

Abstract
Background
Genome-wide association studies (GWAS) have successfully identified a large number of single nucleotide polymorphisms (SNPs) that are associated with a wide range of human diseases. However, many of these disease-associated SNPs are located in non-coding regions and have remained largely unexplained. Recent findings indicate that disease-associated SNPs in human large intergenic non-coding RNA (lincRNA) may lead to susceptibility to diseases through their effects on lincRNA expression. There is, therefore, a need to specifically record these SNPs and annotate them as potential candidates for disease.
Description
We have built LincSNP, an integrated database, to identify and annotate disease-associated SNPs in human lincRNAs. The current release of LincSNP contains approximately 140,000 disease-associated SNPs (or linkage disequilibrium SNPs), which can be mapped to around 5,000 human lincRNAs, together with their comprehensive functional annotations. The database also contains annotated, experimentally supported SNP-lincRNA-disease associations and disease-associated lincRNAs. It provides flexible search options for data extraction and searches can be performed by disease/phenotype name, SNP ID, lincRNA name and chromosome region. In addition, we provide users with a link to download all the data from LincSNP and have developed a web interface for the submission of novel identified SNP-lincRNA-disease associations.
Conclusions
The LincSNP database aims to integrate disease-associated SNPs and human lincRNAs, which will be an important resource for the investigation of the functions and mechanisms of lincRNAs in human disease. The database is available at http://bioinfo.hrbmu.edu.cn/LincSNP.
Keywords: LincRNA, Disease-associated SNPs, GWAS, Non-coding RNA, Database
Background
Identification of genetic variants that underlie complex traits is one of the main tasks of current genetic research [1]. In recent years, genome-wide association studies (GWAS) have identified thousands of genetic variants that are associated with a wide spectrum of diseases (or phenotypes). However, many single nucleotide polymorphisms (SNPs), the most common type of genetic variant, are located in the intergenic regions, which makes it difficult to clarify their functions and involvement in human disease [2]. Recently, a small number of studies have begun to create a relationship between disease-associated SNPs and large intergenic non-coding RNAs (lincRNAs) [3]. These lincRNAs are greater than 200 nucleotides in length and have been shown to play a critical role in many key biological processes [4,5]. The number of human lincRNAs continues to increase and many studies have demonstrated a role in a wide variety of diseases, such as cancer [6,7].
In previous studies, disease-associated SNPs in microRNAs and microRNA target sites have been widely investigated [8,9]. MicroRNAs (miRNAs) represent an abundant class of small non-coding RNAs that regulate gene expression by binding mostly to the 3′-untranslated region of mRNA. This results in RNA degradation or translational repression [10]. The role of miRNA polymorphisms in human diseases has been well established in both experimental and bioinformatic analyses [11,12]. Recently, the linkage of disease-associated SNPs with human lincRNAs has become a new area of interest. For example, a recent study of papillary thyroid carcinoma (PTC) found that a PTC-associated SNP, rs944289, identified by GWAS, is located 3.2 kb upstream of a lincRNA (PTCSC3). This SNP could affect the expression of PTCSC3, which indicates a potential mechanism behind susceptibility to PTC [13]. Another study performed a meta-analysis of two existing results from GWAS. It identified a new SNP, rs3787016, which is associated with prostate cancer and is found in a lincRNA region [14]. A different study found a SNP, rs7763881, in HULC lincRNA, which could contribute to decreased susceptibility to hepatocellular carcinoma in HBV persistent carriers [15]. In addition, GWAS have identified several disease-associated SNPs in a lincRNA called ANRIL[3] and have provided more examples where SNPs affect lincRNA expression [16]. This growing list of related studies will provide a clearer blueprint of the extent and precise mechanism of lincRNA polymorphisms in various human diseases. Therefore, it is necessary to identify and understand these potential functional variants that reside in human lincRNAs.
In our previous work, we have performed an analysis of lincRNA polymorphisms and identified disease-associated SNPs in these regions [17]. Therefore, we anticipate that lincRNAs can be mapped to more disease-associated SNPs. In this study, we developed a practical and user-friendly database called LincSNP (available at http://bioinfo.hrbmu.edu.cn/LincSNP), which is a comprehensive data source for integrating current lincRNA and GWAS SNP annotations. The overall structure of LincSNP is shown in Figure 1. Linkage disequilibrium (LD) analysis has been used and greater than 1.5 million annotated SNPs are stored in the current version of LincSNP. Approximately 140,000 SNPs can be associated with approximately 5,000 human lincRNAs. The database makes an attempt to bridge the gap between disease-associated SNPs and human lincRNAs. This will enhance our understanding of lincRNA function, particularly the potential role of lincRNAs in human disease.
Figure 1.
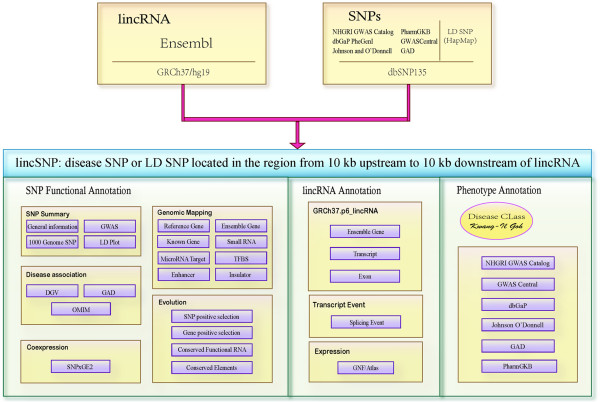
Overall structure of the LincSNP database. LincSNP integrates the annotation of three groups: disease-associated SNPs, lincRNAs and diseases.
Construction and content
Data sources
We downloaded approximately 5,700 human lincRNAs from the Ensembl database, together with their annotations (Ensembl version 68, Homo sapiens assembly GRCh37.p8), such as gene feature, transcripts, exons, transcription events and expression. Disease (phenotype) associated SNPs were integrated using six data sources: dbGAP [18], GAD [19], GWAS Central [20], Johnson and O’Donnell [21], NHGRI GWAS Catalog [22] and PharmGKb [23] (versions shown in Table 1). These data sources have strict criteria to filter published GWAS studies and ensure the dependability of the data for further analysis. Previous studies have shown that many true disease-associated SNPs do not have stringent P-values [24]. Thus, to increase the number, we selected disease-associated SNPs from original publications with moderate thresholds (P-values < 1.0 × 10−3). If data for the same SNP could be obtained from different publications, we selected only the most significant data set [25]. In addition, we manually collected SNP-lincRNA-disease associations from publications taken from the PubMed database (published before April 1st, 2014), where experimental evidence was given. Experimentally supported lincRNA-disease associations from a previous study were also collected and annotated in the LincSNP database [26].
Table 1.
Version of GWAS data sources
| Source | Link | Version |
|---|---|---|
| dbGAP |
http://www.ncbi.nlm.nih.gov/gap |
Aug/2012 |
| GAD |
http://geneticassociationdb.nih.gov/cgi-bin/index.cgi |
Jun/2013 |
| GWAS Central |
http://www.gwascentral.org |
Mar/2013 |
| Johnson and O’Donnell [21] |
http://www.ncbi.nlm.nih.gov/pubmed/19161620 |
Jan/2009 |
| NHGRI GWAS Catalog |
http://www.genome.gov/gwastudies |
Aug/2011 |
| PharmGKb | http://www.pharmgkb.org/index.jsp | Apr/2013 |
In total, 128,407 unique disease-associated SNPs were collected. We also extracted SNPs that had linkage disequilibrium (LD SNP, r2 ≥ 0.5 in at least one population) relationships with disease-associated SNPs from the HapMap CEU, HCB + JPT and YRI populations (release #28). After LD analysis, approximately 1.5 million potential disease-associated SNPs or LD SNPs was collected in LincSNP. We performed comprehensive annotation for these SNPs using currently available annotation information. A detailed list that describes the annotation sources is provided in Table 2.
Table 2.
Sources of disease-associated SNP annotation
| Annotation | Item | Source | Description |
|---|---|---|---|
| SNP Summary |
General Information |
BioQ |
BioQ provides query and documentation tools for genomic relational databases. |
| |
Genome Wide Association |
BioQ |
|
| |
1000 Genome SNP |
BioQ |
|
| |
LD Plot |
BioQ |
|
| Genomic Mapping |
Reference Gene |
UCSC |
Genes and Gene Prediction Tracks\RefSeq Genes\refGene; |
| |
Ensemble Gene |
UCSC |
Genes and Gene Prediction Tracks\Ensembl Genes\ensGene; |
| |
Known Gene |
UCSC |
Genes and Gene Prediction Tracks\UCSC Genes\knownGene; |
| |
Small RNA |
UCSC |
Genes and Gene Prediction Tracks\sno/miRNA\wgRna; |
| |
MicroRNA Target |
UCSC |
Regulation\TS miRNA sites\targetScanS; |
| |
TFBS |
UCSC |
Regulation\TFBS Conserved\tfbsConsSites; |
| |
Enhancer |
VISTA Enhancer |
Tissue specific human enhancers; |
| |
Insulator |
CTCFBSDB |
Vertebrate genomic insulators; |
| Evolution |
SNP and Gene positive selection |
SNP@Evolution |
A hierarchical database of positive selection on the human genome; |
| |
Conserved Functional RNA |
UCSC |
Genes and Gene Prediction Tracks\EvoFold\evofold; |
| |
Conserved Elements |
UCSC |
Comparative Genomics\Conservation\Mammal EI; |
| Gene Co-Expression |
3-way SNP-expression Associations |
SNPxGE2 |
Human SNP–expression associations |
| Disease Association |
OMIM gene |
OMIM |
Online Mendelian Inheritance in Man; |
| |
DGV gene |
DGV |
Structural variation in the human genome; |
| GAD gene | GAD | Genetic Association Database; |
Note: BioQ: http://bioq.saclab.net; UCSC: http://genome.ucsc.edu; VISTA Enhancer: http://enhancer.lbl.gov; CTCFBSDB: http://insulatordb.uthsc.edu; SNP@Evolution: http://bighapmap.big.ac.cn; SNPxGE2: http://lambchop.ads.uga.edu/snpxge2/index.php; OMIM: http://www.ncbi.nlm.nih.gov/omim; DGV: http://dgv.tcag.ca/dgv/app/home; GAD: http://geneticassociationdb.nih.gov/cgi-bin/index.cgi.
Mapping disease-associated SNPs to human lincRNAs
One of the main features of LincSNP is that all disease-associated SNPs from existing studies that can be mapped to human lincRNAs are identified and annotated. It has also been found that disease-associated SNPs in the up- and down-stream regions of human lincRNA may be potential functional variants. They may disrupt some functional elements, such as transcription factor binding sites (TFBSs), and lead to disease by changing the expression of lincRNA [13]. Thus, we identified all disease-associated SNPs located in human lincRNA regions and within 10 kb up- and down-stream of lincRNAs [27]. Of the 128,407 SNPs associated with various diseases or phenotypes, 11,631 were mapped to 3,323 human lincRNAs. After LD analysis, a total of 128,785 LD SNPs (r2 ≥ 0.5) were mapped to 4,906 human lincRNAs.
Database construction
The LincSNP database is composed of a web interface and a MySQL database management system. The MySQL (version 5.1) system was used to store and manage all data in LincSNP. The data processing programs were written in Java (version 1.6.0), the web interfaces were built in JSP and jQuery plugins were used for the interface development. The web services were developed using Apache Struts2 (version 2.1.8), which is a Java web application framework.
We built user-friendly web interfaces that allow users to perform free text searches and download data sets in the LincSNP database. The searchable terms include disease/phenotype name, lincRNA name (Ensembl ID), SNP ID (rs) and chromosome region. We provide alternative query options (disease-associated SNPs located in the lincRNAs or different distances from lincRNAs) so that users have the freedom to query disease-associated SNPs in the lincRNAs or up- and down-steam of lincRNAs. To collect new, experimentally supported SNP-lincRNA-disease associations, we also provide a web interface for users to submit novel data into the database. Search results are returned as a list of SNP IDs (rs) that correspond to a ‘Result information’ page, which can be optionally displayed in a number of sections based on selected fields. These sections include: an ‘SNP’ section for basal information on the inquired SNP and other annotated LD SNPs related to this initial SNP; a ‘lincRNA’ section that contains the annotations for the lincRNAs; and a ‘Phenotype’ section that contains the GWAS information, such as phenotype name, original P-values and references. Details of these sections and subsections are described in the Help page of the LincSNP database.
Utility and discussion
Discovery of the roles of genetic variants in common diseases is currently the subject of intense research. The most common variants are SNPs, which have been used as biomarkers for disease association and susceptibility. Within coding regions, SNPs can affect protein functions directly, by changing the amino acid sequences or by disrupting their regulation. When SNPs are located in non-coding RNA sequence, they may be involved in different mechanisms. Currently, lincRNAs have been identified as a class of non-coding functional transcripts and it is becoming increasingly clear that disease-associated SNPs can reside in these regions. Several previous studies have integrated GWAS and miRNA data and have provided resources for miRNA related SNP studies [11,28]. Therefore, we developed the LincSNP database to provide researchers with a time- and cost-efficient bioinformatic tool to query candidate disease-associated SNPs in human lincRNAs.
Using data from LincSNP, we found that many human lincRNA regions could be mapped to disease-associated SNPs (Figure 2A). Approximately one third of human lincRNAs were mapped to at least one disease-associated SNP and several lincRNAs were mapped to more than six disease-associated SNPs (Figure 2B). In addition, we identified all disease (phenotype) associated SNPs that could be mapped to human lincRNAs and classified the diseases into 21 different classes (unclassified not shown), using the classification scheme from a previous study [29]. We then investigated whether human lincRNAs were more likely to take part in specific disease classes. We found that metabolic, neurological and psychiatric diseases were the top three potential lincRNA related classes (Figure 2C).
Figure 2.
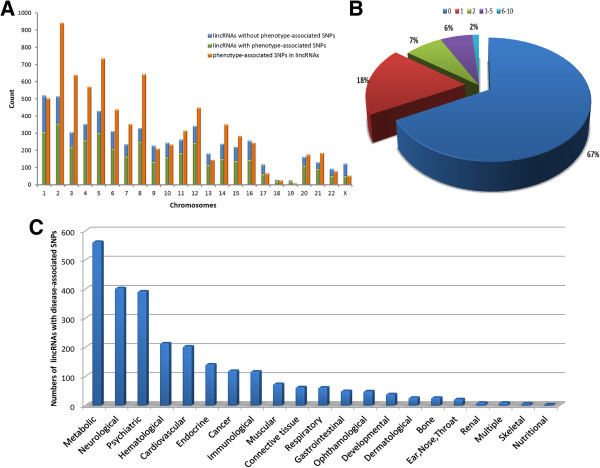
Distribution of disease-associated SNPs in human lincRNAs. (A) The distribution of lincRNAs and disease (phenotype) associated SNPs, classified by chromosomes. Green bars represent the lincRNAs with disease (phenotype) associated SNPs, blue bars represent other lincRNAs and orange bars represent disease-associated SNPs in human lincRNAs. (B) The distribution of lincRNAs with different numbers of disease-associated SNPs. (C) The distribution of disease-associated SNPs in human lincRNAs. These diseases (phenotypes) were classified into 21 classes, in accordance with the criterion published by Goh et al.
One of the potential applications of LincSNP is to predict new disease-associated lincRNAs, based on SNPs that are already known to be associated with certain diseases. For example, a previous GWAS study has identified an SNP (rs12543663) associated with prostate cancer susceptibility [30]. Using the LincSNP database, we found that this SNP is located in a newly annotated lincRNA, PCAT-1 (ENSG00000253438). This lincRNA has recently been demonstrated to be a transcriptional repressor implicated in a subset of prostate cancer patients (Figure 3) [31]. Another application of LincSNP is to find the principles behind specific lincRNAs and diseases. For example, we found that human lincRNAs can be divided into three categories based on our genome-wide disease-associated SNP mapping. Firstly, several lincRNAs, such as ENSG00000256166 and ENSG00000214894, were found to have multiple SNPs that were associated with specific diseases or phenotypes. These two lincRNAs were mapped to many disease-associated SNPs, most of which are associated with immunological disease. Secondly, some lincRNAs are enriched for disease-associated SNPs that are linked to multiple classes of disease. Examples of this are ENSG00000232080, ENSG00000237838 and ENSG00000242996, which were mapped to SNPs associated with metabolic, endocrine, immunological, neurological and other diseases. Lastly, many lincRNAs were not mapped to any disease-associated SNPs, which suggested that SNPs in these lincRNAs were either lethal mutations or had no effect on diseases or phenotypes. These findings will provide novel insight into the roles of lincRNAs in human disease.
Figure 3.
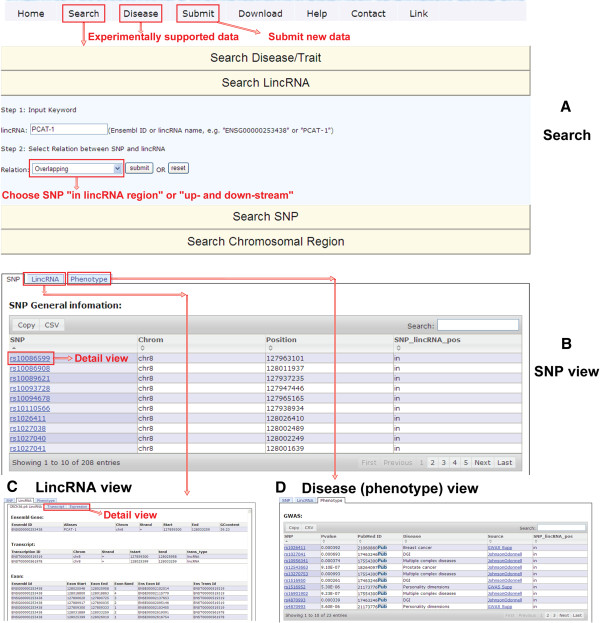
Overview of the LincSNP web interface. (A) An example: screenshot of the main search page, search for lincRNA PCAT-1 (ENSG00000253438). The corresponding result pages are shown. (B) A list of SNPs in or around ENSG00000253438 is shown in the ‘SNP’ section, (C) the ‘lincRNA’ section contains three subsections: ‘GRCh36.p6_lincrna’, ‘Transcript’ and ‘Expression’, and (D) the ‘phenotype’ section provides information on the disease-associated SNPs and sources.
Conclusions
LincSNP is designed as a comprehensive resource for linking disease-associated SNPs to human lincRNAs. We manually collected experimentally supported SNP-lincRNA-disease associations in the LincSNP database. Although the current number is limited, with the growth of interest in human lincRNAs and the availability of high-throughput technologies, the total number of disease-associated lincRNAs and SNPs will undoubtedly continue to grow. We will recruit new disease-associated lincRNAs and SNPs and update the LincSNP database in a timely manner. We will also incorporate new functional annotations and more data sources to improve the utility of this database. These strategies will make the data more comprehensive and improve the performance of LincSNP, to make it increasingly useful for future studies.
Availability and requirements
LincSNP is freely available on the web at http://bioinfo.hrbmu.edu.cn/LincSNP.
Abbreviations
GWAS: Genome-wide association studies; SNP: Single nucleotide polymorphism; lincRNA: Large intergenic non-coding RNA; LD: Linkage disequilibrium; CEU: Samples of Utah residents with Northern and Western European ancestry from the CEPH collection; HCB: Samples of Han Chinese in Beijing; JPT: Samples of Japanese in Tokyo; YRI: Samples of Yoruba people in Ibadan.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
XL conceived of the project. SN, ZZ, HZ, RL and TW participated in the collection and analysis of all data sources. JY and PW designed and implemented the database. SN, ZZ and XL wrote the manuscript. All authors have read and approved the final manuscript.
Contributor Information
Shangwei Ning, Email: ningsw@ems.hrbmu.edu.cn.
Zuxianglan Zhao, Email: zhaozuxianglan@126.com.
Jingrun Ye, Email: yejingrun87@163.com.
Peng Wang, Email: wpgqy@163.com.
Hui Zhi, Email: zhihui013201@gmail.com.
Ronghong Li, Email: ronghong_li@hotmail.com.
Tingting Wang, Email: always_smileatme@163.com.
Xia Li, Email: lixia@hrbmu.edu.cn.
Acknowledgements
This work was supported by the National High Technology Research and Development Program of China [863 Program, Grant Nos. 2014AA021102], the National Natural Science Foundation of China [Grant Nos. 91129710, 61073136 and 61170154], and the Undergraduate Innovation Funds of Harbin Medical University [Grant Nos. YJSCX2012-226HLJ].
References
- Freimer NB, Sabatti C. Human genetics: variants in common diseases. Nature. 2007;445(7130):828–830. doi: 10.1038/nature05568. [DOI] [PubMed] [Google Scholar]
- Ward LD, Kellis M. Interpreting noncoding genetic variation in complex traits and human disease. Nat Biotechnol. 2012;30(11):1095–1106. doi: 10.1038/nbt.2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasmant E, Sabbagh A, Vidaud M, Bieche I. ANRIL, a long, noncoding RNA, is an unexpected major hotspot in GWAS. Faseb J. 2010;25(2):444–448. doi: 10.1096/fj.10-172452. [DOI] [PubMed] [Google Scholar]
- Ponting CP, Oliver PL, Reik W. Evolution and functions of long noncoding RNAs. Cell. 2009;136(4):629–641. doi: 10.1016/j.cell.2009.02.006. [DOI] [PubMed] [Google Scholar]
- Wilusz JE, Sunwoo H, Spector DL. Long noncoding RNAs: functional surprises from the RNA world. Genes Dev. 2009;23(13):1494–1504. doi: 10.1101/gad.1800909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai MC, Spitale RC, Chang HY. Long intergenic noncoding RNAs: new links in cancer progression. Cancer Res. 2011;71(1):3–7. doi: 10.1158/0008-5472.CAN-10-2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wapinski O, Chang HY. Long noncoding RNAs and human disease. Trends Cell Biol. 2011;21(6):354–361. doi: 10.1016/j.tcb.2011.04.001. [DOI] [PubMed] [Google Scholar]
- Sethupathy P, Collins FS. MicroRNA target site polymorphisms and human disease. Trends Genet. 2008;24(10):489–497. doi: 10.1016/j.tig.2008.07.004. [DOI] [PubMed] [Google Scholar]
- Ryan BM, Robles AI, Harris CC. Genetic variation in microRNA networks: the implications for cancer research. Nat Rev Cancer. 2010;10(6):389–402. doi: 10.1038/nrc2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136(2):215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Z, Bruno AE. The influence of 3’UTRs on MicroRNA function inferred from human SNP data. Comp Funct Genom. 2011;2011:910769. doi: 10.1155/2011/910769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders MA, Liang H, Li WH. Human polymorphism at microRNAs and microRNA target sites. Proc Natl Acad Sci U S A. 2007;104(9):3300–3305. doi: 10.1073/pnas.0611347104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jendrzejewski J, He H, Radomska HS, Li W, Tomsic J, Liyanarachchi S, Davuluri RV, Nagy R, de la Chapelle A. The polymorphism rs944289 predisposes to papillary thyroid carcinoma through a large intergenic noncoding RNA gene of tumor suppressor type. Proc Natl Acad Sci U S A. 2012;109(22):8646–8651. doi: 10.1073/pnas.1205654109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin G, Sun J, Isaacs SD, Wiley KE, Kim ST, Chu LW, Zhang Z, Zhao H, Zheng SL, Isaacs WB, Xu J. Human polymorphisms at long non-coding RNAs (lncRNAs) and association with prostate cancer risk. Carcinogenesis. 2011;32(11):1655–1659. doi: 10.1093/carcin/bgr187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Pan S, Liu L, Zhai X, Liu J, Wen J, Zhang Y, Chen J, Shen H, Hu Z. A genetic variant in long non-coding RNA HULC contributes to risk of HBV-related hepatocellular carcinoma in a Chinese population. PLoS One. 2012;7(4):e35145. doi: 10.1371/journal.pone.0035145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar V, Westra HJ, Karjalainen J, Zhernakova DV, Esko T, Hrdlickova B, Almeida R, Zhernakova A, Reinmaa E, Vosa U, Hofker MH, Fehrmann RS, Fu J, Withoff S, Metspalu A, Franke L, Wijmenga C. Human disease-associated genetic variation impacts large intergenic non-coding RNA expression. PLoS Genet. 2013;9(1):e1003201. doi: 10.1371/journal.pgen.1003201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunnington MS, Santibanez Koref M, Mayosi BM, Burn J, Keavney B. Chromosome 9p21 SNPs associated with multiple disease phenotypes correlate with ANRIL expression. PLoS Genet. 2010;6(4):e1000899. doi: 10.1371/journal.pgen.1000899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mailman MD, Feolo M, Jin Y, Kimura M, Tryka K, Bagoutdinov R, Hao L, Kiang A, Paschall J, Phan L, Popova N, Pretel S, Ziyabari L, Lee M, Shao Y, Wang ZY, Sirotkin K, Ward M, Kholodov M, Zbicz K, Beck J, Kimelman M, Shevelev S, Preuss D, Yaschenko E, Graeff A, Ostell J, Sherry ST. The NCBI dbGaP database of genotypes and phenotypes. Nat Genet. 2007;39(10):1181–1186. doi: 10.1038/ng1007-1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker KG, Barnes KC, Bright TJ, Wang SA. The genetic association database. Nat Genet. 2004;36(5):431–432. doi: 10.1038/ng0504-431. [DOI] [PubMed] [Google Scholar]
- Thorisson GA, Lancaster O, Free RC, Hastings RK, Sarmah P, Dash D, Brahmachari SK, Brookes AJ. HGVbaseG2P: a central genetic association database. Nucleic Acids Res. 2009;37(Database issue):D797–D802. doi: 10.1093/nar/gkn748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson AD, O’Donnell CJ. An open access database of genome-wide association results. BMC Med Genet. 2009;10:6. doi: 10.1186/1471-2350-10-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hindorff LA, Sethupathy P, Junkins HA, Ramos EM, Mehta JP, Collins FS, Manolio TA. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci U S A. 2009;106(23):9362–9367. doi: 10.1073/pnas.0903103106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altman RB. PharmGKB: a logical home for knowledge relating genotype to drug response phenotype. Nat Genet. 2007;39(4):426. doi: 10.1038/ng0407-426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- C WTCC. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447(7145):661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begum F, Ghosh D, Tseng GC, Feingold E. Comprehensive literature review and statistical considerations for GWAS meta-analysis. Nucleic Acids Res. 2012;40(9):3777–3784. doi: 10.1093/nar/gkr1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Wang Z, Wang D, Qiu C, Liu M, Chen X, Zhang Q, Yan G, Cui Q. LncRNADisease: a database for long-non-coding RNA-associated diseases. Nucleic Acids Res. 2013;41(Database issue):D983–D986. doi: 10.1093/nar/gks1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JH, Li JH, Jiang S, Zhou H, Qu LH. ChIPBase: a database for decoding the transcriptional regulation of long non-coding RNA and microRNA genes from ChIP-Seq data. Nucleic Acids Res. 2013;41(Database issue):D177–D187. doi: 10.1093/nar/gks1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziebarth JD, Bhattacharya A, Chen A, Cui Y. PolymiRTS Database 2.0: linking polymorphisms in microRNA target sites with human diseases and complex traits. Nucleic Acids Res. 2012;40(Database issue):D216–D221. doi: 10.1093/nar/gkr1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goh KI, Cusick ME, Valle D, Childs B, Vidal M, Barabasi AL. The human disease network. Proc Natl Acad Sci U S A. 2007;104(21):8685–8690. doi: 10.1073/pnas.0701361104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eeles RA, Kote-Jarai Z, Giles GG, Olama AA, Guy M, Jugurnauth SK, Mulholland S, Leongamornlert DA, Edwards SM, Morrison J, Field HI, Southey MC, Severi G, Donovan JL, Hamdy FC, Dearnaley DP, Muir KR, Smith C, Bagnato M, Ardern-Jones AT, Hall AL, O'Brien LT, Gehr-Swain BN, Wilkinson RA, Cox A, Lewis S, Brown PM, Jhavar SG, Tymrakiewicz M, Lophatananon A. et al. Multiple newly identified loci associated with prostate cancer susceptibility. Nat Genet. 2008;40(3):316–321. doi: 10.1038/ng.90. [DOI] [PubMed] [Google Scholar]
- Prensner JR, Iyer MK, Balbin OA, Dhanasekaran SM, Cao Q, Brenner JC, Laxman B, Asangani IA, Grasso CS, Kominsky HD, Cao X, Jing X, Wang X, Siddiqui J, Wei JT, Robinson D, Iyer HK, Palanisamy N, Maher CA, Chinnaiyan AM. Transcriptome sequencing across a prostate cancer cohort identifies PCAT-1, an unannotated lincRNA implicated in disease progression. Nat Biotechnol. 2011;29(8):742–749. doi: 10.1038/nbt.1914. [DOI] [PMC free article] [PubMed] [Google Scholar]