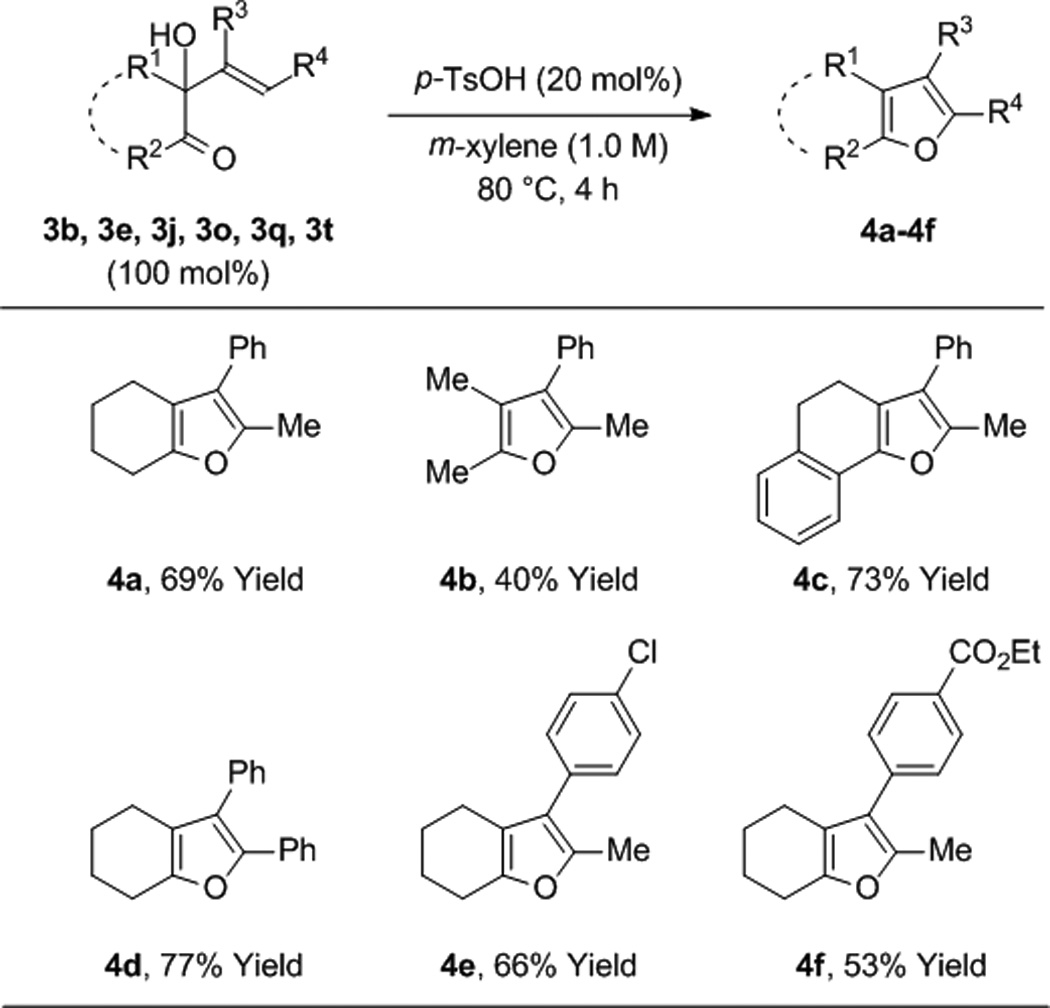 |
Yields are of material isolated by silica gel chromatography. See Supporting Information for further details.
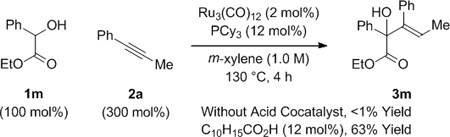 |
(eq. 2) |
A plausible catalytic mechanism is illustrated in the coupling of trans-1,2-cyclohexane diol 1b and 1-phenylpropyne 2a to form adduct 3b (Scheme 1). A mononuclear ruthenium(0) complex[17] promotes the oxidative coupling of dione tetradehydro-1b and 1-phenylpropyne 2a to form oxaruthenacycle I.[18,19] As corroborated by LRMS of crude reaction mixtures, which reveal the presence of alkene, the requisite dione tetradehydro-1b may be formed through successive Ru3(CO)12 catalyzed transfer of hydrogen from cyclohexanediol 1b to the alkyne.[20–22] The protonation of oxaruthenacycle I by cyclohexanediol 1b or hydroxyketone didehydro-1b to form the ruthenium alkoxide III, is postulated to be slow compared to protonolytic cleavage of oxaruthenacycle I by 1-adamantanecarboxylic acid to form ruthenium carboxylate II. Exchange of the ruthenium carboxylate II with 1b or didehydro-1b forms ruthenium alkoxide III, which upon β-hydride elimination delivers didehydro-1b or tetradehydro-1b, respectively and ruthenium hydride IV. Subsequent C-H reductive elimination furnishes adduct 3b and returns ruthenium to its zero-valent form. The indicated model accounts for the observed regioselectivity, including the inversion of alkyne regioselectivity found in the coupling of diol 1l (Figure 2). The steric demand of the metal center promotes oxidative coupling at the less bulky carbonyl moiety of the transient dione and controls alkyne orientation (A and C, Figure 2). The inversion of alkyne regioselectivity evident in the formation of 3l can be explained by the increase in steric repulsion between the dione and alkyne substituent (B and D, Figure 2). The proposed structure of these complexes is analogous to that of related metallacycles obtained upon ruthenium(0) mediated ketone-diene oxidative coupling.[8e]
Scheme 1.
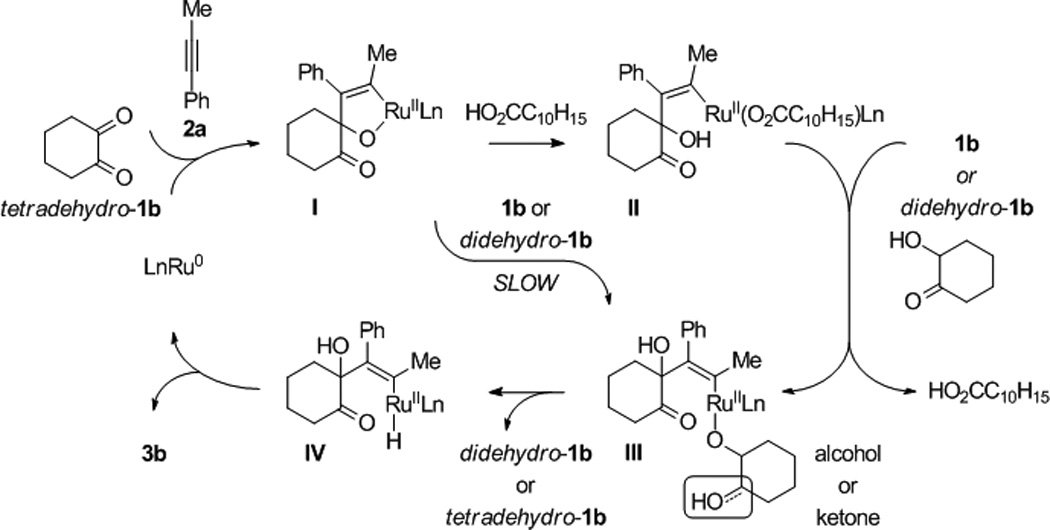
Postulated mechanism for ruthenium(0) catalyzed diol-alkyne hydrohydroxyalkylation as illustrated in the coupling of 1,2-cyclohexane diol 1b and 1-phenyl-1-propyne 2a.
Figure 2.
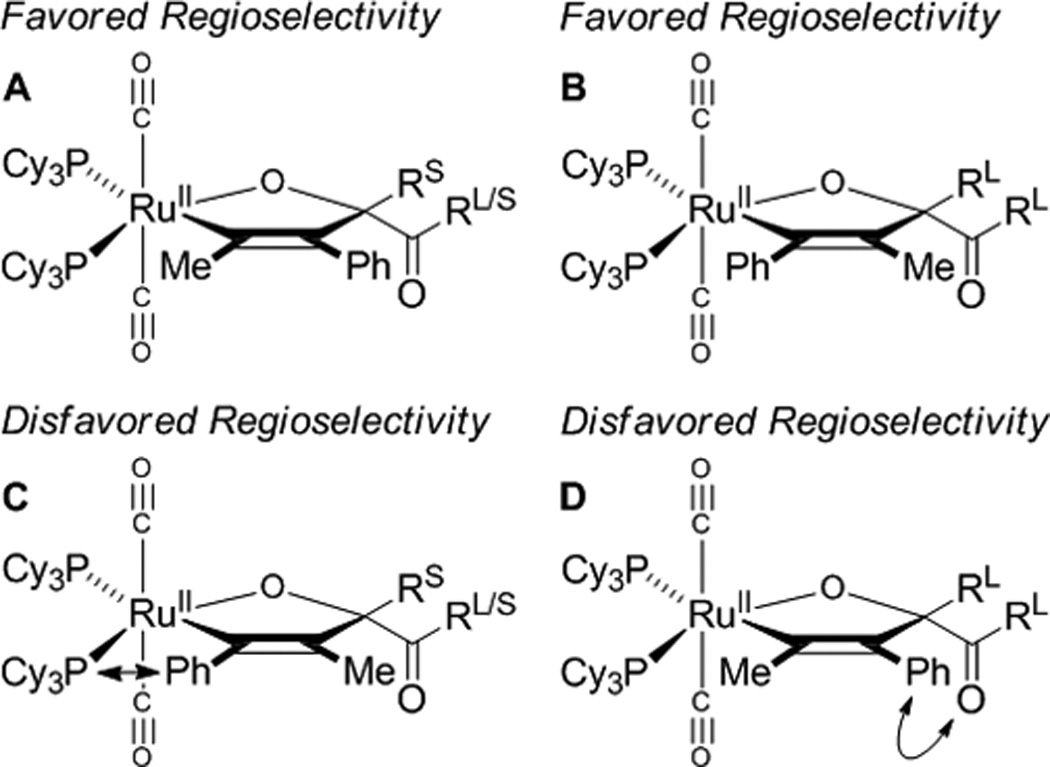
Model to account for the observed regioselectivity of alkyne coupling.
In summary, we report the direct ruthenium catalyzed C-C coupling of alkynes and vicinal diols to form β,γ-unsaturated ketones with complete levels of regioselectivity and good to complete control of alkene geometry. Exposure of the reaction products to substoichiometric quantities of p-toluenesulfonic acid induces cyclodehydration to form tetrasubstituted furans. The present alkyne-diol hydrohydroxyalkylations contribute to a growing body of merged redox-construction events that bypass the use of premetallated reagents and, hence, stoichiometric quantities of metallic byproducts.
Supplementary Material
Footnotes
Acknowledgment is made to the Robert A. Welch Foundation (F-0038) and the NIH-NIGMS (RO1-GM069445) for partial financial support.
References
- 1.For selected reviews on metal catalyzed reductive C-C coupling of carbonyl compounds, see: Montgomery J. Angew. Chem. 2004;116:3980. Angew. Chem. Int. Ed. 2004;43:3890. Montgomery J, Sormunen GJ. Top. Curr. Chem. 2007;279:1. Hirao T. Top. Curr. Chem. 2007;279:53. Nishiyama H, Shiomi T. Top. Curr. Chem. 2007;279:105. Kimura M, Tamaru Y. Top. Curr. Chem. 2007;279:173. Iida H, Krische MJ. Top. Curr. Chem. 2007;279:77. Ngai M-Y, Kong JR, Krische MJ. J. Org. Chem. 2007;72:1063. doi: 10.1021/jo061895m. Skucas E, Ngai M-Y, Komanduri V, Krische MJ. Acc. Chem. Res. 2007;40:1394. doi: 10.1021/ar7001123.
- 2.The catalytic reductive cyclization of nonconjugated alkenes and alkynes onto unactivated ketones has been achieved, but analogous intermolecular reactions are unknown: Kablaoui NM, Buchwald SL. J. Am. Chem. Soc. 1996;118:3182. Kablaoui NM, Buchwald SL. J. Am. Chem. Soc. 1995;117:6785. Crowe WE, Rachita MJ. J. Am. Chem. Soc. 1995;117:6787. Saito N, Sugimura Y, Sato Y. Org. Lett. 2010;12:3494. doi: 10.1021/ol101329d.
- 3.For catalytic reductive aldol cyclizations onto ketones, see: Huddleston RR, Krische MJ. Org. Lett. 2003;5:1143. doi: 10.1021/ol0300219. Koech PK, Krische MJ. Org. Lett. 2004;6:691. doi: 10.1021/ol030136c. Miura K, Yamada Y, Tomita M, Hosomi A. Synlett. 2004:1985. Lam HW, Joensuu PM. Org. Lett. 2005;7:4225. doi: 10.1021/ol051649h. Lam HW, Murray GJ, Firth JD. Org. Lett. 2005;7:5743. doi: 10.1021/ol052599j. Lam HW, Joensuu PM, Murray GJ, Fordyce EAF, Prieto O, Luebbers T. Org. Lett. 2006;8:3729. doi: 10.1021/ol061329d. Joensuu PM, Murray GJ, Fordyce EAF, Luebbers T, Lam HW. J. Am. Chem. Soc. 2008;130:7328. doi: 10.1021/ja0775624. Margalef IV, Rupnicki L, Lam HW. Tetrahedron. 2008;64:7896. Deschamp J, Riant O. Org. Lett. 2009;11:1217. doi: 10.1021/ol802879f.
- 4.For catalytic intermolecular reductive aldol reactions onto ketones, see: Revis A, Hilty TK. Tetrahedron Lett. 1987;28:4809. Deschamp J, Chuzel O, Hannedouche J, Riant O. Angew. Chem. 2006;118:1314. doi: 10.1002/anie.200503791. Angew. Chem. Int. Ed. 2006;45:1292. Zhao D, Oisaki K, Kanai M, Shibasaki M. Tetrahedron Lett. 2006;47:1403. Zhao D, Oisaki K, Kanai M, Shibasaki M. J. Am. Chem. Soc. 2006;128:14440. doi: 10.1021/ja0652565. Lumby RJR, Joensuu PM, Lam HW. Org. Lett. 2007;9:4367. doi: 10.1021/ol701980e. Shiomi T, Nishiyama H. Org. Lett. 2007;9:1651. doi: 10.1021/ol070251d. Lumby RJR, Joensuu PM, Lam HW. Tetrahedron. 2008;64:7729. Lipshutz BH, Amorelli B, Unger JB. J. Am. Chem. Soc. 2008;130:14378. doi: 10.1021/ja8045475. Kato M, Oki H, Ogata K, Fukuzawa S-i. Synlett. 2009:1299.
- 5.For intermolecular reductive coupling of 1,3-dienes to unactivated ketones, see: Kimura M, Fujimatsu H, Ezoe A, Shibata K, Shimizu M, Matsumoto S, Tamaru Y. Angew. Chem. 1999;111:410. doi: 10.1002/(SICI)1521-3773(19990201)38:3<397::AID-ANIE397>3.0.CO;2-Y. Angew. Chem. Int. Ed. 1999;38:397. Cho HY, Yu Z, Morken JP. Org. Lett. 2011;13:5267. doi: 10.1021/ol202138v.
- 6.For intermolecular reductive coupling of 1,3-enynes to α-ketoesters or aryl ketones, see: Miller KM, Jamison TF. Org. Lett. 2005;7:3077. doi: 10.1021/ol051075g. Komanduri V, Krische MJ. J. Am. Chem. Soc. 2006;128:16448. doi: 10.1021/ja0673027. Kong JR, Ngai M-Y, Krische MJ. J. Am. Chem. Soc. 2006;128:718. doi: 10.1021/ja056474l.
- 7.For intermolecular reductive coupling of nonconjugated alkynes to α-ketoesters, see: Ngai M-Y, Barchuk A, Krische MJ. J. Am. Chem. Soc. 2007;129:280. doi: 10.1021/ja0670815. Kong JR, Krische MJ. J. Am. Chem. Soc. 2006;128:16040. doi: 10.1021/ja0664786.
- 8.For ruthenium(0) catalyzed hydrohydroxyalkylation of secondary alcohols, see: Leung JC, Geary LM, Chen T-Y, Zbieg JR, Krische MJ. J. Am. Chem. Soc. 2012;134:15700. doi: 10.1021/ja3075049. Chen T-Y, Krische MJ. Org. Lett. 2013;15:2994. doi: 10.1021/ol401184k. Yamaguchi E, Mowat J, Luong T, Krische MJ. Angew. Chem. 2013;125:8586. doi: 10.1002/anie.201303552. Angew. Chem. Int. Ed. 2013;52:8428. Geary LM, Glasspoole BW, Kim MM, Krische MJ. J. Am. Chem. Soc. 2013;135:3796. doi: 10.1021/ja400691t. Park BY, Montgomery TP, Garza V, Krische MJ. J. Am. Chem. Soc. 2013;135:16320. doi: 10.1021/ja4087193. McInturff EL, Mowat J, Waldeck AR, Krische MJ. J. Am. Chem. Soc. 2013;135:17230. doi: 10.1021/ja410533y.
- 9.For ruthenium(II) catalyzed hydrohydroxyalkylation of alkynes employing primary alcohols and corresponding aldehyde reductive couplings, see: Patman RL, Chaulagain MR, Williams VM, Krische MJ. J. Am. Chem. Soc. 2009;131:2066. doi: 10.1021/ja809456u. Williams VM, Leung JC, Patman RL, Krische MJ. Tetrahedron. 2009;65:5024. doi: 10.1016/j.tet.2009.03.068. Bausch CC, Patman RL, Breit B, Krische MJ. Angew. Chem. 2011;123:5805. doi: 10.1002/anie.201101496. Angew. Chem. Int. Ed. 2011;50:5687. Leung JC, Patman RL, Sam B, Krische MJ. Chem. Eur. J. 2011;17:12437. doi: 10.1002/chem.201101554.
- 10.For ruthenium(II) catalyzed hydrohydroxyalkylation of dienes employing primary alcohols and corresponding aldehyde reductive couplings, see: Shibahara F, Bower JF, Krische MJ. J. Am. Chem. Soc. 2008;130:6338. doi: 10.1021/ja801213x. Shibahara F, Bower JF, Krische MJ. J. Am. Chem. Soc. 2008;130:14120. doi: 10.1021/ja805356j. Zbieg JR, Yamaguchi E, McInturff EL, Krische MJ. Science. 2012;336:324. doi: 10.1126/science.1219274. McInturff EL, Yamaguchi E, Krische MJ. J. Am. Chem. Soc. 2012;134:20628. doi: 10.1021/ja311208a.
- 11.For selected reviews on the direct redox-triggered C-C coupling of alcohols, see: Bower JF, Krische MJ. Top. Organomet. Chem. 2011;43:107. doi: 10.1007/978-3-642-15334-1_5. Hassan A, Krische MJ. Org. Proc. Res. Devel. 2011;15:1236. doi: 10.1021/op200195m. Moran J, Krische MJ. Pure Appl. Chem. 2012;84:1729. doi: 10.1351/PAC-CON-11-10-18.
- 12.For seminal reports of intra- and intermolecular nickel catalyzed alkyne-aldehyde reductive coupling, respectively, see: Oblinger E, Montgomery J. J. Am. Chem. Soc. 1997;119:9065. Huang W-S, Chan J, Jamison TF. Org. Lett. 2000;2:4221. doi: 10.1021/ol006781q.
- 13.For an example of Ru3(CO)12 catalyzed alkyne [2+2+2]cycloaddition, see: Kawatsura M, Yamamoto M, Namioka J, Kajita K, Hirakawa T, Itoh T. Org. Lett. 2011;13:1001. doi: 10.1021/ol1030734.
- 14.For a review of ruthenium catalyzed alkyne [2+2+2]cycloaddition, see: Yamamoto Y, Itoh K. In: Ruthenium in Organic Synthesis. Murahashi S-I, editor. Weinheim: Wiley-VCH; 2004. pp. 95–128.
- 15.To our knowledge, only one related acid catalyzed cyclodehydration to form furans is reported: Basavaiah D, Roy S, Das U. Tetrahedron. 2010;66:5612.
- 16.a) Musashi Y, Sakaki S. J. Am. Chem. Soc. 2002;124:7588. doi: 10.1021/ja020063c. [DOI] [PubMed] [Google Scholar]; b) Williams VM, Kong J-R, Ko B-J, Mantri Y, Brodbelt JS, Baik M-H, Krische MJ. J. Am. Chem. Soc. 2009;131:16054. doi: 10.1021/ja906225n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ru3(CO)12 reacts with dppe in benzene solvent to provide Ru(CO)3(dppe): Sanchez-Delgado RA, Bradley JS, Wilkinson G. J. Chem. Soc. Dalton Trans. 1976:399.
- 18.A related ruthenacycle obtained upon diene-ketone oxidative coupling has been isolated and characterized by single-crystal X-ray diffraction (see reference 8e).
- 19.Intermolecular nickel mediated alkyne–aldehyde oxidative coupling delivers isolable nickeladihydrofurans that have been characterized by single-crystal X-ray diffraction: Ogoshi S, Arai T, Ohashi M, Kurosawa H. Chem. Commun. 2008:1347. doi: 10.1039/b717261c.
- 20.For Ru3(CO)12 catalyzed oxidation of alcohols employing olefins and alkynes as hydrogen acceptors, see: Blum Y, Reshef D, Shvo Y. Tetrahedron Lett. 1981;22:1541. Shvo Y, Blum Y, Reshef D, Menzin M. J. Organomet. Chem. 1982;226:C21. Meijer RH, Ligthart GBWL, Meuldijk J, Vekemans JAJM, Hulshof LA, Mills AM, Kooijman H, Spek AL. Tetrahedron. 2004;60:1065.
- 21.For mechanistically related Ru3(CO)12 catalyzed transfer hydrogenation of ketones mediated by isopropanol, see: Johnson TC, Totty WG, Wills M. Org. Lett. 2012;14:5230. doi: 10.1021/ol302354z.
- 22.For mechanistically related Ru3(CO)12 catalyzed secondary alcohol amination via alcohol mediated hydrogen transfer, see: Bähn S, Tillack A, Imm S, Mevius K, Michalik D, Hollmann D, Neubert L, Beller M. ChemSusChem. 2009;2:551. doi: 10.1002/cssc.200900034. Pingen D, Müller C, Vogt D. Angew. Chem. 2010;122:8307. doi: 10.1002/anie.201002583. Angew. Chem. Int. Ed. 2010;49:8130. Zhang M, Imm S, Bähn S, Neumann H, Beller M. Angew. Chem. 2011;123:11393. doi: 10.1002/anie.201104309. Angew. Chem. Int. Ed. 2011;50:11197.
- 23.Related models to account for regioselectivity in nickel catalyzed alkyne-carbonyl reductive couplings have been proposed: Malik HA, Sormunen GJ, Montgomery J. J. Am. Soc. Chem. 2010;132:6304. doi: 10.1021/ja102262v. Liu P, Montgomery J, Houk KN. J. Am. Soc. Chem. 2011;133:6956. doi: 10.1021/ja202007s.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.