

Abstract
Tandemly arrayed genes (TAGs) are an important genomic component. However, most previous studies have focused on individual TAG families, and a broader characterization of their genomic distribution is not yet available. In this study, we examined the distribution of TAGs in the Arabidopsis thaliana genome and examined TAG density with relation to recombination rates. Recombination rates along A. thaliana chromosomes were estimated by comparing a genetic map with the genome sequence. Average recombination rates in A. thaliana are high, and rates vary more than threefold among chromosomal regions. Comparisons between TAG density and recombination indicate a positive correlation on chromosomes 1, 2, and 3. Moreover, there is a consistent centromeric effect. Relative to single-copy genes, TAGs are proportionally less frequent in centromeres than on chromosomal arms. We also examined several factors that have been proposed to affect the sequence evolution of TAG members. Sequence divergence is related to the number of members in the TAG, but genomic location has no obvious effect on TAG sequence divergence, nor does the presence of unrelated genes within a TAG. Overall, the distribution of TAGs in the genome is not consistent with theoretical models predicting the accumulation of repeats in regions of low recombination but may be consistent with stabilizing selection models of TAG evolution.
The evolution, maintenance, and organization of repetitive DNA have been the focus of many theoretical and empirical studies. Theoretical studies generally assume that DNA repeats are non-coding, that increases in array size are deleterious (i.e., the fitness of an individual is inversely related to the number of repeats it harbors), and that the number of repeats is modified, at least in part, by unequal crossing over (UCO). Under these conditions, high recombination regions of a genome should harbor little repetitive DNA relative to low recombination regions (Charlesworth et al. 1986; Stephan 1986). This is true both because selection is more efficient in high recombination regions and because high recombination regions may have a higher probability of UCO events, thereby providing more opportunities to generate favorable, repeat-poor alleles. This simple prediction is confounded by other evolutionary and mechanistic factors but appears to fit several empirical observations. For example, both transposable elements and satellite DNAs tend to accumulate preferentially in low recombination genomic regions (see John and Miklos 1979; Arabidopsis Genome Initiative 2000; Bartolomé et al. 2002).
Despite several recent studies of repetitive DNA in sequenced genomes, the organization and evolution of tandemly arrayed coding regions has not been studied carefully. Tandemly arrayed genes (or TAGs) comprise a large proportion of sequenced genomes; for example, 10% and 17% of the total predicted genes in the Caenorhabditis elegans and Arabidopsis thaliana genomes, respectively, are members of a TAG (Semple and Wolfe 1999; Arabidopsis Genome Initiative 2000). TAGs are also an important functional genomic component and are likely evolutionarily important because they are a reservoir of genetic redundancy that can be co-opted for new gene functions (Ohno 1970) or new expression patterns (Force et al. 1999). However, the evolutionary forces acting on tandem duplications are unclear. For example, Ohno (1970) hypothesized that tandem duplication of coding regions may often be deleterious because duplication disrupts gene dosage and may also initiate additional UCO events that cause further fluctuations in gene dosage. If tandem duplication is primarily deleterious, TAGs—such as other repetitive sequences—are expected to accumulate in centromeric regions where recombination is sparse and selection against deleterious mutations is ineffective.
Here we characterize the genomic distribution of tandemly repeated protein coding regions in the A. thaliana genome and explore the relationship between the distribution of TAGs and chromosomal recombination rates. To make this comparison, we first estimate recombination rates along the physical length of A. thaliana chromosomes by comparing genetic and physical maps. Given these estimates, we address the following questions. (1) How many TAGs are in the A. thaliana genome, and how are they distributed across the genome? (2) Do TAGs preferentially accumulate in low recombination regions, as predicted by theory? (3) Is there a relationship between TAG location and sequence divergence among TAG members?
RESULTS
Genetic Map and Recombination Rates
The relationship between genetic and physical length is shown for all chromosomes (Fig. 1). Chromosomes 2 and 4 are acrocentric, whereas chromosomes 1, 3, and 5 are metacentric. The density of genetic markers used to estimate recombination rates ranged from 1.66 to 3.09 markers/Mb and from 0.39 to 0.58 markers/cM. Average recombination rates, calculated by comparing Mb and cM distances, as has been done previously (Arabidopsis Genome Initiative 2000), were 4.25, 4.49, 3.88, 6.45, and 5.04 cM/Mb for chromosomes 1 through 5, respectively.
Figure 1.

Genetic and physical maps of all chromosomes and the distribution of TAGs with respect to the maps. For each chromosome, circles represent the genetic and physical position of markers, the histogram represents the number of TAG genes in the region, and the bar represents the centromeric region.
The contrast between physical and genetic lengths was used to estimate recombination rates by two methods: a global method and a local method (Fig. 2). With both methods, all five chromosomes exhibited reduced rates of recombination near the centromere. In noncentromeric regions, recombination rate estimates varied as a function of chromosomal location. For example, estimates of telomeric recombination rates exceeded 6 cM/Mb for chromosomes 4 and 5, but telomeric recombination rates were less pronounced for chromosome 1, in which the highest recombination rates were found in the middle of chromosomal arms (Fig. 2). For all chromosomes, rate estimates varied roughly threefold among chromosomal regions, with as much as a fivefold range in recombination rates between regions of chromosome 4. For all chromosomes except chromosome 3, estimates of recombination rates by the local approach were significantly correlated with estimates of global estimates from the fifth order polynomial, but not the fourth order polynomial (data not shown). We thus rely on fifth order inferences for the remainder of the study.
Figure 2.

Recombination rates estimated by the global and local approaches. + represents global rate estimates by a fifth-order polynomial; circles represent local estimates. The bar represents the centromeric region and estimate recombination rates in this region.
For chromosome 3, the global and local estimates have a large discrepancy (Fig. 2). Given that the graph of the physical and genetic maps of chromosome 3 is similar to other chromosomes (Fig. 1) and also given estimated recombination rates for other chromosomes (Fig. 2), the differences between local and global estimates for chromosome 3 likely reflect a poor fit of the global polynomial function. Low correlations between local and global estimates persisted for this chromosome when global rates were estimated with higher- and lower-order polynomials (data not shown). Because polynomial curve fitting is more sensitive to individual outliers than is the local approach, it is possible that there are misplaced markers on chromosome 3 that adversely influence global rate estimates. In contrast, local estimates are imprecise only for the regions in which misplaced markers reside, but these markers do not affect estimates across the entire chromosome. Based on these considerations, it is likely that local estimates of recombination are more accurate for chromosome 3. Nevertheless, we include both estimates in subsequent analyses.
The Distribution of TAGs on the Chromosomes
The number of TAGs identified depends on the TAG definition. The definition is affected by two factors: the E-value threshold of BLASTP and the number of spacers allowed within a TAG. Figure 3 shows the effect of varying these two factors on the number of identified TAGs. Not surprisingly, for all chromosomes there were more TAGs with higher E-values. Similarly, the number of TAGs increased with the number of spacers in the array. For any given E-value, the increase in the number of TAGs was greatest between zero and one spacer; after this, the number of TAGs increased slowly as a function of the number of spacers (Fig. 3). Given these observations, we analyzed two data sets that represent a broad range of TAG definitions. The first data set was based on a relatively strict TAG definition: an E-value of 10-30 and no spacers. Hereafter, this data set is called the “10-30/0” data set to reflect the 10-30 E-value and zero spacers. The second data set was based on a less strict TAG definition: an E-value of 10-10 and one spacer, which we called the “10-10/1” data set.
Figure 3.

The number of TAGs as a function of spacers and E-values. The numbers represent the chromosomes. For each chromosome, red numbers represent TAGs identified with an E-value 10-10, blue numbers represent TAGs identified with an E-value of 10-20, and black numbers represent TAGs identified with an E-value of 10-30.
Detailed information on TAG identification for these two data sets is listed (Table 1). Both data sets indicated that TAGs comprise a substantial amount of the A. thaliana genome. For the 10-30/0 data set, there were 1237 TAG arrays consisting of 3207 genes. The proportion of TAGs out of all genes was ∼12.6%. For the 10-10/1 data set, there were 1587 TAG arrays consisting of 4249 genes, and the proportion of TAGs out of all genes was ∼16.6%. On each chromosome, the TAG proportion ranged from 10.9% to 13.9% in the 10-30/0 data set and from 15.0% to 18.2% in the 10-10/1 data set (Table 1). It should be mentioned that the Arabidopsis Genome Initiative (2000) documented 1528 TAGs using an E-value of 10-20 and one spacer; with the same criteria, we found only 1476 TAGs. Differences between our results and the Arabidopsis Genome Initiative (2000) probably reflect alterations in genome sequence annotation since the Arabidopsis Genome Initiative analysis.
Table 1.
The Statistics of TAGs in the Genome for Two Data Sets
|
Data set 10–10/1a
|
Data set 10–30/0b
|
|||||||
|---|---|---|---|---|---|---|---|---|
| Chromosome | Arraysc | #TAGd | %TAGe | Arraysc | #TAGd | %TAGe | Total genes | Total Mb |
| 1 | 432 | 1145 | 17.3 | 330 | 860 | 13.0 | 6606 | 29.6 |
| 2 | 247 | 617 | 15.0 | 184 | 451 | 10.9 | 4122 | 19.6 |
| 3 | 302 | 854 | 16.5 | 235 | 646 | 12.5 | 5163 | 23.3 |
| 4 | 250 | 695 | 18.2 | 196 | 529 | 13.9 | 3809 | 17.5 |
| 5 | 356 | 938 | 16.0 | 292 | 721 | 12.3 | 5845 | 26.3 |
| All | 1587 | 4249 | 16.6 | 1237 | 3207 | 12.6 | 25,545 | 116.3 |
TAG data set based on an E-value = 10–10 and one spacer
TAG data set based on E-value = 10–30 and no spacer
The number of arrays containing TAGS
The total number of TAGs
The percentage of TAGs out of all genes
The number of members within a TAG was distributed similarly on all five chromosomes for both data sets (data not shown). The distribution of TAG sizes for the entire genome is shown for the 10-30/0 data set (Fig. 4). Most TAGs had two(∼69%) or three (∼18%) members in the array; TAGs with more members constituted only ∼13% of all TAGs.
Figure 4.

The distribution of the number of genes in a TAG for the data set with E-value 10-30 and no spacer.
The Distribution of TAGs in the Context of Recombination Rates
The physical distribution of TAGs is graphically represented for the 10-30/0 data set (Fig. 5). For all chromosomes and both data sets, there were few TAGs around the centromere and apparent clusters of TAGs elsewhere. Centromeres have both low levels of recombination and a dearth of coding regions relative to chromosomal arms (Arabidopsis Genome Initiative 2000). If recombination affects the distribution of TAGs, then TAGs should demonstrate a “centromeric effect” beyond that of nontandemly arrayed genes (non-TAGs). To investigate this prediction, we contrasted the TAG density (the proportion of TAGs relative to non-TAGs; see Methods), between centromeres and chromosomal arms (Table 2). The null hypothesis that TAGs are found in equal proportion between centromeric and noncentromeric regions was rejected for both data sets with chromosomes 2 and 3 and for four of five chromosomes with the 10-30/0 data set. Moreover, the prediction holds over all chromosomes for both data sets (P = 0.03 and P < 0.001, respectively).
Figure 5.

The physical distribution of TAGs along each chromosome. For each chromosome, the vertical line represents the physical location of TAGs, the horizontal line represents the chromosome itself, and the open box represents the centromere.
Table 2.
Fisher's Exact Tests, Based on the Number of TAGs and Non-TAG Genes in Centromeric and Noncentromeric Regions
|
Centromeric
|
Noncentromeric
|
||||
|---|---|---|---|---|---|
| Chromosome | No. TAGa | No. geneb | No. TAG | No. gene | P Value |
| Data set 10–10/1 | |||||
| 1 | 98 | 469 | 1047 | 4992 | 0.51 |
| 2 | 55 | 480 | 562 | 3025 | <0.001 |
| 3 | 46 | 368 | 808 | 3941 | <0.001 |
| 4 | 109 | 410 | 586 | 2704 | 0.96 |
| 5 | 73 | 379 | 865 | 4528 | 0.56 |
| All | 381 | 2106 | 3868 | 19,190 | 0.03 |
| Data set 10–30/0 | |||||
| 1 | 49 | 518 | 811 | 5228 | <0.001 |
| 2 | 34 | 501 | 417 | 3170 | <0.001 |
| 3 | 22 | 392 | 624 | 4125 | <0.001 |
| 4 | 64 | 455 | 465 | 2825 | 0.15 |
| 5 | 40 | 412 | 681 | 4712 | <0.01 |
| All | 209 | 2278 | 2998 | 20,060 | <0.001 |
Number of TAGs
Number of non-TAGs
The overall result based on the 10-10/1 data set is not significant after Bonferroni correction for 12 tests (six tests on each of two data sets) and an experiment-wide significance level of 5%. However, the Bonferroni correction is overly conservative for nonindependent tests, and appropriate P-values are difficult to determine for cases such as these in which many of the tests are not independent. Despite this caveat, the overall trend is clear: Relative to non-TAG genes, there are proportionally fewer TAGs in centromeric regions than in non centromeric regions. This trend opposes predictions based on models of satellite DNA evolution (see Charlesworth et al. 1986).
The TAG distribution with respect to the genetic map of each chromosome is shown for the 10-30/0 data set (Fig. 1). This figure provides a graphical description of the relationship between the TAG density and recombination rate, but we also investigated formally the correlation between TAG density and estimated recombination rates. With global recombination estimates, the correlation with TAG density was positive and significant for chromosomes 1 and 2 (Table 3). The correlation was also positive, but not significant, for chromosome 3 and for data summed across all five chromosomes. In contrast, the correlation between TAG density and recombination rates was negative, although not significantly so, for both chromosomes 4 and 5. These results did not vary qualitatively when based on different numbers of partitions across the chromosomes (see Methods; data not shown).
Table 3.
Correlations Between TAG Density and Recombination Rates Estimated by Both the Local and Global Approaches
|
Local estimates
|
||||||
|---|---|---|---|---|---|---|
|
All data
|
Excluding telomere and centromere
|
Global estimates
|
||||
| Chromosome | ra | Pb | r | P | r | P |
| Data 10–10/1 | ||||||
| 1 | 0.38 | 0.11 | 0.70 | 0.02 | 0.54 | 0.05 |
| 2 | 0.59 | 0.03 | 0.53 | 0.09 | 0.70 | 0.01 |
| 3 | 0.80 | 0.01 | 0.90 | 0.01 | 0.01 | 0.48 |
| 4 | –0.32 | 0.85 | –0.30 | 0.79 | –0.21 | 0.71 |
| 5 | –0.09 | 0.61 | 0.01 | 0.47 | –0.25 | 0.75 |
| All | 0.37 | 0.003 | 0.40 | 0.002 | 0.10 | 0.24 |
| Data 10–30/0 | ||||||
| 1 | 0.30 | 0.18 | 0.51 | 0.09 | 0.58 | 0.03 |
| 2 | 0.59 | 0.03 | 0.49 | 0.11 | 0.62 | 0.03 |
| 3 | 0.77 | <0.01 | 0.91 | 0.01 | 0.07 | 0.41 |
| 4 | –0.38 | 0.89 | –0.41 | 0.87 | –0.09 | 0.58 |
| 5 | 0.01 | 0.47 | –0.08 | 0.58 | –0.35 | 0.84 |
| All | 0.37 | 0.002 | 0.34 | 0.01 | 0.12 | 0.20 |
Significant results are shown in bold.
Pearson correlation coefficient between TAG density and recombination rate estimates
All P values calculated by 10,000 bootstrap resamplings
With local recombination rate estimates, positive correlations were also detected on chromosomes 1, 2, and 3 and summed over chromosomes. For both data sets, the positive correlations were significant for chromosomes 2 and 3 and summed across chromosomes. In contrast, TAG density and recombination was negatively correlated for chromosome 4.
One advantage of the local method of rate estimation is that recombination rates can be partitioned by chromosomal region. This is important because telomeric and centromeric regions could be driving observed correlations. The telomeres could drive correlations both because telomeres have unusual recombinational patterns (Wintle et al. 1997) and because telomeres tend to have high estimated recombination rates in A. thaliana (Fig. 2). Centromeres could drive correlations because they lack recombination and contain a relative dearth of TAGs (Table 2). To see whether our correlation results were driven by either of these chromosomal regions, we excluded these regions and recalculated correlations. The results were qualitatively consistent whether the regions were excluded individually (data not shown) or excluded together (Table 3). In short, without centromeric and telomeric data: (1) Correlations between recombination rate and TAG density remained positive on chromosomes 1, 2 and 3, even though some P-values drifted slightly above or below the 5% significance level, (2) the correlations on chromosomes 4 and 5 remained negative, and (3) the correlation across all five chromosomes remained positive and significant.
TAG Sequence Evolution
We used D, the average pairwise distance among members of a TAG, to describe divergence among TAG members (Table 4). The average divergence among TAG members varied little among chromosomes but did vary among data sets as a function of E-value (Table 4). A small number of TAG families were too diverged for reliable analyses; for example, for data set 10-30/0, ∼10.6% of TAG families had D > 1, indicating that TAG members were too diverged for reliable analysis. We therefore limited our analyses to TAGs with D < 1, but inclusion of TAG families with higher D made little qualitative difference to results.
Table 4.
The Mean and Range of D for TAGs
| Chromosome | Data set 10-10/1 Range (mean) | Data set 10-30/0 Range (mean) |
|---|---|---|
| 1 | 0-3.59 (0.67) | 0-2.25 (0.49) |
| 2 | 0-2.80 (0.65) | 0-1.98 (0.53) |
| 3 | 0-2.82 (0.64) | 0-1.60 (0.51) |
| 4 | 0-2.80 (0.76) | 0-1.83 (0.54) |
| 5 | 0-3.06 (0.68) | 0-2.17 (0.53) |
The first analysis with D was to examine the effect of TAG size on sequence divergence, because simulation studies have indicated that, under a UCO model of TAG generation, divergence among TAG members increases with the number genes (Smith 1974). To test whether TAG member divergence was positively correlated with array size, we grouped TAGs based on the number of members in the TAG, combined observations when the group contained fewer than ten observations, and performed Kruskal-Wallis nonparametric rank tests. For both data sets summed over all chromosomes, Kruskal-Wallis rank tests were statistically significant (Table 5), indicating that TAGs with more members have a significantly higher average degree of sequence divergence.
Table 5.
The Effect of TAG Size and the TAG Location on D
|
TAG size
|
TAG location
|
|||
|---|---|---|---|---|
| Factor chromosome | Va | P value | Vb | P value |
| Data set 10-10/1 | ||||
| 1 | 23.55 | <0.001 | 1685 | 0.02 |
| 2 | 8.58 | 0.04 | 533 | 0.90 |
| 3 | 13.70 | <0.01 | 573 | 0.36 |
| 4 | 17.64 | <0.001 | 800 | 0.15 |
| 5 | 15.27 | <0.01 | 868 | 0.59 |
| All | 31.24 | <0.001 | 21,828 | 0.11 |
| Data set 10-30/0 | ||||
| 1 | 15.18 | <0.01 | 1003 | 0.15 |
| 2 | 6.76 | 0.03 | 453 | 0.61 |
| 3 | 20.88 | <0.001 | 477 | 0.72 |
| 4 | 8.30 | 0.02 | 676 | 0.60 |
| 5 | 14.12 | <0.01 | 1101 | 0.40 |
| All | 48.45 | <0.001 | 18,247 | 0.97 |
The Kruskal-Wallis rank sum statistic
The Wilcoxon rank sum statistic
If TAG homogenization via UCO and gene conversion is a continual process, one might expect TAGs near centromeres, where recombination is rare, to be more diverged than are TAGs elsewhere. To test this idea, we contrasted TAGs in centromeric regions with TAGs in noncentromeric regions by applying Wilcoxon's rank test. Because sequence divergence is a function of TAG size, the test was performed only for TAGs with two members. (TAGs with more members were not suitable for tests due to the small number of observations.) For chromosome 1, the 10-1/1 data set shows statistical significance for the contrast between centromeric and noncentromeric regions, but the test was not significant for the other four chromosomes or for the 10-30/0 data set (Table 5). Thus, there is no clear centromeric effect with regard to sequence homogenization among members within a TAG.
Finally, previous studies have indicated that spacers might hinder homogenization among TAG members (Zimmer et al. 1980). Under this hypothesis, one expects that TAGs with spacers are more divergent than are TAGs without spacers. We therefore compared D between TAGs without spacers and with one spacer for TAG size 2, using the 10-10/1 data set. (There were insufficient samples for other TAG sizes.) There was no significant difference between the two types of TAGs for all chromosomes (data not shown); thus, on a genomic scale the presence of spacers does not appear to hinder TAG homogenization.
DISCUSSION
Recombination in Arabidopsis thaliana
Our “local” and “global” estimates of recombination reveal similar patterns of recombination rates along chromosomes, except chromosome 3 (Fig. 2). Three points can be made about these estimates. First, centromeric regions exhibit low recombination rates for all five chromosomes. Estimated centromeric recombination rates vary from ∼2 to ∼4 cM/Mb. These estimates are probably still higher than actual recombination rates, however, because empirical crossing experiments show that there is little or no recombination in these regions (Haupt et al. 2001). Our relatively high estimates could reflect the paucity of markers in centromeric regions. For example, chromosome 1 has no markers within the defined centromeric regions (Fig. 1), and hence, we had to use markers that border the centromeric region to estimate a centromeric rate.
Second, recombination rates are generally elevated in telomeric regions (Fig. 2). This pattern has also been seen in humans (Yu et al. 2001) and mouse (Nachman and Churchill 1996). In the highest recombination regions, estimated rates exceed ∼6 cM/Mb on chromosomes 2 and 3 and ∼8 cM/Mb on chromosomes 4 and 5. Finally, the average recombination rate across the A. thaliana genome is ∼4.8 cM/Mb, and this estimate is within the range of the previous estimates of recombination rates based on tetrad analysis (Copenhaver et al. 1999). This estimated average recombination rate is more than three times higher than the average recombination rate in the human genome (∼1.5 cM/Mb; Payseur and Nachman 2000), six times higher than the average rate in maize (∼0.7 cM/Mb; Fu et al. 2002), and 1.7-fold that of the D. melanogaster genome (∼2.9 cM/Mb; Betancourt and Presgraves 2002). It thus appears that physical (as opposed to effective) recombination rates are highly elevated in the selfing species A. thaliana relative to other model organisms. It has been predicted that selfing organisms should have elevated rates of recombination (Charlesworth and Charlesworth 1979). Our observations match this prediction, but additional contrasts between selfing and nonselfing species are merited.
We also have two cautionary notes. First, calculating an “average” recombination rate can mask substantial variation in local recombination rates among genomic regions. To date, there have been no studies on the scale at which recombination rates vary in A. thaliana, but recombination hot-spots do exist in plants. For example, Fu et al. (2002) found that recombination rates near the bronze (bz) locus can be 40 to 80 times higher than the genome average. Second, recombination rates likely vary through time, particularly in a plant like A. thaliana, whose closest congener (A. lyrata) is outcrossing and differs in chromosome number. There is unfortunately no information on the rate at which recombination rates change through time, but such information would be a valuable contribution to understanding plant genome evolution.
The Evolution and Distribution of TAGs in A. thaliana
TAGs comprise a substantial proportion of the A. thaliana genome; depending on the TAG definition, ≥10% of the genes in the genome are members of a tandem array (Table 1). The distribution of these TAGs is governed, in part, by a centromeric effect, because TAGs are disproportionally underrepresented in centromeric regions relative to non-TAG genes (Table 2). There are thus two centromeric effects with respect to coding regions: a dearth of coding genes in general (Arabidopsis Genome Initiative 2000) and disproportionally fewer TAGs relative to non-TAG genes.
In addition, our analyses consistently identify a positive correlation between TAG density and recombination rate on chromosomes 1, 2, and 3 and also on data summed across chromosomes. The statistical support for these positive correlations varies only slightly with the data—that is, with the 10-10/1 data set, the 10-30/0 data set, or data that exclude telomeric and centromeric regions (Table 3)—but can vary substantially with the method used to estimate recombination rates. This is especially true for chromosome 3, for which we have reason to believe that global recombination rates are inaccurate (see above). If the chromosome 3 global rate estimates are inaccurate, this could explain differences between local and global rate correlations that sum across chromosomes (Table 3).
We do not detect a positive correlation between recombination rates and TAG density on chromosomes 4 and 5—in fact, the correlation is slightly negative (Table 3)—but the overarching picture is one in which TAGs are relatively sparse in low recombination regions of the genome. This picture contradicts theoretical predictions based on models that assume fitness decreases with an increasing number of repeats. Although most of these models were proposed for satellite DNAs—for which it is reasonable to assume either neutral or slightly deleterious effects of repeat amplification—Ohno (1970) specifically proposed that tandem duplication of genic regions is often deleterious for two reasons: (1) Tandem duplication may disrupt dosage balance, and (2) continued UCO is not evolutionarily stable because it leads to copy number fluctuation. Under a deleterious model, TAGs should accumulate in centromeric and low recombination regions. We find no such effect, with a clear trend in the opposite direction.
There are at least two possibilities as to why the TAG distribution in A. thaliana differs from these predictions. The first possibility is that our estimates of recombination rates are either inaccurate or do not reflect long-term recombination rates. Regarding the latter, the difference between our observations and theoretical predictions can only be rectified if one surmises that our observed regions of low recombination are, in fact, regions of historically high recombination (and vice versa). This seems highly unlikely. It also seems unlikely that our results are due entirely to poor estimates of recombination rates, as noisy estimates should not generate significant positive correlations by chance alone on three individual chromosomes.
A second possibility is that TAGs do not generally fit neutral and deleterious models. A few investigators have examined models of repeat evolution that include stabilizing selection (Crow and Kimura 1970; Ohta 1981; Takahata 1981). For example, Crow and Kimura (1970, pp. 294-296) examined the evolution of TAGs under stabilizing selection by assuming that both too few and too many copies of any particular gene are deleterious. This model predicts that the mean number of repeats (k̄) among individuals at a TAG is proportional to the square root of the rate at which the new variants are generated (u); in other words, k̄ ∞ u1/2. This model predicts that k̄ should increase with recombination rate, if three things hold true: (1) The size distribution of tandem mutational events (in terms of the number of genes) is similar across genomic regions, (2) u is a function of the rate of UCO, and (3) the rate of UCO is, in turn, related to recombination rate. To properly evaluate this model requires a population sample that measures the size of TAGs across individuals, and we do not have such information. Nonetheless, our demonstration that TAG density correlates positively with recombination is superficially consistent with this model. Note, however, that the effect of recombination under this model should be quite small (approximately twofold), because recombination varies only three- to fivefold among A. thaliana genomic regions.
The stabilizing selection model has intuitive appeal, because it posits that gene loss is as detrimental as gene gain. In this context it is interesting to note that ∼87% of TAGs in A. thaliana contain only two or three members (Fig. 2), and this estimate varies little over the initial definition used to identify TAGs. Although by no means definitive, this distribution indicates that there could be strong limits on the number of genes within many of the TAGs in the genome.
Under stabilizing selection and other models, one might also assume that TAGs in regions of high recombination should be homogenized by concerted evolution more often than TAGs in low recombination regions. We do not observe this effect either on individual chromosomes (Table 5) or by combining data across chromosomes (e.g., P-value for data set 10-30/0 = 0.96). However, this should not be interpreted as evidence against stabilizing selection, because D can be affected by many additional factors, including different times of TAG origin in different chromosomal regions, a lack of complete correlation between recombination and gene conversion rates, and the possibility that gene conversion is not ongoing in some TAGs.
It is likely that both sequence homogenization and TAG density are a complex function of many factors, including TAG size, TAG function, both intra- and inter-strand recombination (Walsh 1987), the relationship between recombination and UCO, the relationship between UCO and gene conversion, natural selection and, finally, the rate at which these factors change over time. Our data are not consistent with models that predict the accumulation of repeated sequences in low recombination regions and appear to be more consistent with a stabilizing selection model. However, the joint effects of natural selection and recombination on the distribution and maintenance of TAGs has yet to be elucidated fully. Given that TAGs are a large and important component of sequenced genomes, their distribution and evolution merit further research.
METHODS
Identification of TAGs
All predicted A. thaliana open reading frames (ORFs) were downloaded from the Web site ftp://ncbi.nlm.nih.gov/genbank/genomes/A_thaliana in September 2001. According to the genome annotation, there were 6606, 4122, 5136, 3809, and 5845 putative proteins on chromosomes 1 through 5, respectively. BLASTP (Altschul et al. 1997) was performed on each chromosome against itself by using the BLOSUM45 substitution matrix and applying the SEG filter. The number of TAG families varied depending on the E-values used as the search threshold. We therefore examined three E-values (10-10, 10-20, and 10-30) for subsequent TAG identification.
To identify TAGs, the BLASTP hits were first indexed by their chromosomal locations. Not surprisingly, in some cases, we found “nonhomologous genes” between BLAST hits. These non-homologous genes, hereafter called “spacers”, do not hit query sequences under the specified BLAST search threshold. We define a “perfect” TAG as TAGs with no spacers within the array. However, this perfect criterion is probably too stringent given the possibility of subsequent interruption after tandem duplication. We thus relaxed the criterion by allowing spacers within the array. Specifically, we defined a TAG as two or more copies of duplicated genes in an array, and we set the number of allowed spacer genes to range from 0-10. It should be mentioned that the Arabidopsis Genome Initiative (2000) defined TAGs as an array of duplicated genes with two or more copies and one or fewer spacer.
Estimation of Recombination Rates Along Chromosomes
We obtained genetic markers from the Lister and Dean 1993 RI map, with genetic map positions from the TAIR database (ftp://tairpub:tairpub@ftp.arabidopsis.org/home/tair/Maps/mapviewer_data). The physical locations of these genetic markers in the genome sequence were obtained from http://www.arabidopsis.org/servlets/Search?action=new_search&type=marker. Initially, there were altogether 87, 73, 81, 118, and 77 markers on chromosomes 1 through 5, respectively. After plotting genetic positions against physical positions, we found that some markers were not in collinear order, probably reflecting genetic map error. Because noncollinear markers create problems for estimating recombination rates, we parsed our data by computing the longest common subsequences between physical and genetic positions using Algorithm::Diff (Gusfield 1997). After this parsing procedure, we were left with 49, 51, 47, 54, and 53 markers on chromosomes 1 through 5, respectively.
Two approaches were used to estimate recombination rates: global estimation and local estimation. For the global approach, fourth and fifth order polynomials were fitted to all marker points; the derivative of the polynomial represents estimated recombination rates (see Kliman and Hey 1993). For the local approach, we partitioned the chromosomes into centromeric and noncentromeric regions based on previous studies of chromosome structure (Haupt et al. 2001). The centromeric regions were 4.40, 4.35, 4.20, 3.55, and 4.41 Mb in length for chromosomes 1 through 5, respectively, within which there are little or much reduced rates of recombination (Copenhaver et al. 1999; Haupt et al. 2001). For each chromosome, a linear function was fitted to all of the markers in the centromeric regions, using least squares. The slope of the line was taken as the estimate of recombination rates throughout the centromere. For the remaining chromosomal regions, recombination rates were estimated by the same principle, except we estimated the slope in non-overlapping windows that contained five genetic markers.
Estimation of TAG Density and Statistical Analyses of the TAG Distribution
To explore the relationship between recombination rates and the distribution of TAGs along chromosomes, we defined the “TAG density” as the number of TAG members out of the total number of genes within a region. By considering the TAG density relative to non-TAG genes, this measurement inherently corrects for gene distribution effects that are independent of forces that specifically govern the generation and maintenance of TAGs.
We examined the relationship of TAG density and recombination by two statistical approaches. First, we performed Fisher's exact test to examine whether the relative density for centromeric TAGs is equal to those of other chromosomal regions. This test was based on a priori knowledge that the centromere has lower recombination rates than do chromosomal arms (Copenhaver 1999; Haupt et al. 2001). The second analysis examined the correlation between recombination rates and TAG density. To calculate the correlation, we partitioned each chromosome into 10 or 15 equally sized segments. Recombination rates were obtained for each of these partitions, and we calculated the Pearson correlation coefficient between density and recombination. The significance of the correlation was determined by 10,000 bootstraps, in which each bootstrap sampled TAG density with replacements and assigned densities to partitions.
TAG Sequence Evolution
Protein sequences within a TAG were aligned by using T_COFFEE (Notredame et al. 2000), using default parameters. Protein distances were calculated based on Dayhoff's PAM substitution model (Dayhoff et al. 1978) by using Phylip 3.5 (Felsenstein 1990). The computed distance is in units of the expected fraction of amino acids changed. The average pairwise distance of each TAG was calculated using the following formula:
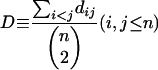 |
where dij is the distance between TAG member i and j; n is the size of a TAG (i.e. the number of genes within a TAG). We used D as a measure of the extent of TAG member sequence divergence.
Acknowledgments
We are very grateful to two anonymous reviewers, both of whom helped improve the manuscript substantively. We also thank S. Wright, A. Mclysaght, M. Tenaillon, T. Long, and W. Fitch for discussions and comments. This study was supported by USDA no. 98-35301-6153 and NSF no. DBI-9872631 and an NSF dissertation grant to L.Z.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.1318503.
References
- Altschul, S.F., Madden, T.L., Schaffer, A.A., Zhang, J., Zhang, Z., Miller, W., and Lipman, D.J. 1997. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 25: 3389-3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arabidopsis Genome Initiative. 2000. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 408: 796-815. [DOI] [PubMed] [Google Scholar]
- Bartolomé, C., Maside, X., and Charlesworth, B. 2002. On the abundance and distribution of transposable elements in the genome of Drosophila melanogaster. Mol. Biol. Evol. 19: 926-937. [DOI] [PubMed] [Google Scholar]
- Betancourt, A.J. and Presgraves, D.C. 2002. Linkage limits the power of natural selection in Drosophila. Proc. Natl. Acad. Sci. 99: 13616-13620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth, B. and Charlesworth, D. 1979. The evolutionary genetics of sexual systems in flowering plants. Proc. R. Soc. Lond. B 205: 513-530. [DOI] [PubMed] [Google Scholar]
- Charlesworth, B., Langley, C.H., and Stephan, W. 1986. The evolution of restricted recombination and the accumulation of repeated DNA sequences. Genetics 112: 947-962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copenhaver, G.N., Kuromori, K., Benito, T., Kaul, M.I., Lin, S., Bevan, X.Y., Murphy, M., Harris, G., Parnell, B., McCombie, L.D., et al. 1999. Genetic definition and sequence analysis of Arabidopsis centromeres. Science 286: 2468-2474. [DOI] [PubMed] [Google Scholar]
- Crow, J. and Kimura, M. 1970. An introduction to population genetics theory. Harper and Row, New York.
- Dayhoff, M.O., Schwartz, R.M., and Orcutt, B.C. 1978. A model for evolutionary change in proteins. In Atlas of protein sequence and structure (ed. M.O. Dayhoff), pp. 345-352. National Biochemical Research Foundation, Washington, DC.
- Felsenstein, J. 1990. PHYLIP manual. University Herbarium, University of California, Berkeley, CA.
- Force, A., Lynch, M., Pickett, F.B., Amores, A., Yan, Y.L., and Postlethwait, J. 1999. Preservation of duplicate genes by complementary degenerative mutations. Genetics 151: 1531-1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu, H.H., Zheng, Z.W., and Dooner, H.K. 2002. Recombination rates between adjacent genic and retrotransposon regions in maize vary by two orders of magnitude. Proc. Natl. Acad. Sci. 99: 1082-1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gusfield, D. 1997. Algorithms on strings trees and sequences. Cambridge University Press, Cambridge, UK.
- Haupt, W., Fischer, T.C., Winderl, S., Fransz, P., and Torres-Ruiz, R.A. 2001. The centromere1 (CEN1) region of Arabidopsis thaliana: Architecture and functional impact of chromatin. Plant J. 27: 285-296. [DOI] [PubMed] [Google Scholar]
- John, B. and Miklos, G.L.G. 1979. Functional aspects of satellite DNA and heterochromatin. Int. Rev. Cytol. 58: 1-114. [DOI] [PubMed] [Google Scholar]
- Kliman, R.M. and Hey, J. 1993. Reduced natural selection associated with low recombination in Drosophila melanogaster. Mol. Biol. Evol. 10: 1239-1258. [DOI] [PubMed] [Google Scholar]
- Nachman, M.W. and Churchill, G.A. 1996. Heterogeneity in rates of recombination across the mouse genome. Genetics 142: 537-548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notredame, C., Higgins, D.G., and Heringa, J. 2000. T-Coffee: A novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 302: 205-217. [DOI] [PubMed] [Google Scholar]
- Ohno, S. 1970. Evolution by gene duplication. Springer-Verlag, Heidelberg, Germany.
- Ohta, T. 1981. Genetic variation in small multigene families. Genet. Res. 37: 133-149. [DOI] [PubMed] [Google Scholar]
- Payseur, B.A. and Nachman, M.W. 2000. Microsatellite variation and recombination rate in the human genome. Genetics 156: 1285-1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semple, C. and Wolfe, K.H. 1999. Gene duplication and gene conversion in the Caenorhabditis elegans genome. J. Mol. Evol. 48: 555-564. [DOI] [PubMed] [Google Scholar]
- Smith, G.P. 1974. Unequal crossover and the evolution of multigene families. Cold Spring Harb. Symp. Quant. Biol. 38: 507-513. [DOI] [PubMed] [Google Scholar]
- Stephan, W. 1986. Recombination and the evolution of satellite DNA. Genet. Res. 47: 167-174. [DOI] [PubMed] [Google Scholar]
- Takahata, N. 1981. A mathematical study on the distribution of the number of repeated genes per chromosome. Genet. Res. 38: 97-102. [Google Scholar]
- Walsh, J.B. 1987. Persistence of tandem arrays: Implications for satellite and simple-sequence DNAs. Genetics 115: 553-567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wintle, R.F., Nygaard, T.G., Herbrick, J.A., Kvaloy, K., and Cox, D.W. 1997. Genetic polymorphism and recombination in the subtelomeric region of chromosome 14q. Genomics 40: 409-414. [DOI] [PubMed] [Google Scholar]
- Yu, A., Zhao, C., Fan, Y., Jang, W., Mungall, A.J., Deloukas, P., Olsen, A., Doggett, N.A., Ghebranious, N., Broman, K.W., et al. 2001. Comparison of human genetic and sequence-based physical maps. Nature 409: 951-953. [DOI] [PubMed] [Google Scholar]
- Zimmer, E.A., Martin, S.L., Beverley, S.M., Kan, Y.W., and Wilson, A.C. 1980. Rapid duplication and loss of genes coding for the α chains of hemoglobin. Proc. Natl. Acad. Sci. 77: 2158-2162. [DOI] [PMC free article] [PubMed] [Google Scholar]
WEB SITE REFERENCES
- ftp://tairpub:tairpub@ftp.arabidopsis.org/home/tair/Maps/mapviewer_data; marker information from the Lister and Dean 1993 RI map and genetic map positions.
- http://www.arabidopsis.org/servlets/Search?action=new_search&type=marker; information about the physical locations of genetic markers in the genome sequence.