

Abstract
Akt has emerged as a critical target for the development of anti-cancer therapies. It has been found to be amplified, overexpressed, or constitutively activated in numerous human malignancies with oncogenesis derived from the simultaneous promotion of cell survival and suppression of apoptosis. A valuable alternative to the more common ATP-mimetic based chemotherapies is a substrate-mimetic approach, which has the potential advantage of inherent specificity of the substrate-binding pocket. In this paper we present the development of high affinity non-peptidic, substrate-mimetic inhibitors based on the minimum GSK3β substrate sequence. Optimization of initial peptidic leads resulted in the development of several classes of small molecule inhibitors, which have comparable potency to the initial peptidomimetics, while eliminating the remaining amino acid residues. We have identified the first non-peptidic substrate-mimetic lead inhibitors of Akt 29a–b, which have affinities of 17 and 12 μM, respectively. This strategy has potential to provide a useful set of molecular probes to assist in the validation of Akt as a potential target for anti-cancer drug design.
Keywords: Akt, Protein kinase B, Substrate-mimetic, Cancer, Inhibitor
1. Introduction
Akt/protein kinase B (PKB) has been shown to be a widely expressed Ser/Thr protein kinase whose persistent activation leads to human oncogenesis.1–13 Its role in cancer and chemoresistance is accomplished by the concomitant promotion of cell growth, migration, and angiogenesis as well as the suppression of the apoptotic pathway.14–16 There has been significant interest in Akt for its structural and functional properties as well as its implications in the area of cancer therapy.
The Akt family consists of three members, Akt1 (PKBα), Akt2 (PKBβ), and Akt3 (PKBγ). Numerous cellular stimuli result in Akt activation including molecules that regulate tyrosine kinase activity and G-protein-linked receptors, cAMP/PKA agonists, and phosphatase inhibitors.5,17 Direct activation of Akt is mediated by phosphoinositide-3 kinase (PI3K) which generates phosphatidylinositol-3,4,5-triphosphate (PIP3), a lipid second messenger which binds to the PH domain of Akt and translocates it to the intracellular side of the plasma membrane.18–20 Akt then undergoes dual phosphorylation by membrane associated protein kinases PDK1 and ‘PDK2’ on Thr308 in the activation loop and Ser473 in the C-terminal hydrophobic motif, respectively.21 This dual phosphorylation induces a conformational change in the enzyme to its activated form, which incorporates an ATP binding site as well as a substrate-binding site.
Akt directly phosphorylates substrates that are involved in the regulation of cellular functions such as cellular proliferation, transcription, migration, apoptosis, cellular differentiation, and metabolism.22,23 One of the first cellular substrates discovered for Akt was glycogen synthase kinase 3 (GSK3), which plays a key role in metabolism and insulin signaling pathways. GSK3 is negatively regulated by Akt via insulin stimulated phosphorylation of the enzyme that results in the inactivation of the enzyme and consequent activation of glycogen synthase.5 The disregulation of Akt kinase activity has been detected in a number of human malignancies including ovarian, breast, thyroid, and colon cancers.24 Amplification or overexpression of Akt results in the up-regulation of cell growth and the down-regulation of apoptosis. The cellular levels of PIP3 regulate the activity of PDK-1, which is responsible for Akt activation. The levels of these phosphoinositides are dependent on the activity of PI3K and a set of phosphatases PTEN and SHIP.25 Tumor suppressor PTEN negatively regulates the activity of Akt by converting PIP3 back to PIP2. PTEN deletions and mutations are prevalent in a variety of human cancers.17 Inhibition of Akt activity has been shown to suppress cell growth and induce apoptosis in tumor cell lines derived from various organs possessing constitutively activated Akt.24
The majority of small molecule kinase inhibitors to date target the ATP binding pocket and there have been few reports targeting the substrate-binding site. ATP mimetics have met with much success, however selective binding within this pocket remains challenging as these inhibitors compete with the many ATP utilizing enzymes possessing similar contact regions as well as with high cellular concentrations of ATP.26–29 Substrate-mimetics offer a promising strategy for the design of selective inhibitors of Akt as they can potentially exploit sequence specificity.30–34 The substrate-binding region has evolved to recognize specific substrate sequences and therefore offers a larger number of potential interactions for a properly designed inhibitor than the corresponding ATP pocket. The inherent design challenges present in substrate-mimetics are the large binding pocket and extended binding conformation of many natural substrates. We recently described the development of substrate-mimetic inhibitors of Akt based on the consensus sequence and the structure of an enzyme bound substrate.35 These preliminary studies demonstrated that limited structural modification of the initial peptidic substrate can overcome these challenges and provide peptidomimetic inhibitors with increasing lipophilicity, rigidity, and potency as well as decreasing the size and peptidic nature of the inhibitors.
Our substrate-mimetic design was based on the truncated GSK3β substrate sequence, GRPRTTSF, utilizing the recently published X-ray crystal structure of an activated Akt ternary complex with GSK3β and an ATP analogue.36 Our design approach focused on reducing the entropy cost of the extended binding conformation, accessing a large unoccupied hydrophobic pocket adjacent to the C-terminus, and eliminating nonessential amino acid residues. From this we identified inhibitor 1 with in vitro inhibition of IC50 = 14 μM (Fig. 1).35
Figure 1.

Flexible ligand docking GOLD37 of leading peptidomimetic inhibitor 1 (previously reported)35 (green) against the solvent-accessible surface area of Akt (Gradient color scale- Red hydrophobic, Blue Hydrophilic).
Herein we describe the first non-peptidic, substrate-mimetic inhibitor of Akt developed through systematic rigidification and replacement of the remaining amino acid residues. Further structural refinement included the incorporation of essential binding groups into organic scaffolds to increase rigidity and to deliver improved potency and selectivity.
2. Results and discussion
cModifications of our earlier preliminary peptidomimetic structures focused on three main areas: the N-terminal hydro-philic domain, the central region, and C-terminal substituents. The efficiencies of the inhibitors in disrupting Akt function was tested utilizing a fluorescence polarization assay system.38 The least amount of sequence homology in Akt substrates exists within the dipeptide sequence adjacent to the phosphorylated serine/threonine residue. In 1 this region is replaced by a 4-aminobenzoic acid (Abz) spacer. The contacts within this region are mainly hydrophobic, therefore a variety of hydrophobic substituents projected from the central phenyl spacer was explored (Table 1). Incorporation of a phenyl substituent at R2 provides 5 with a slight increase in activity when compared to previously reported inhibitor 2.35 Docking studies suggested that the phenyl substituent is able to access the Thr pocket previously exploited in the design of inhibitor 1. Truncation of the N-terminus of the inhibitors (7–9) resulted in a modest decrease in affinity, but a desirable decrease in molecular weight and peptidic character of the inhibitors. The study of the central portion of the inhibitor confirmed the importance of the projection of substituents into the Thr binding pocket.
Table 1.
Investigations of central hydrophobic contacts
| Compound |
|
IC50 (μM)a | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 2b | Ac- | GR | P | R | -H | -H | V | F | -Bn | 28±8 |
| 3 | Ac- | GR | P | R | -CH3 | -H | V | F | -Bn | 50±10 |
| 4 | Ac- | GR | P | R | -Ph | -H | V | F | -Bn | >500 |
| 5 | Ac- | GR | P | R | -H | -Ph | V | F | -Bn | 16±5 |
| 6 | Ac- | GR | P | R | -H | -Nap | V | F | -Bn | 78±14 |
| 7 | Ac- | R | -Ph | -H | V | F | -Bn | 288±100 | ||
| 8 | Ac- | R | -H | -Ph | V | F | -Bn | 173±31 | ||
| 9 b | Ac- | R | -H | -H | V | F | -Bn | 119±36 | ||
Reported values are an average of three independent-binding curves utilizing an IMAP® Akt Assay Kit (Molecular Devices). Reactions were conducted in wells with 20.0 μL of 10 mM Tris–HCl (pH 7.2), 10 mM MgCl2, 0.1% BSA, 0.05% NaN3, 1 mM DTT, 100 nM 5FAM-GRPRTSSFAEG-COOH, 5 μM ATP, Akt1, and inhibitor (DMSO stock solution). Reaction mixtures were incubated for 1 h at room temperature and then quenched with 60 μL of the IMAP-binding solution. The reactions were equilibrated for 1 h at room temperature then data points were collected and analyzed.
Previously reported inhibitors.35
Flexible ligand docking (GOLD)37 of lead peptidomimetics identified several potential conformationally restrained replacements for the Val-Phe-Bn C-terminal sequence, which remove two of the three remaining amino acids. A simple cyclic constraint such as quinazolines 15a–b project appended hydrophobic groups into adjacent hydrophobic pockets while maintaining the N-terminal and central inhibitor/Akt interactions. Inhibitor 15a has similar affinity (IC50 = 112 μM) to the corresponding inhibitor 9 containing the Val-Phe dipeptide, but contains two fewer stereocenters. Careful consideration of the potential binding site contacts made by the three key areas of the peptidomimetic inhibitors provided guidance in the design of non-peptidic substrate-mimetics.
Inhibitor 21ba was designed using GOLD37 to incorporate important binding elements from the previous studies (Fig. 2). The guanidine group is directly projected into the Arg pocket via an ethylenediamine linker that extends the correct distance between the aromatic spacer and the arginine binding pocket of Akt. The Thr pocket can be accessed by direct projection of substituents from Abz, shown here as a simple phenyl substituent. Finally, the 4-aminoaniline provides a C-terminal rigid scaffold to project various hydrophobic substituents into the pockets of Akt, with 21ba possessing two benzyl substituents. This small molecule substrate-mimetic of Akt has an IC50 of 84 μM, which is comparable or better than our previous peptidomimetic inhibitors, and is significantly more rigid and impervious to proteases.
Figure 2.

Flexible ligand docking GOLD37 of inhibitor 21ba (green) against the solvent-accessible surface area of Akt (Gradient color scale—red hydrophobic, blue hydrophilic).
This non-peptidic scaffold design easily allowed an extensive exploration of the different binding groups, beginning with the C-terminal hydrophobic interactions in series 21aa–21bi (Table 2, Scheme 2). This series suggests that the two pockets are extensive and able to accommodate large hydrophobic substituents (21bd, 21bf, 21bh). Inhibitor 21bi with a 4-cyanobenzyl functional group is the most potent inhibitor in this series having an IC50 of 19 μM. Secondly, a range of substituents were added to explore the role of contacts within the Thr pocket via the projection of functionality directly off Abz to provide inhibitors 21aa and 22aa–fa (Table 2, Scheme 3). Inhibitor 21aa, which lacks the phenyl substituent and the ability to make contacts within this region, is slightly less potent than the biphenyl derivative. This suggests that optimization at this position could lead to increased potency. The addition of H-bond donors and acceptors here did not lead to increased affinity (22aa and 22ba), however, larger hydrophobic groups, such as 2-naphthyl, led to a twofold increase in affinity with inhibitor 22fa having an IC50 of 44 μM.
Table 2.
Optimization of C-terminal and central substituents
| Compound | R1 | R2 | IC50 (μM)a |
|---|---|---|---|
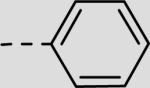
| 21aa | –H |
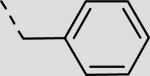
|
96 ± 20 |
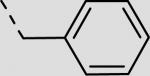
| 21ba |
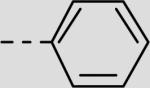
|
|
84 ± 21 |
| 21bb |
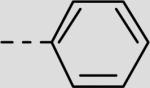
|
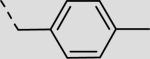
|
215 ± 73 |
| 21bc |
|
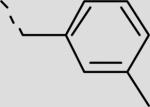
|
152 ± 44 |
| 21bd |
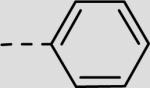
|
|
84 ± 7 |
| 21be |
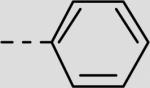
|
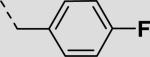
|
55 ± 13 |
| 21bf |
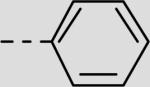
|
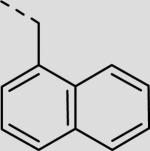
|
43 ± 5 |
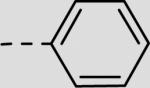
| 21bg |
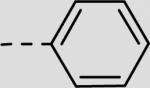
|
|
28 ± 8 |
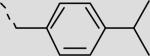
| 21bh |
|
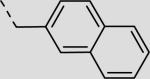
|
23 ± 4 |
| 21bi |
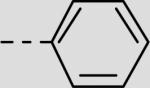
|
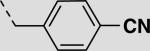
|
19 ± 10 |
| 21cg |
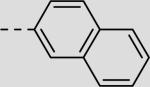
|
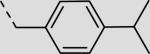
|
38 ± 4 |
| 21ci |
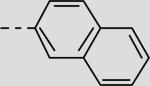
|
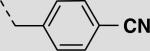
|
17 ± 2 |
| 22aa |
|
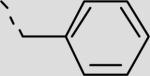
|
253 ± 31 |
| 22ba |
|
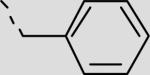
|
158 ± 19 |
| 22ca |
|
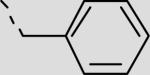
|
83 ± 10 |
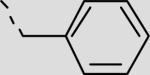
| 22da |
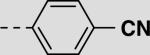
|
|
78 ± 9 |
| 22ea |
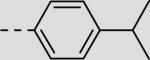
|
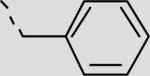
|
52 ± 8 |
| 22fa |
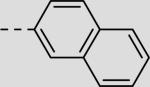
|
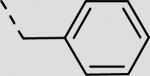
|
44 ± 6 |
Reported values are an average of three independent-binding curves utilizing an IMAP® Akt Assay Kit (Molecular Devices). Reactions were conducted in wells with 20.0 μL of 10 mM Tris–HCl (pH 7.2), 10 mM MgCl2, 0.1% BSA, 0.05% NaN3, 1 mM DTT, 100 nM 5FAM-GRPRTSSFAEG-COOH, 5 μM ATP, Akt1, and inhibitor (DMSO stock solution). Reaction mixtures were incubated for 1 h at room temperature and then quenched with 60 μL of the IMAP-binding solution. The reactions were equilibrated for 1 h at room temperature then data points were collected and analyzed. (Inhibitors 21aa–ci synthesized via Scheme 2, Inhibitors 22aa–22fa synthesized via Scheme 3).
Scheme 2.

Reagents and conditions: (a) R1B(OH)2, PdCl2(dppf), K2CO3, DMF, 100°C, 71–82% (16a = methyl 4-aminobenzoate); (b) i—N-Boc-2-aminoacetaldehyde, AcOH, MeOH, 4 Å mol. sieves; ii—NaCNBH3, 68–100%; (c) i—LiOH, THF, H2O; ii—TFA, 70–100%; (d) BisBocPCH, Et3N, MeOH, 61–71%; (e) RBr, K2CO3, NaI, DMF, 61–100%; (f) SnCl2 2H2O, EtOAc, reflux, 19–92%; (g) i—DIC, cat DMAP, DCM; ii—TFA, 26–76%.
Scheme 3.

Reagents and conditions: (a) LiOH, THF, H2O, 100%; (b) DIC, cat DMAP, DCM; (c) R1B(OH)2, PdCl2(dppf), K2CO3, DMF, 100°C, 62–78%; (d) i—N-Boc-2-aminoacetaldehyde, AcOH, MeOH, 4 Å mol. sieves; ii—NaCNBH3; (e) TFA, 41–69%; (f) BisBocPCH, Et3N, MeOH, 51–88%; (g) TFA, 40–99%.
The previous series of non-peptidic substrate-mimetic inhibitors provided valuable information concerning the nature of the three binding pockets within the active site of Akt. To further optimize our inhibitors, the best substituents at the two positions were combined in an effort to increase potency (21cg and 21ci). Inhibitor 21ci, which incorporates the best C-terminal functionality, 4-cyanobenzyl, and the best central element, 2-naphthyl, is the most potent non-peptidic inhibitor of this scaffold series with an IC50 of 17 μM, a slight improvement from phenyl derivative 21bi. To increase the stability and rigidity of 21cg and 21ci, the amide analogs 29a–b were synthesized, which also led to a further increase in potency (IC50's = 17 μM and 12 μM, respectively) (Scheme 4).
Scheme 4.

Reagents and conditions: (a) Boc-Gly-OH, DIC, cat. DMAP, DCM, 45%; (b) i—LiOH, THF, H2O; ii—TFA, 90%; (c) 20g or 20i, BisBocPCH, Et3N, MeOH, 54%; (d) i—DIC, cat DMAP, DCM; ii—TFA, 57%.
The initial non-peptidic substrate-mimetic design was successful and optimization of the scaffold provided inhibitors 29a–b that are comparable to our previous lead 1. Further optimizations focused on increasing rigidity by the addition of a ring constraint through an indole-aryl scaffold 36a–b (Fig. 3). The indole derivative 36a is comparable to 21aa as both lack access to the Thr pocket and possess C-terminal benzyl substituents. The inclusion of an indole scaffold provided a slight decrease in affinity in 36a (Scheme 5). Consistent with the previous scaffold, the addition of the C-terminal 4-cyanobenzyl substituent in 36b provided a fourfold increase in affinity from 126 to 32 μM.
Figure 3.

Flexible ligand docking GOLD37 of inhibitor 36a (green) against the solvent-accessible surface area of Akt (Gradient color scale—red hydrophobic, blue Hydrophilic).
Scheme 5.

Reagents and conditions: (a) Boc-Gly-OH, DIC, cat. DMAP, DCM, 100%; (b) i—TMS–acetylene, CuI, Pd(PPh3)2Cl2, Et3N; ii—K2CO3, MeOH, 74%; (c) CuI, Pd(PPh3)2Cl2, 2:1 Et3N/THF, 82%; (d) CuOAc2, DMF, 120 C, 3 days, 66%; (e) H2 (1 atm), Pd/C, MeOH, 92%; (f) RBr, K2CO3, DMF, 100%, 78%; (g) i—1:1 TFA/DCM; ii—BisBocPCH, Et3N, MeOH, 71%, 62%; (h) 1:1 TFA/DCM, 32%, 20%.
3. Synthesis
Peptidomimetics 2–9 were synthesized via solid phase peptide synthesis. Suzuki couplings employing various boronic acids and aryl bromides were performed to provide intermediates that displayed hydrophobic substituents from the aromatic spacer (Abz). The simple quinazoline scaffolds were readily derived from commercially available starting materials. The synthesis of the quina zolines cores 10a–b was accomplished by the cyclization of 4-nitroanthranilic acid by the reaction with sodium isocyanate or cyclization employing a carbon dioxide atmosphere with catalytic DBU (1,8-diazabicyclo[5.4.0]undec-7-ene) from 4- and 5-nitro precursors respectively (Scheme 1).39–41 Alkylation was followed by reduction of the nitro group followed by coupling with 4-nitrobenzoyl chloride via anilide formation to provide 13a–b. Reduction to the aniline, coupling with AcArg(Pmc)-OH, and deprotection of the guanidine protecting groups afforded 15a–b.
Scheme 1.

Reagents and conditions: (a) i—NaNCO; ii—NaOH; iii—HCl, 53%; (b) CO2 1 atm, DBU, THF, 82%; (c) K2CO3, BnBr, DMF, 81%, 91%; (d) Pd/C, H2 35 psi, CH3OH, 88%, 81%; (e) i—LDA THF; ii—4-nitro benzoyl chloride –78–50 °C, 76%, 81%; (f) Pd/C, H2 35 psi, 1:1 CH3OH/CH2Cl2, 80%, 86%; (g) AcArg(Pmc)-OH, DIC, CH2Cl2, cat. DMAP; (h) TFA, 5% thioanilsole, 1% TIPS, 1% H2O, 4%, 5%.
A convergent synthesis starting from methyl-4-amino-2-bro mobenzoate or methyl-4-aminobenzoate and 4-nitroaniline provided non-peptidic inhibitors 21aa–ci (Scheme 2). Suzuki coupling of the bromoaniline with the corresponding boronic acid employing PdCl2(dppf) as a catalyst (16a commercially available) followed by reductive amination utilizing N-Boc-aminoacetalde-hyde provided 17a–c. A series of deprotections followed by guanidinylation of the resulting amine afforded the N-terminal portions of the inhibitor 18a–c. The C-terminal hydrophobic portion of the molecule was synthesized via alkylation of 4-nitroani-line with the corresponding bromide and subsequent reduction of the nitro group utilizing tin (II) chloride to afford 20a–i. Coupling of 18a–c and 20a–i followed by Boc deprotection under acidic conditions gave final inhibitors 21aa–ci. Inhibitors 29a–b were derived from a similar synthesis, but in place of the reductive amination step, 16c was reacted with Boc-Gly-OH to provide the amide intermediate 27 which was manipulated in a similar manner to provide 29a–b.
The synthesis of inhibitors 22aa–fa used a late stage Suzuki coupling to provide faster access to a number of derivatives at the R1 position, while keeping R2 as a benzyl substituent (Scheme 3). Commercially available methyl-4-amino-3-bromobenzoate was saponified under basic conditions followed by amide bond formation with 20a to provide 24a. This intermediate was then reacted with different boronic acid derivatives PdCl2(dppf) as a catalyst to provide 25aa–fa. A series of functional group transformations similar to Scheme 2 provided inhibitors 22aa–fa. Amide analogs 29a–b was synthesized in a similar manner (Scheme 4).
The indole scaffold was readily derived from commercially available 4-iodoaniline and Boc-Gly-OH, which were reacted to form iodo-amide 30 (Scheme 5). Sonagashira cross-coupling of 30 and ethynyl–trimethyl-silane (TMS–acetylene) followed by removal of the silyl protecting group afforded terminal alkyne 31. A consecutive Sonagashira cross-coupling with 2-iodo-4-nitroani-line followed by cycloisomerization employing catalytic copper (II) acetate42 afforded indole scaffold 33. Reduction of the nitro to the amine followed by alkylation with the corresponding bromide provided 35a–b. A series of functional group transformations similar to Schemes 1 and 2 provided inhibitors 36a–b.
4. Conclusion
Akt is an attractive target for developing novel cancer therapies and there exists a need for isoform-specific inhibitors to validate them as potential targets for anti-cancer drug design. The substrate-mimetic approach is a valuable alternative to ATP-mimetic based chemotherapies due to the inherent specificity of the substrate-binding pocket. Preliminary peptidomimetic substrate-mimetic studies provided the foundation for the development of small molecule inhibitors by elucidating critical inhibitor/Akt interactions. The published X-ray crystal structure36 along with flexible ligand docking (GOLD)37 and extensive structure–activity relationships allowed the design of the first non-peptidic substrate-mimetic inhibitor of Akt. Through optimizations of the various substituents we improved the affinity of the non-peptidic inhibitors to provide more potent inhibitors than the initial peptidomimetics. These substrate-mimetics are more rigid, lipophilic inhibitors that lack amino acids and are significantly smaller in size than the GSK3β peptide.
5. Experimental
5.1. Synthesis and spectroscopic data for representative compounds
5.1.1. General
All air and/or moisture sensitive reactions were carried out under a positive pressure of nitrogen in flame-dried glassware. Solvents dimethylformamide (DMF), tetrahydrofuran (THF), dichloromethane, and toluene were obtained from commercial sources and dried on an Innovative Technology SPS-400 dry solvent system. Flash column chromatography was performed on silica gel (40–63 μm) under a pressure of about 4 psi. Analytical thin layer chromatography (TLC) was carried out on glass plates coated with 0.25 mm of silica gel with 254 nm fluorescent indicator (Ba ker, Si250F). 1H and 13C spectra were recorded on a Bruker AM-500 or Bruker AM-400 spectrometer. Chemical shifts are reported in δ (ppm) relative to tetramethylsilane. All coupling constants are described in Hz. Analysis and purification by revere phase HPLC (rpHPLC) were performed using either a Waters 2487 dual λ UV detector with a Waters 1525EF binary pump using a Phenomenex Luna 5 μ C18(2) 250 × 21 mm column run at 20 mL/min (preparative), or a Waters 2487 dual λ UV detector with a Waters 1525 binary pump using a Microsorb-MV 300 Å C18 250 × 4.6 mm column run at 1 mL/min (analytical), using gradient mixtures of water with 0.1% trifluoroacetic acid (TFA) (A) and 10:1 acetonitrile/water (B) with 0.1% TFA. Compound purity was confirmed by analytical rpHPLC using a linear gradient from 100% A to 100% B with changing solvent composition over either: (I) 20 minutes or (II) a linear gradient from 90% A to 80% B over 25 min or (III) a linear gradient from 100% A to 60% B over 20 min. All gradients started after an initial 2 min of 100% A. Preparative HPLC purifications were performed using Method I and product fractions were always lyophilized to dryness. High resolution mass (HRMS) determinations were performed at the University of Illinois at Urbana-Cham paign Mass Spectrometry Center on a Varian MAT-CH-5.
5.1.2. General coupling/deprotection procedure
The required acid (1.0 equiv) was added in one portion to a solution of DIC (1.0 equiv) in CH2Cl2 (5 mL/mmol), and the resulting solution stirred at room temperature for 5 min under a nitrogen atmosphere. The required amine (2.0 equiv) was then dissolved in a solution of CH2Cl2 (5 mL/mmol) and added to the activated acid drop-wise in one portion followed by the addition of DMAP (catalytic). The resulting solution was stirred for 20 h, then diluted with CH2Cl2 (100 mL/mmol) and washed successively with sat. NaHCO3 (100 mL/mmol) and brine (100 mL/mmol). The organic layer was dried over Na2SO4, and the solvent was removed under reduced pressure to afford the desired amide. The amide was then dissolved in TFA (1 mL/mmol) and stirred at room temperature for 1 h. The TFA was evaporated under reduced pressure and the crude product was dissolved in acetonitrile/H2O and purified by preparatory HPLC column chromatography to afford the desired amide as the TFA salt.
5.1.3. 6-(2-Guanidino-ethylamino)-biphenyl-3-carboxylic acid {4-[bis-(4-cyano-benzyl)-amino]-phenyl}-amide (21bi)
Reaction of 18b (0.050 g, 0.100 mmol) and 20i (0.034 g, 0.100 mmol) according to procedure above, yield after preparatory HPLC (47%, tan powder). 1H NMR (DMSO, 500 MHz) δ 9.63 (1H, s, NH), 7.82 (1H, dd, J = 8.7 and 2.1 Hz, Ar-H), 7.79 (4H, d, J = 8.1 Hz, Ar-H), 7.66 (1H, d, J = 2.1 Hz, Ar-H), 7.63 (1H, b, NH), 7.52–7.01 (14H, m, Ar-H and NH), 6.81 (1H, d, J = 8.7 Hz, Ar-H), 6.61 (2H, d, J = 9.1 Hz, Ar-H), 5.19 (1H, b, NH), 4.78 (4H, s, CH2), 3.31–3.27 (4H, m, CH2); 13C NMR (DMSO, 500 MHz) δ 164.2, 156.9, 147.0, 145.3, 143.5, 138.4, 132.3, 129.7, 129.0, 128., 128.7, 127.6, 127.3, 125.9, 122.3, 122.0, 118.8, 112.5, 109.4, 109.2, 54.2, 41.4, 39.7. HRMS (ESI) m/z calculated for C38H34N8OH+ 619.2934. Found 619.2933. rpHPLC tR: condition (I) 13.930 (II) 13.193 min, purity 100%.
5.1.4. N-{4-[Bis-(4-cyano-benzyl)-amino]-phenyl}-4-(2-guanidino-ethylamino)-3-naphthalen-2-yl-benzamide (21ci)
Reaction of 18c (0.040 g, 0.073 mmol) and 20i according to procedure above, yield after preparatory HPLC (37%, white powder). 1H NMR (MeOD, 500 MHz) δ 7.98 (1H, d, J = 8.4 Hz, Ar-H), 7.92– 7.86 (4H, m, Ar-H), 7.77 (1H, d, J = 1.9 Hz, Ar-H), 7.67–7.66 (4H, m, Ar-H), 7.56–7.36 (9H, m, Ar-H), 6.89 (1H, d, J = 8.7 Hz, Ar-H), 6.67 (2H, d, J = 8.9 Hz, Ar-H), 4.77 (4H, s, CH2), 3.44 (2H, t, J = 6.0 Hz, CH2), 3.38 (2H, t, J = 6.0 Hz, CH2); 13C NMR (MeOD, 500 MHz) δ 168.5, 158.9, 149.3, 146.5, 137.4, 135.3, 134.3, 133.6, 131.4, 130.6, 130.1, 129.8, 129.4, 129.1, 129.0, 128.8, 128.7, 128.4, 127.6, 127.4, 124.4, 124.0, 119.8, 114.6, 111.8, 110.6, 56.2, 43.0, 41.8. HRMS (ESI) m/z calculated for C42H36N8OH+ 669.3090 Found 669.3109. rpHPLC tR: condition (I) 14.128 (II) 13.549 min, purity 100%.
5.1.5. N-{4-[Bis-(4-cyano-benzyl)-amino]-phenyl}-4-(2-guanidino-acetylamino)-3-naphthalen-2-yl-benzamide (29b)
Reaction of 28 (0.041 g, 0.073 mmol) and 20i according to procedure above, yield after preparatory HPLC (58%, white powder). 1H NMR (MeOD, 500 MHz) δ 8.01–7.91 (7H, m, Ar-H), 7.68–7.66 (4H, m, Ar-H), 7.57–7.41 (9H, m, Ar-H), 6.70 (2H, d, J = 9.1 Hz, Ar-H), 4.79 (4H, s, CH2), 3.95 (2H, s, CH2); 13C NMR (MeOD, 500 MHz) δ 168.3, 167.6, 159.4, 146.7, 146.4, 138.4, 136.8, 135.1, 134.4, 133.6, 131.3, 130.2, 129.6, 129.5, 129.3, 129.0, 128.8, 128.5, 127.9, 127.7, 126.0, 125.9, 125.9, 124.3, 119.8, 114.5, 111.8, 56.1, 45.2. HRMS (ESI) m/z calculated for C42H34N8O2H+ 683.2883. Found 683.2872. rpHPLC tR: condition (I) 13.799 (II) 12.997 min, purity 100%.
5.1.6. {[4-(5-Nitro-1H-indol-2-yl)-phenylcarbamoyl]-methyl}-carbamic acid tert-butyl ester (33)
In a pressure vessel, 32 (0.940 g, 2.29 mmol) and copper (II) acetate (0.083 g, 0.46 mmol) were dissolved in dry DMF (10 mL). The reaction was placed under a nitrogen atmosphere and purged three times under high vacuum to provide a complete nitrogen atmosphere. The vessel was then sealed and stirred for 72 h at 120 °C. The reaction was then cooled to room temperature and the DMF was evaporated under reduced pressure and the residue was dissolved in EtOAc (100 mL) and washed with brine (1 × 100 mL). The organic suspension was then filtered to afford the desired in dole 33 as a yellow solid (0.498 g, 66%). 1H NMR (DMSO, 400 MHz) δ 12.24 (1H, s, NH), 10.13 (1H, s, NH), 8.51 (1H, d, J = 2.2 Hz, Ar-H), 7.99 (1H, dd, J = 9.0 and 2.2 Hz, Ar-H), 7.85 (2H, d, J = 8.7 Hz, Ar-H), 7.72 (2H, d, J = 8.7 Hz, Ar-H), 7.53 (1H, d, J = 9.0 Hz, Ar-H), 7.11–7.08 (2H, m, Ar-H and NH), 3.75 (2H, d, J = 6.1 Hz, CH2), 1.40 (9H, s, CH3); 13C NMR (DMSO, 400 MHz) δ 168.3, 155.9, 141.3, 140.8, 140.2, 139.1, 128.0, 126.0, 125.7, 119.2, 116.6, 116.6, 111.4, 99.9, 78.0, 43.7, 28.1. HRMS (ESI) m/z calculated for C21H22N4O5H+ 411.1668. Found 411.1675.
5.1.7. {[4-(5-Amino-1H-indol-2-yl)-phenylcarbamoyl]-methyl}-carbamic acidtert-butyl ester (34)
To a solution of 33 (0.200 g, 0.49 mmol) in anhydrous CH3OH(5 mL) was added 10% Pd/C (0.010 g). The reaction was placed under a hydrogen balloon (1 atm) and purged three times under high vacuum to provide a complete hydrogen atmosphere then stirred at room temperature for 4 hours. The reaction mixture was then filtered through a celite pad and the filtrate was evaporated under reduced pressure. The crude product was purified by FCC eluting with 80% EtOAc: 20% hexanes to afford 34 as a gray solid (0.170 g, 92%). 1H NMR (DMF, 500 MHz) δ 11.03 (1H, s, NH), 10.12 (1H, s, NH), 7.84–7.77 (4H, m, Ar-H), 7.14 (1H, d, J = 8.3 Hz, Ar-H), 6.99 (1H, br, NH), 6.82 (1H, s, Ar-H), 6.62–6.61 (2H, m, Ar-H), 4.58 (2H, br, NH2), 3.94 (2H, s, CH2), 1.44 (9H, s, CH3); 13C NMR (DMF, 500 MHz) δ 169.5, 157.5, 143.0, 139.5, 138.8, 132.7, 131.4, 129.4, 126.2, 120.6, 113.4, 112.3, 104.4, 98.1, 79.4, 45.4, 29.0. HRMS (ESI) m/z calculated for C21H24N4O3H+ 381.1927. Found 381.1928.
5.1.8. [(4-{5-[Bis-(4-cyano-benzyl)-amino]-1H-indol-2-yl}- phenylcarbamoyl)-methyl]-carbamic acid tert-butyl ester (35b)
To a solution of 34 (0.107 g, 0.281 mmol in dry DMF (1.5 mL) was added anhydrous potassium carbonate (0.077 g, 0.562 mmol) and 4-(bromomethyl)benzonitrile (0.110 g, 0.562 mmol). The resulting reaction was placed under a nitrogen atmosphere and stirred at room temperature for 3 h. The DMF was evaporated under reduced pressure and the residue was dissolved in EtOAc (30 mL) and washed with 10% citric acid (30 mL) and brine (30 mL) and the organic layer was dried over Na2SO4 and filtered. The solvent was removed under reduced pressure and the crude product was purified by FCC on silica gel eluting with 60% EtOAc: 40% hexanes (78%, light blue solid). 1H NMR (DMSO, 500 MHz) δ 11.11 (1H, s, NH), 9.99 (1H, s, NH), 7.78 (4H, d, J = 8.0 Hz, Ar-H), 7.70 (2H, d, J = 8.5 Hz, Ar-H), 7.62 (2H, d, J = 8.5 Hz, Ar-H), 7.49 (4H, d, J = 8.0 Hz, Ar-H), 7.17 (1H, d, J = 8.8 Hz, Ar-H), 7.05 (1H, t, J = 5.9 Hz, NH), 6.72–6.66 (2H, m, Ar-H), 6.55 (1H, s, Ar-H), 4.71 (4H, s, CH2), 3.73 (2H, d, J = 5.9 Hz, CH2), 1.40 (9H, s, CH3); 13C NMR (DMSO, 500 MHz) δ 168.09, 155.82, 145.80, 141.57, 137.93, 137.76, 132.17, 131.33, 129.36, 127.92, 127.23, 125.11, 119.14, 118.81, 111.57, 109.30, 104.00, 97.27, 79.06, 77.94, 55.52, 43.69, 28.11. HRMS (ESI) m/z calculated for C37H34N6O3H+ 611.2771. Found 611.2758.
5.1.9.N-(4-{5-[Bis-(4-cyano-benzyl)-amino]-1H-indol-2-yl}-phenyl)-2-guanidino-acetamide (36b)
A solution of 35b (0.120 g, 0.200 mmol) in 1:1 CH2Cl2:TFA was stirred at room temperature for 1 h to remove the Boc protecting group. The solvent was removed under reduced pressure and the crude amine was dried under high vacuum for 1 h. The amine was then dissolved in anhydrous CH3OH (5 mL) and triethylamine (279 μL, 2.00 mmol) and N,N’-bis(tert-butoxycarbonyl)-1H-pyra zole-1-carboxyamidine (0.093 g, 0.300 mmol) were added and the resulting solution was stirred for 24 h. The CH3OH was evaporated under reduced pressure and the concentrates were dissolved in EtOAc (20 mL) and washed consecutively with 10% citric acid (20 mL) and brine (20 mL). The organic layer was dried over sodium sulfate (Na2SO4) and the solvent was removed under reduced pressure. The amine was purified by FCC eluting with 40% EtOAc: 60% hexanes to yield N,N’-Bis-tert-butoxycarbonyl-N”-(4-{5-[Bis-(4-cyano-benzyl)-amino]-1H-indol-2-yl}-phenyl)-2-guanidino-acet-amide (62%, light blue oil). 1H NMR (CDCl3, 400 MHz) δ 11.37 (1H, s, NH), 9.80 (1H, s, NH), 8.98 (1H, t, J = 5.0 Hz, NH), 8.48 (1H, s, NH), 7.58–7.26 (12H, m, Ar-H), 7.15 (1H, d, J = 8.7 Hz, Ar-H), 6.86 (1H, s, Ar-H), 6.68 (1H, d, J = 8.2 Hz, Ar-H), 6.55 (1H, s, Ar-H), 4.59 (4H, s, CH2), 4.13 (2H, d, J = 5.0 Hz, CH2), 1.52 (9H, s, CH3), 1.50 (9H, s,CH3); 13C NMR (CDCl3, 400 MHz) δ 166.7, 162.4, 156.9, 152.7, 144.7, 142.9, 138.7, 137.6, 132.4, 131.7, 130.1, 127.9, 127.8, 125.5, 119.7, 118.9, 112.4, 111.7, 110.8, 105.3, 98.6, 84.0, 80.1, 56.1, 46.5, 28.3, 28.0. HRMS (ESI) m/z calculated for C43H44N8O5H+ 753.3513. Found 753.3510. A solution of N,N’-Bis-tert-butoxycarbonyl-N”-(4-{5-[Bis-(4-cyano-benzyl)-amino]-1H-indol-2-yl}-phenyl)-2- guanidino-acetamide (0.120 g, 0.200 mmol) in 1:1 CH2Cl2:TFA was stirred at room temperature for 1 h to remove the Boc protecting group. The solvent was removed under reduced pressure and the crude product was purified by HPLC. Yield of 36b after preparatory HPLC (20%, pink TFA salt). 1H NMR (DMSO, 500 MHz, 330 K) δ 10.98 (1H, s, NH), 10.08 (1H, s, NH), 7.75–7.71 (6H, m, Ar-H), 7.63 (2H, d, J = 8.7 Hz, 1H), 7.51–7.50 (5H, m, Ar-H and NH), 7.20–7.18 (4H, Ar-H and NH), 6.82 (1H, d, J = 2 Hz, Ar-H), 6.74 (1H, dd, J = 8.8 and 2 Hz, Ar-H), 6.56 (1, s, Ar-H), 4.68 (4H, s, CH2), 4.07 (2H, d, J = 6.1 Hz, CH2); 13C NMR (DMSO, 500 MHz, 330 K) δ 165.6, 157.4, 145.4, 141.6, 137.5, 131.8, 131.5, 129.1, 127.8, 127.5, 124.9, 119.2, 118.3, 111.9, 111.8, 111.3, 109.2, 104.7, 97.3, 55.6, 43.9. HRMS (ESI) m/z calculated for C33H28N8OH+ 553.2464. Found 553.2491. rpHPLC tR: condition (I) 11.432 (II) 8.610 min, purity 97%.
Supplementary Material
Acknowledgments
K.J.K. thanks Sanofi-Aventis for their funding of the American Chemical Society division of Medicinal Chemistry predoctoral fellowship. We also thank the National Institutes of Health (1R01 CA107078-01) for financial support of this work.
Footnotes
Supplementary data
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.bmc.2008.09.058.
References and notes
- 1.Staal SP. Proc. Natl. Acad. Sci. U. S. A. 1987;84:5034. doi: 10.1073/pnas.84.14.5034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Staal SP, Hartley JW. J. Exp. Med. 1988;167:1259. doi: 10.1084/jem.167.3.1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Staal SP, Hartley JW, Rowe WP. Proc. Natl. Acad. Sci. U.S.A. 1977;74:3065. doi: 10.1073/pnas.74.7.3065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Staal SP, Huebner K, Croce CM, Parsa NZ, Testa JR. Genomics. 1988;2:96. doi: 10.1016/0888-7543(88)90114-0. [DOI] [PubMed] [Google Scholar]
- 5.Brazil DP, Hemmings BA. Trends Biochem. Sci. 2001;26:657. doi: 10.1016/s0968-0004(01)01958-2. [DOI] [PubMed] [Google Scholar]
- 6.Jones PF, Jakubowicz T, Hemmings BA. Cell Regul. 1991;2:1001. doi: 10.1091/mbc.2.12.1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jones PF, Jakubowicz T, Pitossi FJ, Maurer F, Hemmings BA. Proc. Natl. Acad. Sci. U.S.A. 1991;88:4171. doi: 10.1073/pnas.88.10.4171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Coffer PJ, Woodgett JR. Eur. J. Biochem. 1991;201:475. doi: 10.1111/j.1432-1033.1991.tb16305.x. [DOI] [PubMed] [Google Scholar]
- 9.Bellacosa A, Testa JR, Staal SP, Tsichlis PN. Science. 1991;254:274. doi: 10.1126/science.254.5029.274. [DOI] [PubMed] [Google Scholar]
- 10.Bellacosa A, Defeo D, Godwin AK, Bell DW, Cheng JQ, Altomare DA, Wan MH, Dubeau L, Scambia G, Masciullo V, Ferrandina G, Panici PB, Mancuso S, Neri G, Testa JR. Int. J. Cancer. 1995;64:280. doi: 10.1002/ijc.2910640412. [DOI] [PubMed] [Google Scholar]
- 11.Cheng JQ, Godwin AK, Bellacosa A, Taguchi T, Franke TF, Hamilton TC, Tsichlis PN, Testa JR. Proc. Natl. Acad. Sci. U.S.A. 1992;89:9267. doi: 10.1073/pnas.89.19.9267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cheng JQ, Ruggeri B, Klein WM, Sonoda G, Altomare DA, Watson DK, Testa JR. Proc. Natl. Acad. Sci. U.S.A. 1996;93:3636. doi: 10.1073/pnas.93.8.3636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nakatani K, Thompson DA, Barthel A, Sakaue H, Liu W, Weigel RJ, Roth RA. J. Biol. Chem. 1999;274:21528. doi: 10.1074/jbc.274.31.21528. [DOI] [PubMed] [Google Scholar]
- 14.Downward J. Nat. Cell Biol. 1999;1:E33. doi: 10.1038/10026. [DOI] [PubMed] [Google Scholar]
- 15.Ozes ON, Mayo UD, Gustin JA, Pfeffer SR, Pfeffer LM, Donner DB. Nature (London, UK) 1999;401:82. doi: 10.1038/43466. [DOI] [PubMed] [Google Scholar]
- 16.Pugazhenthi S, Nesterova A, Sable C, Heidenreich KA, Boxer LM, Heasley LE, Reusch JEB. J. Biol. Chem. 2000;275:10761. doi: 10.1074/jbc.275.15.10761. [DOI] [PubMed] [Google Scholar]
- 17.Datta SR, Brunet A, Greenberg ME. Genes Dev. 1999;13:2905. doi: 10.1101/gad.13.22.2905. [DOI] [PubMed] [Google Scholar]
- 18.Downward J. Curr. Opin. Cell Biol. 1998;10:262. doi: 10.1016/s0955-0674(98)80149-x. [DOI] [PubMed] [Google Scholar]
- 19.Chan TO, Rittenhouse SE, Tsichlis PN. Annu. Rev. Biochem. 1999;68:965. doi: 10.1146/annurev.biochem.68.1.965. [DOI] [PubMed] [Google Scholar]
- 20.Kandel ES, Hay N. Exp. Cell Res. 2000;254:196. doi: 10.1006/excr.1999.4690. [DOI] [PubMed] [Google Scholar]
- 21.Cantrell DA. J. Cell Sci. 2001;114:1439. doi: 10.1242/jcs.114.8.1439. [DOI] [PubMed] [Google Scholar]
- 22.Becker S, Groner B, Muller CW. Nature. 1998;394:145. doi: 10.1038/28101. [DOI] [PubMed] [Google Scholar]
- 23.Brodbeck D, Cron P, Hemmings BA. J. Biol. Chem. 1999;274:9133. doi: 10.1074/jbc.274.14.9133. [DOI] [PubMed] [Google Scholar]
- 24.Yang L, Dan HC, Sun M, Liu Q, Sun X.-m., Feldman RI, Hamilton AD, Polokoff M, Nicosia SV, Herlyn M, Sebti SM, Cheng JQ. Cancer Res. 2004;64:4394. doi: 10.1158/0008-5472.CAN-04-0343. [DOI] [PubMed] [Google Scholar]
- 25.Vivanco I, Sawyers CL. Nat. Rev. Cancer. 2002;2:489. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- 26.Li Q, Li T, Zhu G-D, Gong J, Claibone A, Dalton C, Luo Y, Johnson EF, Shi Y, Liu X. Bioorg. Med. Chem. Lett. 2006;16:1679. doi: 10.1016/j.bmcl.2005.12.017. [DOI] [PubMed] [Google Scholar]
- 27.Reuveni H, Livnah N, Geiger T, Kleid S, Ohne O, Cohen I, Benhar M, Gellerman G, Levitzki A. Biochemistry. 2002;41:10304. doi: 10.1021/bi0202530. [DOI] [PubMed] [Google Scholar]
- 28.Breitenlechner CB, Wegge T, Berillon L, Graul K, Marzenell K, Friebe W-G, Thomas U, Schumacher R, Huber R, Engh RA, Masjost B. J. Med. Chem. 2004;47:1375. doi: 10.1021/jm0310479. [DOI] [PubMed] [Google Scholar]
- 29.Woods KW, Fischer JP, Claiborne A, Li T, Thomas SA, Zhu GD, Diebold RB, Liu XS, Shi Y, Klinghofer V, Han EK, Guan R, Magnone SR, Johnson EF, Bouska JJ, Olson AM, de Jong R, Oltersdorf T, Luo Y, Rosenberg SH, Giranda VL, Li Q. Bioorg. Med. Chem. 2006;14:6832. doi: 10.1016/j.bmc.2006.06.047. [DOI] [PubMed] [Google Scholar]
- 30.Livnah N, Levitzki A, Senderovitz H, Yechezkel T, Salitra Y, Litman P, Ohne O. Develogen Israel Ltd.; Israel. Application: WO: 2004. p. 143. [Google Scholar]
- 31.Livnah N, Yechezkel T, Salitra Y, Perlmutter B, Ohne O, Cohen I, Litman P, Senderowitz H. Peptor Ltd.; Israel, Application: WO: 2003. p. 64. [Google Scholar]
- 32.Luo Y, Smith RA, Guan R, Liu XS, Klinghofer V, Shen JW, Hutchins C, Richardson P, Holzman T, Rosenberg SH, Giranda VL. Biochemistry. 2004;43:1254. doi: 10.1021/bi034515p. [DOI] [PubMed] [Google Scholar]
- 33.Parang K, Cole PA. Pharmacol. Ther. 2002;93:145. doi: 10.1016/s0163-7258(02)00184-5. [DOI] [PubMed] [Google Scholar]
- 34.Litman P, Ohne O, Ben-Yaakov S, Shemesh-Darvish L, Yechezkel T, Salitra Y, Rubnov S, Cohen I, Senderowitz H, Kidron D, Livnah O, Levitzki A, Livnah N. Biochemistry. 2007;46:4716. doi: 10.1021/bi061928s. [DOI] [PubMed] [Google Scholar]
- 35.Kayser KJ, Glenn MP, Sebti SM, Cheng JQ, Hamilton AD. Bioorg. Med. Chem. Lett. 2007;17:2068. doi: 10.1016/j.bmcl.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 36.Yang J, Cron P, Good VM, Thompson V, Hemmings BA, Barford D. Nat. Struct. Biol. 2002;9:940. doi: 10.1038/nsb870. [DOI] [PubMed] [Google Scholar]
- 37.Jones G, Willett P, Glen RC, Leach AR, Taylor RJ. Mol. Biol. 1997;267:727. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- 38.Molecular Devices, IMAP® technology. http://www.moleculardevices.com/pages/reagents/imap.html.
- 39.Goto S, Tsuboi H, Kanoda M, Mukai K, Kagara K. Org. Process Res. Dev. 2003;7:700. [Google Scholar]
- 40.Andrus Merritt B, Mettath Sashikumar N, Song C. J. Org. Chem. 2002;67:8284. doi: 10.1021/jo026217o. [DOI] [PubMed] [Google Scholar]
- 41.Mizuno T, Ishino Y. Tetrahedron. 2002;58:3155. [Google Scholar]
- 42.Hiroya K, Itoh S, Sakamoto T. J. Org. Chem. 2004;69:1126. doi: 10.1021/jo035528b. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.