

Abstract
Mast cells play a central role in allergy through secretion of both preformed and newly synthesized mediators. Mast cell mediator secretion is controlled by a complex network of signaling events. Despite intensive studies, signaling pathways in the regulation of mast cell mediator secretion remain incompletely defined. Here, we examined the role of calpain in IgE-dependent mast cell activation. IgE-mediated activation of mouse bone marrow-derived mast cells (BMMCs) enhanced calpain activity. Inhibition of calpain activity by a number of calpain inhibitors reduced IgE-mediated mast cell degranulation both in vitro and in vivo. Calpain inhibitors blocked IgE-mediated TNF and IL-6 production in vitro and reduced late-phase allergic response in vivo. Importantly, mouse calpain-1 null BMMCs showed reduced IgE-mediated mast cell degranulation in vitro and in vivo, diminished cytokine and chemokine production in vitro, and impaired late-phase allergic response in vivo. Further studies revealed that calpain-1-deficiency led to specific attenuation of IκB-NF-κB pathway and IKK-SNAP23 pathway, while calcium flux, MAP kinases, Akt, and NFAT pathway proceed normally in IgE-activated calpain-1 null mast cells. Thus, calpain-1 is identified as a novel regulator in IgE-mediated mast cell activation and could serve as a potential therapeutic target for the management of allergic inflammation.
Introduction
Mast cells play an essential role in allergy through IgE-dependent activation. Many of the mast cell’s effects are mediated through secretion of mast cell mediators (1, 2). Antigen binding to IgE induces aggregation of FcεRI that in turn initiates a cascade of molecular events leading to mast cell degranulation and production of newly synthesized mediators such as cytokines and chemokines (1, 2). Despite many studies, signaling molecules mediating IgE-induced mast cell activation remains incompletely defined. Two major mechanisms of protein modification, reversible protein phosphorylation controlled by protein kinases and phosphatases, and irreversible protein cleavage induced by proteases, play a fundamental role in intracellular signal transduction pathways (3).
Irreversible protein cleavage by proteases is an essential tool utilized by cells to transduce signals (3). One of the best-characterized regulatory proteases is cysteine protease family including calpains and caspases. Mast cells also utilize proteolysis mechanism to transduce signals from membrane to nucleus (4). However, little is known about the nature of the proteases involved in FcεRI-induced signaling cascade. Calpains are unique cysteine proteases that require Ca2+ for their activity (3, 5–7). Calpains cause limited cleavage of substrates generally localizing near the membrane and cytoskeleton compartments (3, 5–7). The calpain family is composed of 15 distinct proteins designated as calpain 1 to 15 (3, 5–9). Among the calpain family, the major calpain species are calpain-1 (μ-calpain) and calpain-2 (m-calpain), which require micro- and millimolar concentrations of Ca2+ for activation in vitro, respectively (3, 5, 7, 9). A precise role of calpain activity in IgE-mediated mast cell activation is not known. Mast cell adhesion to fibronectin was reduced by chemical inhibitors that non-specifically inhibit calpains (10) suggesting that mast cells may contain functional calpains. It is well known that FcεRI aggregation on mast cells induces a rapid increase of intracellular Ca2+ levels, which is essential for mast cell activation (11–14). Furthermore, a number of calpain substrates are important signaling molecules involved in FcεRI-induced mast cell activation, such as IκB (15, 16), PKC (3, 17), and SNAP23 (18), and active calpain alters the balance between protein kinase and phosphatase activities (19). Therefore, we hypothesized that calpain activation participates in IgE-induced mast cell activation.
Here, we demonstrate that IgE-mediated mast cell activation increased calpain activity in the mouse bone marrow derived mast cells. Calpain inhibitors blocked IgE-dependent mast cell degranulation and cytokine production in vitro and in vivo. Calpain-1 null mouse mast cells showed impaired IgE-mediated mast cell degranulation and cytokine production in vitro and in vivo. Calpain-1-deficiency led to reduced IκB-NFκB pathway activation without affecting mitogen activated protein (MAP) kinases or nuclear factor of activated T-cells (NFAT) pathways in mast cells. Moreover, calpain-1-deficiency suppressed IKK and SNAP23 (Ser120) phosphorylation. Thus, calpain-1 emerges as an integral component of the IgE-mediated signal transduction pathway in mast cells. To our knowledge, this is the first evidence for a functional role of calpain-1 in the regulation of IgE-mediated mast cell activation.
Materials and Methods
Animals
Calpain-1 null mice were generated as previously described (20). The calpain-1 null mice used in this study were back-crossed at least 25 generations using the wild-type C57BL/6 mice (21). Normal C57BL/6 mice were purchased from Charles River Laboratories (Wilmington, MA). Mast cell-deficient W-sh mice were obtained from The Jackson Laboratory (B6Cg-kit W-sh/HNihJacBsmJ NistltF4). Protocols were approved by the University Committee on Laboratory Animals, Dalhousie University, in accordance with the guidelines of the Canadian Council on Animal Care.
Antibodies and Reagents
Antibodies to p38 MAPK, actin, and HRP-linked secondary Abs were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Antibody to calpain-1 and IKKβ, calpain inhibitors (III, V, IX, XI, and XII), and IKK inhibitors (SC-514, BMS-345541, and sulindac) were purchased from EMD Chemicals (Gibbstown, NJ). FITC-conjugated rat anti-mouse IgE (IgG1), FITC-rat IgG1 and FITC-conjugated rat anti-mouse CD117 (c-kit) were purchased from Cedarlane Laboratories Ltd. (Hornby, ON). Antibody to SNAP23 was purchased from Novus Biological, Inc. (Littleton, CO). The phosphorylation-site-specific anti-SNAP23 antibodies (Ser120 and Ser95) were previously characterized (22). Anti-DNP IgE mAb was purchased from Sigma-Aldrich (St. Louis, MO). Other antibodies were purchased from Cell Signaling Technology Inc. (Beverly, MA). Trinitrophenyl (TNP)-bovine serum albumin (BSA) and dinitrophenyl (DNP)-BSA were purchased from Biosearch Technologies (Novato, CA).
Mast cell culture, activation, degranulation and Intracellular calcium
Mouse bone marrow-derived mast cells (BMMCs) were cultured as previously described in complete medium (RPMI 1640 medium containing 10% fetal bovine serum, 10% WEHI-3B conditioned medium, 50 units/ml each of penicillin and streptomycin, 50 μM 2-mercaptoethanol, and 200 nM prostaglandin E2) (23, 24). Prostaglandin E2 was included in the culture media to improve mast cell induction (24). After 4–6 weeks in culture, mast cell purity was greater than 98%. BMMCs were passively sensitized with IgE from TIB-141 cells (American Type Culture Collection) for 18 h. Cells were then activated by stimulation with 10 ng/ml TNP-BSA. Mast cell degranulation was determined by measuring β-hexosaminidase release. Intracellular calcium was measured using Fura-2 AM (Invitrogen) as a probe.
Cytokine protein array and ELISA
Protein arrays (Mouse Inflammation Array 1, RayBiotech, Inc., Norcross, GA) were performed according to the manufacturer’s protocol. Enzyme-Linked ImmunoSorbent Assay (ELISA) was used to measure cytokines or chemokines according to the manufacturer’s protocol (Minneapolis, MN). Prostaglandin D2 (PGD2) and leukotriene C4 (LTC4) enzyme immunoassay kit were obtained from Cayman (Ann Arbor, MI). For the studies on IKK-IκB-NF-κB signaling, IgE-sensitized BMMCs were pre-incubated with IKKβ inhibitors (100 μM SC-514, 10 μM BMS-345541, or 1 mM sulindac) for 2 hours and then stimulated with 10 ng/ml TNP-BSA for 4 hours, cell free supernatant were collected for ELISA. Histamine levels were measured in the plasma by ELISA (Beckman Coulter, Brea, CA, USA).
Calpain activity assay
Calpain activity was measured using a commercial assay kit (Calbiochem, EMD Chemicals, Gibbstown, NJ) according to the manufacturer’s protocol. This assay entails detection of the cleavage of the calpain substrate Suc-Leu-Leu-Val-Tyr-7-amino-4-methylcoumarin (Suc-LLVY-AMC) by fluorescence. Briefly, IgE-sensitized and TNP-BSA (10 ng/ml) stimulated BMMC or unstimulated BMMCs were lysed in lysis buffer (provided in the kit). Whole cell lysates were then used to perform the calpain activity assay. After incubation in the dark at room temperature for 15 min, fluorescence was measured at 360 nm excitation and 450 nm emission using a spectrophotometer.
Electrophoretic mobility shift assay (EMSA), Western blotting and luciferase assay
Nuclear protein extracts were obtained using a nuclear extract kit (Active Motif, Carlsbad, CA) according to the manufacturer’s protocol. EMSA and Western blotting were performed as previously described (25). The following double-stranded oligonucleotides (Sigma-Aldrich) were used in EMSA: NFAT binding consensus sequence (N) on mouse IL-13 promoter 5′-AAG GTG TTT CCC CAA GCC TTT CCC-3′, and NF-κB binding consensus sequences on mouse IL-6 promoter 5′-TTATCAAATGTGGGATTTTCCCAT-3′. Luciferase assay was performed as previously described (26).
Local Reconstitution of mast cell-deficient W-sh mice
W-sh mice were reconstituted with mast cells by injection of 20 μl BMMCs at a density of 2.5 × 107 cells/ml in RPMI 1640 (5 × 105 cells/injection) into the ear and footpad. Wild-type BMMCs were injected into the right side ear and footpad, whereas calpain-1 null BMMCs were injected into the left side ear and footpad. Alternatively, individual W-sh mouse was reconstituted with either wild-type BMMC or calpain-1 null BMMC. Mast cell reconstitution was carried out by injection of 20 μl wild-type or calpain-1 null BMMCs at a density of 2.5 × 107 cells/ml in RPMI 1640 (5 × 105 cells/injection) into left ear and footpad, whereas right ear and footpad were injected with 20 μl saline as control. Five weeks later, mast cell-reconstituted W-sh mice were used for the study of IgE-dependent late-phase cutaneous reactions (thickness of ear or footpad) or IgE-mediated passive cutaneous anaphylaxis (Evans blue dye leakage). Ear tissues were collected and fixed in Carnoy’s fixative. Specimens were embedded in paraffin and sectioned onto microscope slides, and stained with 1% Alcian blue and 1% Safranin O. All images were taken using equivalent setting on a Nikon E600 microscope equipped with a 20× and 40× objective lens using ACT-1 software (Nikon).
Whole body reconstitution of mast cell-deficient W-sh mice and passive systemic anaphylaxis (PSA)
Five million wild-type or calpain-1 null BMMCs in 200 μl of PBS were injected i.v. via tail vein into 5–6 week old W-sh mice. Five weeks later, mast cell reconstituted W-sh mice were used for passive systemic anaphylaxis (PSA). Briefly, about 200 μl blood were collected from each mouse. Then mice were sensitized with 10 μg anti-DNP IgE in 100 μl saline by i.v. injection. Twenty four hours after anti-DNP IgE injection, mice were injected with 1.5 mg DNP in 100 μl saline, and blood were collected at 10 min post DNP injection. Histamine levels in the plasma were measured by ELISA according the to manufacturer’s instruction (Beckman Coulter, Brea, CA, USA). Ear tissues were collected, fixed, and stained as above described.
IgE-dependent late-phase cutaneous reactions
Mice were sensitized with anti-DNP IgE and challenged with DNFB as previously described (25, 26). The thickness of the footpad or ear was measured after 24 hrs. For calpain inhibitor studies, 24 h after sensitization, the C57BL/6 mice were injected (i.p.) with 500 μl of 5 mM calpain inhibitor III or 500 μl of 1:4000 DMSO/saline as control. The cutaneous reaction was elicited one hour later by the application of DNFB. The thickness of the left ear or left hind paw (treated with acetone/olive oil only) was used as baseline values. For the studies on mast cell-reconstituted W-sh mice, the thickness of ear and footpad was measured before (as baseline values) and after the DNFB treatment. The value of DNFB-induced change in tissue thickness was obtained after baseline subtraction.
IgE-mediated passive cutaneous anaphylaxis (vascular permeability)
Mice were sensitized with anti-DNP IgE and challenged with DNP-BSA in Evans blue dye. Vascular permeability was determined by measuring Evans blue dye leakage (25, 26). For calpain inhibitor studies, 24 h after IgE sensitization, the C57BL/6 mice were injected (i.p.) with 500 μl of 5 mM calpain inhibitor III or 500 μl of 1:4000 DMSO/saline as a control. One hour later, mice were then challenged with DNP-BSA. For the studies using W-sh mice, data from W-sh mice without mast cell reconstitution were used as a baseline.
Statistics
Data are presented as mean ± S.E. of the indicated number of experiments. Differences between the left and right ears or footpads of the same animal were evaluated using a paired Student t test. Differences between different groups of animals or different mast cell populations (wild-type vs calpain-1 null BMMC) were evaluated using an unpaired Student t test. One-way ANOVA analysis and the post hoc Tukey’s tests were performed in experiments where there were multiple concentrations of calpain inhibitors used in one cell population. Two-way ANOVA followed with Tukey’s tests were performed in experiments where there are multiple groups (multiple time-points or concentrations of TNP-BSA) and there were two cell populations (wild-type and calpain-1 null BMMCs). Differences were considered significant at *p < 0.05, **p < 0.01, ***p < 0.001.
Results
Calpain activity is required for IgE-mediated mast cell degranulation
Calpains are widely expressed and distributed in tissues and organs, and participate in a wide range of cellular processes, including signal transduction pathways (3, 7, 27). However, to the best of our knowledge, direct evidence of calpain in FcεRI-mediated mast cell activation has not been demonstrated previously. To determine whether calpain activity is required for IgE-mediated mast cell degranulation, a series of inhibitors were employed. Anti-TNP IgE sensitized BMMCs were treated with calpain inhibitors and then stimulated with TNP-BSA. Mast cell degranulation was assessed by β-hexosaminidase release. All tested calpain inhibitors (III, V, IX, XI, and XII) suppressed IgE-mediated mast cell degranulation in a dose-dependent manner (Fig. 1A–E). We further tested effects of calpain inhibitor on mast cell degranulation in vivo. Mice were sensitized with anti-DNP IgE intradermally in ear tissue for 24 h, followed by i.p. injection of calpain inhibitor III (or DMSO as a control) for 1 h, and then i.v. challenge with DNP-BSA in a 1% solution of Evans blue dye via tail vein injection. Thirty minutes later, ear tissue was collected, and Evans blue dye was extracted to assess vascular permeability (Fig. 1F). Mice treated with calpain inhibitor III showed reduced IgE-mediated vascular permeability (Evans blue dye leakage) compared with the control group treated with DMSO. These results suggest that calpain participates in the IgE-mediated mast cell degranulation in vitro and in vivo.
Figure 1. Calpain activity is required for FcεRI-dependent mast cell degranulation both in vitro and in vivo.

A–E, Wild-type BMMCs were sensitized with anti-TNP IgE and treated with various calpain inhibitors for 30 min, and then stimulated with TNP-BSA for 20 min. Mast cell degranulation was measured by β-hexosaminidase release. Calpain inhibitor III (A), V (B), IX (C), XI (D), and XII (E) significantly inhibited mast cell degranulation in a dose-dependent manner. F, C57BL/6 mice were sensitized with anti-DNP IgE for 24 h by intradermal injection, followed by intraperitoneal injection with 500 μl of 5 mM calpain inhibitor III for 1 hour. Mice were then challenged by i.v. injection of DNP-BSA in Evans blue dye solution. Thirty minutes later, ear tissue was collected and Evans blue dye was extracted to assess vascular permeability by spectrophotometry. n = 4 (A, C, D, and E) or 5 (B) cultures of BMMCs, or n = 6 mice (F). Data are means ± SEM, *p< 0.05, **p < 0.01.
Calpain activity is required for IgE-mediated late-phase cutaneous anaphylaxis and mast cell cytokine production
Activated mast cells participate in the development of late-phase allergic reactions via cytokines production (28). To examine whether calpain contributes to IgE-mediated late-phase allergic reaction, C57BL/6 mice were sensitized with anti-DNP IgE intravenously for 24 h, followed by i.p. injection of calpain inhibitor III or DMSO control for one hour. Mice were then challenged with 0.3% DNFB solution or control solution in the ear and foot for 24h. Calpain inhibitor III treated mice displayed a deficit in IgE-dependent inflammation, as assessed by ear and foot thickness (Fig. 2A–D). Thus, inhibition of calpain led to reduced IgE-dependent late-phase cutaneous anaphylaxis.
Figure 2. Calpain is required for IgE-mediated late-phase cutaneous anaphylaxis and cytokine production.

A–D, C57BL/6 mice were passively sensitized by i.v. injection of anti-DNP IgE mAb. Twenty-four hours later, mice received 500 μl of 5 mM calpain inhibitor III (CI3) or dimethyl sulfoxide (DMSO) control (i.p.) for 1 hour, and then challenged by applying 0.3% DNFB in acetone/olive oil (4:1) to the right ear and hind paw, whereas acetone/olive oil control solution (vehicle) to the left ear and hind paw. Twenty-four hours later, thickness of ear (A) and foot (C), and weight of ear (B) and foot (D) was measured. Data are expressed as mean of actual measurements of thickness (mm) or weight (mg) ± SEM. n = 10 mice, *p< 0.05, **p < 0.01 by comparison with DMSO group. E–F, After sensitization with anti-TNP IgE, BMMCs from C57BL/6 mice were treated various concentrations of calpain inhibitor V for 1 hour, and then challenged with TNP-BSA for 5 h. Cell free supernatants were collected and subjected to ELISA analysis for IL-6 (E) and TNF (F). Data are mean ± SEM. n = 5 cultures of BMMCs, *p< 0.05, **p < 0.01.
Mast cell-derived cytokines contribute to the late-phase allergic reactions. To determine whether calpain activity regulates IgE-mediated mast cell cytokine production, BMMCs from C57BL/6 mice were sensitized with the anti-TNP IgE, and treated with calpain inhibitor V for one hour. Mast cells were then stimulated with TNP-BSA for 5 h. Cell-free supernatants were analyzed by ELISA for IL-6 (Fig. 2E) and TNF (Fig. 2F). TNP-BSA-induced IL-6 and TNF production were significantly reduced by calpain inhibitor in a dose-dependent manner (Fig. 2E, 2F).
Together, these results suggest that inhibition of calpain activity leads to reduced mast cell degranulation in vitro and in vivo, decreased mast cell cytokine production in vitro, and reduced late-phase anaphylaxis in vivo.
Calpain-1 deficiency leads to reduced IgE-mediated mast cell degranulation, but does not affect mast cell maturation
Among 15 calpain isoforms known to date, calpain-1 is the major form expressed in immune cells (3), and it contributes about 80% of the total calpain activity in mouse platelets (20). To evaluate the specific role of calpain-1 in IgE-mediated mast cell activation, calpain-1 null mice were used. Bone marrow cells were isolated from both wild-type and calpain-1 null mice, and cultured in IL-3 and PGE2-conditioned media for 5 wks (24). Mast cell maturation was examined by granule staining with toluidine blue dye and by flow cytometry for IgE receptor and c-Kit expression. Toluidine blue staining showed similar metachromatic staining and morphology of mast cells from wild-type and calpain-1 null mice (Fig. 3A). Similarly, no difference in the expression of IgE receptor (Fig. 3B) and c-Kit (Fig. 3C) was observed between wild-type and calpain-1 null mast cells. These results suggest that mast cells develop normally in vitro in the absence of calpain-1. The absence of calpain-1 protein in mast cells was confirmed by Western blotting using the anti-calpain-1 antibody (Fig. 3D).
Figure 3. BMMCs from calpain-1 null mice display deficit in IgE-dependent mast cell degranulation but not mast cell maturation.

A, Cells were stained with toluidine blue to determine mast cell morphology. A representative photomicrograph is shown (original magnification = × 100). Specimens were viewed on a Nikon Eclipse E600 microscope equipped with a DMX1200 camera, a CFI Plan-Fluor DDL 100 ×/1.30 oil-immersion objective lens. Acquisition was performed with Nikon ACT-1 software version 2.20. Images were processed using Adobe Photoshop 5.0. B–C, Mast cell maturation were examined by flow cytometry of IgE receptor (B) and c-Kit (C) expression. D, Calpain-1 deficiency was examined by Western blotting of BMMCs lysates. Actin was used as loading control. E, Intracellular Ca2+ flux analysis of wild-type and calpain-1 null BMMCs. Mast cells were sensitized with anti-TNP IgE and stimulated with TNP. Intracellular Ca2+ flux was measured using Fura-2 as a probe. The data are representative of four experiments with similar results. F, Mast cell degranulation was determined by measuring β-hexosaminidase release after sensitization with anti-TNP IgE and stimulation with TNP-BSA for 20 min. Data are mean ± SEM. n = 3 cultures of BMMCs, *p< 0.05 by comparison with calpain-1 null BMMC group. G, Passive cutaneous anaphylaxis. Mast cell-deficient W-sh mice were reconstituted locally in the ears by intradermal injection of wild-type (right ear) or calpain-1 null (left ear) BMMCs. Five weeks later, mast cell-reconstituted W-sh mice were locally sensitized with anti-DNP IgE for 24 h by intradermal injection, and then challenged by i.v. injection of DNP-BSA in Evans blue dye solution. Thirty minutes later, ear tissue was collected and Evans blue dye was extracted to assess vascular permeability by spectrophotometry. Data from W-sh mice without mast cell reconstitution were used as a baseline. Data are mean ± SEM. n = 8 mice, *p< 0.05.
FcεRI aggregation on mast cells induces a rapid increase of intracellular Ca2+ levels (11–14). To examine whether calpain-1 regulates the effects of calcium mobilization, mast cells from wild-type and calpain-1 null mice were sensitized with anti-TNP IgE and preloaded with Fura 2-AM, followed by stimulation with TNP-BSA. Calcium influx following TNP-BSA stimulation was determined. TNP-stimulation induced similar levels of calcium in calpain-1 null and wild-type BMMCs (Fig. 3E).
To examine whether deficiency of calpain-1 affects mast cell degranulation in vitro, BMMCs from wild-type and calpain-1 null mice were sensitized with an anti-TNP IgE and stimulated with TNP-BSA for 20 min. The β-hexosaminidase release was determined. Calpain-1 null mast cells appear to show reduced spontaneous release of β-hexosaminidase under resting condition (Fig. 3F). The TNP-BSA-induced β-hexosaminidase release was significantly reduced in the calpain-1 null mast cells (Fig. 3F).
To determine a functional role of calpain-1 in IgE-mediated mast cell degranulation in vivo, mast cell-deficient W-sh mice were reconstituted with calpain-1 null (left ear) or wild-type (right ear) BMMCs locally at ear for 5 wks. Mast cell-reconstituted W-sh mice were sensitized with anti-DNP IgE intradermally in ear tissue for 24 h, followed by i.v. challenge with DNP-BSA in a 1% Evans Blue solution via tail vein. Thirty minutes later, tissue from both ears were collected for extraction of Evans blue dye to determine vascular permeability by spectrophotometry. Ear tissues from normal W-sh mice were used as background. Vascular permeability (Evans blue dye leakage) was reduced in left ear reconstituted with calpain-1 null BMMCs as compared with right ear reconstituted with wild-type BMMCs (Fig. 3G).
Alternatively, individual mast cell-deficient W-sh mouse was reconstituted with either calpain-1 null or wild-type BMMCs locally at left ear. An equal volume of saline was injected into right ear as control. Five weeks later, mast cell reconstitution was confirmed by alcian blue and safranin O staining (Supplemental Fig. S1A). Similar number of mast cells was observed between wild-type and calpain-1 null BMMC groups (Supplemental Fig. S1B). Mast cell-reconstituted W-sh mice were sensitized with anti-DNP IgE and stimulated with DNP-BSA in a 1% Evans Blue solution as described above. Evan blue dye leakage (vascular permeability) was determined. Vascular permeability was reduced in mice reconstituted with calpain-1 null BMMCs as compared with that in mice reconstituted with wild-type BMMCs (Supplemental Fig. S1C and S1D).
To further confirm a functional role of calpain-1 in IgE-mediated mast cell degranulation in vivo, passive systemic anaphylaxis (PSA) assay was performed in W-sh mice reconstituted with BMMC via tail vein injection (whole body mast cell reconstitution). We first carried out histological examination of ear tissue from these mice to confirm mast cell reconstitution. There was no significant difference in mast cell number between mice reconstituted with calpain-1 null BMMC and mice with wild-type BMMC (Supplemental Fig. S2A and S2B). Importantly, following antigen challenge, histamine level in mice reconstituted with calpain-1 null mice was significantly lower than that in mice reconstituted with wild-type BMMC (Supplemental Fig. S2C).
These results demonstrate that calpain-1 contributes to IgE-mediated mast cell degranulation both in vitro and in vivo.
Calpain-1 contributes to total calpain activity in mast cell
In mouse platelets, calpain-1 contributes about 80% of the total calpain activity (20). We tested calpain activity in BMMCs. Wild-type and calpain-1 null BMMCs were sensitized with anti-TNP IgE and stimulated with TNP-BSA for various time intervals. BMMCs without IgE sensitization or after IgE sensitization without anti-TNP stimulation were served as a control. Whole cell lysates were used to determine calpain activity. Under resting condition, BMMCs express a high level of calpain activity (Fig. 4), suggesting that calpain may be required for mast cell homeostasis. Calpain activity was reduced (approximately 50%) in calpain-1 null BMMCs, suggesting that calpain-1 contributes to approximately 50% of total calpain activity in mast cells under resting condition. After stimulation with TNP-BSA, BMMCs showed enhanced calpain activity, peaking at 0.5–1 h post-stimulation (Fig. 4). These data suggested that calpain activity participates in IgE-FcεRI mediated signaling in mast cells, and calpain-1 contributes to total calpain activity in mast cell.
Figure 4. Calpain activity is enhanced following IgE-dependent mast cell activation.

Wild-type or calpain-1 deficient BMMCs were left without sensitization (no IgE) or sensitized with anti-TNP IgE and stimulated with 10 ng/ml TNP-BSA for various time. Whole cell lysates were subjected to calpain activity test using a calpain activity assay kit. n = 4 cultures of BMMCs, *p< 0.05, ***p < 0.001. ns, not significant.
Calpain-1 deficiency leads to impaired production of IL-6, TNF, CCL1, CCL2, LTC4, and PGD2 in response to IgE-mediated mast cell activation
To determine the contribution of calpain-1 in IgE-mediated mast cell cytokine and chemokine production, wild-type and calpain-1 null BMMCs were sensitized with an anti-TNP IgE and stimulated with TNP-BSA for 6 h. Cell-free supernatants were analyzed for cytokine and chemokine production using the protein array methodology. Results showed that calpain-1 deficiency impaired the production of IL-6, CCL1, and CCL2 in TNP-stimulated BMMCs (Supplemental Fig. S3). To further confirm these findings, cell free supernatants were prepared at various time intervals and analyzed by ELISA. Following TNP-BSA stimulation, production of IL-6 (Fig. 5A), TNF (Fig. 5B), CCL1 (Fig. 5C), and CCL2 (Fig. 5D), was significantly reduced in calpain-1 null BMMCs as compared to wild-type control cells. These data indicate that calpain-1 contributes to IgE-mediated cytokine and chemokine production by mast cells.
Figure 5. Calpain-1 deficiency in mast cells led to decreased IgE-dependent cytokine/chemokine production and reduced late phase anaphylaxis.

A–F, Wild-type (+/+) and calpain-1 null (−/−) mast cells were sensitized with anti-TNP IgE and stimulated with TNP-BSA. Cell-free supernatants were collected for measuring cytokines and chemokines by ELISA. IL-6 (A), TNF (B), CCL1 (C), CCL2 (D), LTC4 (E), and PGD2 (F) production was significantly impaired in calpain-1null mast cells. Data are mean ± SEM. n = 6 (A–D) or 4 (E & F) cultures of BMMCs. *p< 0.05, **p< 0.01 by comparison with calpain-1 −/− group. G–H, Calpain-1 deficiency leads to a deficit in late-phase cutaneous anaphylaxis. Mast cell-deficient W-sh mice were reconstituted locally in the ears and footpads by intradermal injection of wild-type (WT, right ear and footpad) or calpain-1 null (KO, left ear and footpad) BMMCs. Five weeks later, mice were sensitized by intravenous injection of anti-DNP IgE. Twenty-four hours later, a cutaneous reaction was elicited by the application of a solution (20 μl) of 0.3% DNFB in acetone/olive oil (4:1) to the ears and footpads. After another 24 h, the cutaneous reaction was assessed by comparing the ear (G) or footpad (H) thickness before and after DNFB stimulation. Data are mean ± SEM. n = 8 mice, *p< 0.05, ***p< 0.001.
To examine whether calpain-1 contributes to IgE-mediated lipid mediator production, cell free supernatants were used to determine LTC4 and PGD2. Calpain-1 null BMMCs displayed reduced levels of LTC4 (Fig. 5E) and PGD2 (Fig. 5F).
Calpain-1 deficiency reduced IgE-mediated late-phase cutaneous anaphylaxis
To examine a role of calpain-1 in late-phase allergic inflammation, mast cell-deficient W-sh mice were reconstituted with calpain-1 null (left side) or wild-type (right side) BMMCs locally at ear and footpad for 5 wks. Mast cell-reconstituted W-sh mice were sensitized with anti-DNP IgE by i.v. injection 24 h prior to epicutaneous application of 0.3% DNFB solution on the ear and foot for 24 h. Left side ear and footpad, which reconstituted with calpain-1 null BMMCs, displayed a deficit in IgE-mediated inflammation, as assessed by ear and foot thickness before and after DNFB treatment (Fig. 5G, 5H).
Alternatively, individual mast cell-deficient W-sh mouse was reconstituted with either calpain-1 null or wild-type BMMCs locally at the left ear or footpad. An equal volume of saline was injected into the right ear or footpad as a control. Five weeks later, reconstitution of mast cells was confirmed by staining of ear tissues (Supplemental Fig. S1A and S1B). Mast cell-reconstituted W-sh mice were sensitized with anti-DNP IgE and stimulated with DNFB. The thickness of the right ear or footpad was used as baseline. W-sh mice reconstituted with calpain-1 null BMMCs displayed a reduced thickness following DNFB stimulation comparing to mice reconstituted with wild-type BMMC (Supplemental Fig. S1E and S1F). These results suggest that calpain-1 contributes to the development of IgE-dependent late-phase cutaneous anaphylaxis.
IκBα-NF-κB pathway is specifically attenuated in calpain-1 null BMMCs
FcεRI aggregation initiates a cascade of signaling events (1). To examine which signaling pathway is regulated by calpain-1 in mast cells, wild-type and calpain-1 null BMMCs were sensitized with anti-TNP IgE and stimulated with TNP-BSA. Cell lysates were analyzed by Western blotting for activation of IκBα, MAPKs, and Akt. Activation of IκBα was specifically reduced at early (5 min) time point in calpain-1 null BMMCs (Fig. 6A, 6B), whereas MAPKs (ERK, JNK, and p38), Akt, and GSK3β activation was consistent with that of wild-type BMMCs (Fig. 6C). This finding suggests that calpain-1 specifically regulates IκBα in mast cells.
Figure 6. Calpain-1 deficiency leads to decreased IgE-dependent IκBα phosphorylation but not MAPK, Akt, or GSK3β.

Wild-type (+/+) and calpain-1 null (−/−) BMMCs were sensitized with anti-TNP IgE and stimulated with TNP-BSA for various times. Total cell lysates were analyzed by Western blotting for various phosphorylated and total proteins. A–B, Decrease IκB phosphorylation in calpain-1 null BMMCs was found at 5 min after TNP stimulation when compared with that in wild-type cells. Data are mean ± SEM. n = 3 cultures of BMMCs, **p< 0.01 by comparison with calpain-1 null BMMCs. C, The pattern of TNP-induced phosphorylation of Erk1/2, p38, JNK, Akt, and GSK3β is similar between wild-type and calpain-1 null BMMCs.
We next examined the impact of calpain-1 deficiency on IgE-mediated NF-κB activation. Mast cells were transfected with an NF-κB luciferase reporter plasmid and an internal control plasmid. Cells were sensitized with anti-TNP IgE and stimulated with TNP-BSA or left untreated for 5 h. The luciferase activity was then assessed. No differences in NF-κB luciferase activity were observed in untreated wild-type and calpain-1 null cells. However, TNP-BSA stimulation-induced NF-κB luciferase activity was significantly reduced in the calpain-1 null mast cells when compared to that of wild-type mast cells (Fig. 7A). We further examined NF-κB activation using EMSA. Wild-type and calpain-1 null BMMCs were sensitized with an anti-TNP IgE and stimulated with TNP-BSA. Nuclear proteins analyzed by EMSA using a 32P-labeled NF-κB probe (Fig. 7B and 7C). Wild-type BMMCs show significantly enhanced NF-κB activation in response to TNP-BSA stimulation. The NF-κB activation was observed at 5 min, reached a maximum at 60 min, and persisted for up to 6 h post-stimulation. However, there is significantly reduced NF-κB activation in calpain-1 null cells (Fig. 7B and 7C). Together, these results suggest that calpain-1 is required for IgE-mediated NF-κB activation in mast cells. In contrast, the IgE-mediated NFAT activation was not affected by calpain-1 deficiency when measured by either luciferase assay (Fig. 7D) or EMSA (Fig. 7E, 7F).
Figure 7. Calpain-1 deficiency leads to decreased IgE-dependent NF-κB activation but not NFAT.

A & D, Wild-type (+/+) and calpain-1 null (−/−) BMMCs were transfected with a pNF-κB-Luc (A) or pNFAT-Luc (D) and the control reporter plasmid pPL-TK. After transfection (24 h), cells were sensitized with anti-TNP IgE, and then cells were either left untreated (NT) or stimulated with TNP-BSA for 6 h (TNP). Firefly and Renilla activities were sequentially quantified using a dual-luciferase reporter assay system. Data are mean ± SEM. n = 4 different BMMCs, **p< 0.01 by comparison with calpain-1 null BMMCs. B & E, NF-κB binding consensus sequence (B) and NFAT binding consensus sequence (E) were labeled with 32P for use as a probe in EMSA. BMMCs were sensitized overnight with anti-TNP IgE, and then either stimulated with TNP-BSA for indicated times or left untreated (NT). Nuclear proteins were isolated and analyzed by EMSA. A representative from three independent experiments is shown. C & F, Densitometry analysis of NF-κB activation (C) and NFAT activity (F) by EMSA was performed based on three experiments from three different cultures of BMMCs. *p< 0.05, **p< 0.01 by comparison with calpain-1 null BMMCs.
To confirm IκB-NF-κB pathway in calpain-dependent mast cell activation, calpain-1 null and wild-type BMMCs were treated with IKK inhibitors (SC-514, BMS-345541, or sulindac) for 2 hours, and then stimulated with TNP-BSA for 4 hours. Cell free supernatants were analyzed for IL6 and TNF production. Both calpain-1 dependent and independent cytokine production was blocked by IKK inhibitors (Supplemental Fig. S4A and S4B).
To explore whether calpain is involved in broader physiological or pathological conditions, calpain-1 null and wild-type BMMCs were stimulated with 100 ng/ml stem cell factor (SCF) or 3 μg/ml lipopolysaccharide (LPS). Nuclear protein were extracted and used for EMSA assay to examine NF-κB activity. NF-κB activity stimulated by either SCF or LPS was reduced in calpain-1 null BMMCs comparing to that in wild-type BMMCs (supplemental Fig. S4C and S4D).
IKK and SNAP23 phosphorylation is reduced in capain-1 null mast cells
IgE-mediated activation of IKK-SNAP23 pathway has been recognized as a major mechanism in mediating mast cell degranulation (29). To explore whether calpain-1 mediates IKK-SNAP23 activation, wild-type and calpain-1 null BMMCs with or without TNP-BSA stimulation were subjected to Western blotting for phospho-IKK and phospho-SNAP23 (Ser120 or Ser95). TNP-stimulated phosphorylation of IKK and SNAP23 (Ser120) was reduced in calpain-1 null BMMCs (Fig 8). No phosphorylation of SNAP23 on Ser95 was detectable (data not shown).
Figure 8. IgE-mediated phosphorylation of IKK and SNAP23 is reduced in calpain-1 null mast cells.
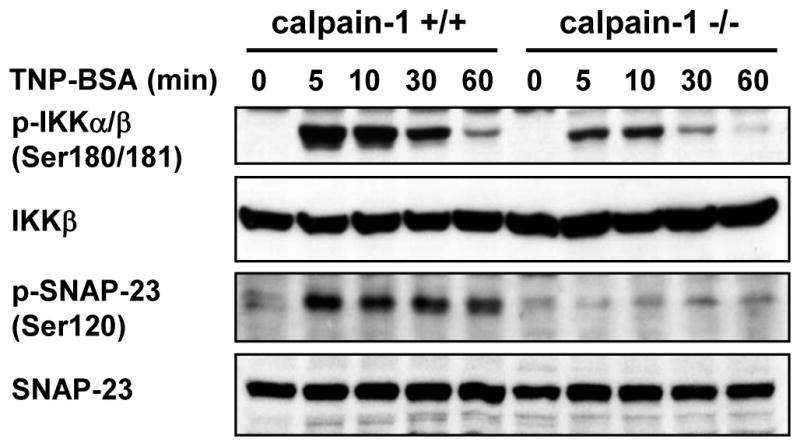
Wild-type (+/+) and calpain-1 null (−/−) BMMCs were sensitized with anti-TNP IgE and stimulated with TNP-BSA for various times. Total cell lysates were analyzed by Western blotting for phosphorylated and total proteins of IKK and SNAP23 (Ser120). Phosphorylation of IKK and SNAP23 were impaired in calpain-1 null BMMCs. A representative photomicrograph is shown from three different cultures of BMMCs.
Discussion
The current study was initiated to elucidate a previously unknown role for calpain enzyme as a regulator of IgE-mediated mast cell activation in vitro and in vivo. Mast cells are central players in allergic inflammation through secretion of mast cell mediators. Mast cell mediators can be broadly divided into preformed mediators and newly synthesized mediators. Preformed mediators such as histamine and β-hexosaminidase are presynthesized and stored in mast cell granules and are secreted by degranulation. Mast cell mediators released by degranulation play a key role in induction of acute allergic response (30, 31). An animal model of FcεRI-mediated passive cutaneous anaphylaxis, which is characterized by an increase of vascular permeability, has been well accepted for the examination of the role of mast cells in acute allergic responses (31, 32). In vitro, inhibition of calpain activity or genetic inactivation of calpain-1 leads to reduced mast cell degranulation upon activation via FcεRI. In vivo, FcεRI-mediated passive cutaneous anaphylaxis was reduced by inhibition of calpain activity or by calpain-1 deficiency. These findings suggest that calpain-1 is required for IgE-mediated mast cell degranulation in vitro and acute allergic response in vivo.
Upon allergen activation, mast cells also produce and secrete another important category of mediators through de novo synthesis, namely, newly synthesized mediators. These include cytokines and chemokines, such as IL-6 and TNF. Mast cell-derived cytokines and chemokines contribute to the development of late-phase allergic reactions (28, 31). An animal model of passive cutaneous allergic reactions characterized by ear or footpad swelling has been well accepted for the evaluation of the late-phase allergic response (28, 32). In vitro, inhibition of calpain activity or calpain-1 deficiency leads to impaired IL-6, TNF, CCL1, CCL2, PGD2, and LTC4 production by IgE-activated mast cells. In vivo, calpain-1 null mast cells elicited a reduced late-phase allergic response in mice. These findings suggest that calpain activity participates in IgE-mediated production of newly synthesized mediators and subsequent late-phase allergic inflammation.
Mast cell mediator secretion is controlled by FcεRI-dependent signaling pathways. The FcεRI-mediated proximal signaling events are well characterized. Cross-linking of the FcεRI activates Lyn and Fyn (1, 2). Lyn phosphorylates tyrosine residues of the immunoreceptor tyrosine-based activation motif in the β and γ subunits of FcεRI (1, 2). Syk is then recruited to the γ subunit and activates a multitude of protein kinases. Fyn is responsible for the subsequent activation of phosphatidylinositol 3 kinase (PI3K) and Akt pathway (1, 2). Concerted actions of Lyn and Fyn initiates several signalling pathways including IκB-NF-κB, nuclear factor of activated T cells (NFAT), mitogen-activated protein kinases (MAPK), PI3K-Akt, and intracellular calcium mobilization thus leading to mast cell degranulation and production of cytokines and chemokines (1, 2). We found that IgE-dependent calcium flux, activation of MAPK, Akt, and NFAT proceeds normally in calpain-1 null mast cells. Thus, it is likely that calpain-1 does not regulate FcεRI-mediated proximal signaling events, but rather specifically targets downstream signaling molecules. This pattern of calpain function in mast cells appears to be different from that in T cells. In T cells, the T cell receptor complex CD3ε, CD3ζ and proximal signaling molecule ZAP-70 can serve as substrates of calpain (3, 33). Thus, the function of calpain is context and cell-type specific.
The reduced IκB and NF-κB activation in calpain-1 null mast cells suggests that calpain-1 and its downstream effectors specifically target IκB-NF-κB pathway in IgE-activated mast cells. IκB is a NF-κB inhibitor. Degradation of IκB frees NF-κB for nuclear translocation. Two mechanisms of IκB degradation have been described, one well characterized through phosphorylation and the subsequent ubiquitin-proteasome pathway (1), and another through calpain activity (3, 17). Thus, it is possible that FcεRI aggregation mediated calcium flux activates calpain system, which together with proteasome pathway participates in IκB degradation leading to NF-κB activation and subsequent production of TNF, IL-6, CCL1, CCL2, LTC4, and PGD2. It is noteworthy that calpain-1 deficiency led to reduced IKK and IκB phosphorylation in mast cells following IgE-mediated activation. This finding suggests that calpain-1 may cleave a substrate upstream of IKK-IκB that is involved in the phosphorylation of IKK and IκB.
Specific mast cell mediator secretion is governed by distinct signaling pathways. It is conceivable that impaired IκB-NF-κB activation is not likely accountable for the reduced degranulation in calpain-1 null mast cells. Signaling pathways leading to degranulation are distinct from those leading to cytokine production (1). Cytokines are largely controlled by transcription factors such as NF-κB (1, 34), while degranulation is controlled by exocytosis machinery which involves a family of the soluble N-ethylmaleimide-sensitive fusion factor attachment protein receptors (SNAREs) (1, 35). Mast cells express various SNAREs including SNAP23 (36) and SNAP25 (37). Calpain is able to cleave SNAP23 in platelets (18) or SNAP25 in neurons (38). However, no SNAP23 cleavage was found in IgE-activated wild-type or calpain-1 null mast cells. Phosphorylation of SNAP23 at Ser120 plays a central role in mast cell degranulation (22, 29). IKK mediates IgE-induced SNAP23 phosphorylation (29). We found that IgE-induced phosphorylation of IKK and SNAP23 (Ser120) was reduced in calpain-1 null mast cells. Thus, it is likely that calpain regulates mast cell degranulation through IKK-SNAP23.
The list of calpain substrates is growing. Calpain cleaves various signaling molecules such as PKC isoforms (including PKCα, β, γ, δ, ε, ζ), G-protein coupled receptors, and phospholipase Cβ in neurons or endothelial cells (3, 39). Calpain modulates the function of cytoskeletal proteins such as FAK, talin, paxillin, fodrin, ezrin, moesin, and the cytoplasmic domain of integrin β subunits in platelets (40). Many of the calpain substrates described in other cell types are present in mast cells. Our finding that calpain deficiency led to impaired secretion of both preformed mediators (degranulation) and newly synthesized mediators (cytokine and chemokine production) suggest that calpain may have multiple targets in IgE-dependent signal transduction in mast cells.
In summary, we found that calpain-1deficiency led to specific inhibition of IκB-NF-κB activation without affecting MAPK, NFAT, and Akt pathways. These data suggest that calpain-1 functions in the downstream signaling events in IgE-mediated signal transduction pathway. Thus, in addition to the well characterized signaling mechanisms via reversible protein phosphorylation and dephosphorylation, the IgE-mediated signal transduction in mast cells also involves calpain activation that is characterized by irreversible protein cleavage. The fact that deficiency of calpain-1 impairs mast cell degranulation and production of cytokines and chemokines in vitro and in vivo positions calpain-1 as a novel regulator and therapeutic target in IgE-mediated allergic inflammation.
Supplementary Material
Acknowledgments
This work was supported by grants from the Canadian Institutes of Health Research, and Izaak Walton Killam Health Center to TJL, and National Institutes of Health (Grant number: NIH-HL089517) to AHC. ZLW is a recipient of a post-doctoral fellowship from Izaak Walton Killam Health Center.
References
- 1.Gilfillan AM, Tkaczyk C. Integrated signalling pathways for mast-cell activation. Nat Rev Immunol. 2006;6:218–230. doi: 10.1038/nri1782. [DOI] [PubMed] [Google Scholar]
- 2.Galli SJ, Tsai M, Piliponsky AM. The development of allergic inflammation. Nature. 2008;454:445–454. doi: 10.1038/nature07204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sato K, Kawashima S. Calpain function in the modulation of signal transduction molecules. Biol Chem. 2001;382:743–751. doi: 10.1515/BC.2001.090. [DOI] [PubMed] [Google Scholar]
- 4.Suzuki K, Nakajima H, Kagami S, Suto A, Ikeda K, Hirose K, Hiwasa T, Takeda K, Saito Y, Akira S, Iwamoto I. Proteolytic processing of Stat6 signaling in mast cells as a negative regulatory mechanism. J Exp Med. 2002;196:27–38. doi: 10.1084/jem.20011682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Campbell RL, Davies PL. Structure-function relationships in calpains. Biochem J. 2012;447:335–351. doi: 10.1042/BJ20120921. [DOI] [PubMed] [Google Scholar]
- 6.Goll DE, V, Thompson F, Li H, Wei W, Cong J. The calpain system. Physiol Rev. 2003;83:731–801. doi: 10.1152/physrev.00029.2002. [DOI] [PubMed] [Google Scholar]
- 7.Storr SJ, Carragher NO, Frame MC, Parr T, Martin SG. The calpain system and cancer. Nat Rev Cancer. 2011;11:364–374. doi: 10.1038/nrc3050. [DOI] [PubMed] [Google Scholar]
- 8.Huang Y, Wang KK. The calpain family and human disease. Trends Mol Med. 2001;7:355–362. doi: 10.1016/s1471-4914(01)02049-4. [DOI] [PubMed] [Google Scholar]
- 9.Sorimachi H, Suzuki K. The structure of calpain. J Biochem (Tokyo) 2001;129:653–664. doi: 10.1093/oxfordjournals.jbchem.a002903. [DOI] [PubMed] [Google Scholar]
- 10.Forsythe P, Befus AD. Inhibition of calpain is a component of nitric oxide-induced down-regulation of human mast cell adhesion. J Immunol. 2003;170:287–293. doi: 10.4049/jimmunol.170.1.287. [DOI] [PubMed] [Google Scholar]
- 11.Fewtrell C, Mohr FC, Ryan TA, Millard PJ. Calcium: an important second messenger in mast cells. Ciba Found Symp. 1989;147:114–127. doi: 10.1002/9780470513866.ch8. discussion 128–132. [DOI] [PubMed] [Google Scholar]
- 12.Hoth M, Fasolato C, Penner R. Ion channels and calcium signaling in mast cells. Ann N Y Acad Sci. 1993;707:198–209. doi: 10.1111/j.1749-6632.1993.tb38053.x. [DOI] [PubMed] [Google Scholar]
- 13.Parekh AB, Penner R. Regulation of store-operated calcium currents in mast cells. Soc Gen Physiol Ser. 1996;51:231–239. [PubMed] [Google Scholar]
- 14.Pearce FL. Calcium and mast cell activation. Br J Clin Pharmacol. 1985;20(Suppl 2):267S–274S. doi: 10.1111/j.1365-2125.1985.tb02812.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shumway SD, Maki M, Miyamoto S. The PEST domain of IkappaBalpha is necessary and sufficient for in vitro degradation by mu-calpain. J Biol Chem. 1999;274:30874–30881. doi: 10.1074/jbc.274.43.30874. [DOI] [PubMed] [Google Scholar]
- 16.Pianetti S, Arsura M, Romieu-Mourez R, Coffey RJ, Sonenshein GE. Her-2/neu overexpression induces NF-kappaB via a PI3-kinase/Akt pathway involving calpain-mediated degradation of IkappaB-alpha that can be inhibited by the tumor suppressor PTEN. Oncogene. 2001;20:1287–1299. doi: 10.1038/sj.onc.1204257. [DOI] [PubMed] [Google Scholar]
- 17.Kang MY, Zhang Y, Matkovich SJ, Diwan A, Chishti AH, Dorn GW., 2nd Receptor-independent cardiac protein kinase Calpha activation by calpain-mediated truncation of regulatory domains. Circ Res. 2010;107:903–912. doi: 10.1161/CIRCRESAHA.110.220772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rutledge TW, Whiteheart SW. SNAP-23 is a target for calpain cleavage in activated platelets. J Biol Chem. 2002;277:37009–37015. doi: 10.1074/jbc.M204526200. [DOI] [PubMed] [Google Scholar]
- 19.Robles E, Huttenlocher A, Gomez TM. Filopodial calcium transients regulate growth cone motility and guidance through local activation of calpain. Neuron. 2003;38:597–609. doi: 10.1016/s0896-6273(03)00260-5. [DOI] [PubMed] [Google Scholar]
- 20.Azam M, Andrabi SS, Sahr KE, Kamath L, Kuliopulos A, Chishti AH. Disruption of the mouse mu-calpain gene reveals an essential role in platelet function. Mol Cell Biol. 2001;21:2213–2220. doi: 10.1128/MCB.21.6.2213-2220.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kuchay SM, Kim N, Grunz EA, Fay WP, Chishti AH. Double knockouts reveal that protein tyrosine phosphatase 1B is a physiological target of calpain-1 in platelets. Mol Cell Biol. 2007;27:6038–6052. doi: 10.1128/MCB.00522-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hepp R, Puri N, Hohenstein AC, Crawford GL, Whiteheart SW, Roche PA. Phosphorylation of SNAP-23 regulates exocytosis from mast cells. J Biol Chem. 2005;280:6610–6620. doi: 10.1074/jbc.M412126200. [DOI] [PubMed] [Google Scholar]
- 23.Boudreau RT, Garduno R, Lin TJ. Protein phosphatase 2A and protein kinase Calpha are physically associated and are involved in Pseudomonas aeruginosa-induced interleukin 6 production by mast cells. J Biol Chem. 2002;277:5322–5329. doi: 10.1074/jbc.M108623200. [DOI] [PubMed] [Google Scholar]
- 24.Hu ZQ, Asano K, Seki H, Shimamura T. An essential role of prostaglandin E on mouse mast cell induction. J Immunol. 1995;155:2134–2142. [PubMed] [Google Scholar]
- 25.Wu Z, Macneil AJ, Junkins R, Li B, Berman JN, Lin TJ. Mast Cell Fc{varepsilon}RI-Induced Early Growth Response 2 Regulates CC Chemokine Ligand 1-Dependent CD4+ T Cell Migration. J Immunol. 2013;190:4500–4507. doi: 10.4049/jimmunol.1203158. [DOI] [PubMed] [Google Scholar]
- 26.Yang YJ, Chen W, Edgar A, Li B, Molkentin JD, Berman JN, Lin TJ. Rcan1 negatively regulates Fc epsilonRI-mediated signaling and mast cell function. J Exp Med. 2009;206:195–207. doi: 10.1084/jem.20081140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Donkor IO. Calpain inhibitors: a survey of compounds reported in the patent and scientific literature. Expert Opin Ther Pat. 2011;21:601–636. doi: 10.1517/13543776.2011.568480. [DOI] [PubMed] [Google Scholar]
- 28.Nagai H, Abe T, Yamaguchi I, Mito K, Tsunematsu M, Kimata M, Inagaki N. Role of mast cells in the onset of IgE-mediated late-phase cutaneous response in mice. J Allergy Clin Immunol. 2000;106:S91–98. doi: 10.1067/mai.2000.106778. [DOI] [PubMed] [Google Scholar]
- 29.Suzuki K, I, Verma M. Phosphorylation of SNAP-23 by IkappaB kinase 2 regulates mast cell degranulation. Cell. 2008;134:485–495. doi: 10.1016/j.cell.2008.05.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ramos BF, Zhang Y, Angkachatchai V, Jakschik BA. Mast cell mediators regulate vascular permeability changes in Arthus reaction. J Pharmacol Exp Ther. 1992;262:559–565. [PubMed] [Google Scholar]
- 31.Moore KG, Dannenberg AM., Jr Immediate and delayed (late-phase) dermal contact sensitivity reactions in guinea pigs. Passive transfer by IgG1 antibodies, initiation by mast cell degranulation, and suppression by soybean proteinase inhibitor. Int Arch Allergy Immunol. 1993;101:72–81. doi: 10.1159/000236501. [DOI] [PubMed] [Google Scholar]
- 32.Malaviya R, Uckun FM. Role of STAT6 in IgE receptor/FcepsilonRI-mediated late phase allergic responses of mast cells. J Immunol. 2002;168:421–426. doi: 10.4049/jimmunol.168.1.421. [DOI] [PubMed] [Google Scholar]
- 33.Penna D, Muller S, Martinon F, Demotz S, Iwashima M, Valitutti S. Degradation of ZAP-70 following antigenic stimulation in human T lymphocytes: role of calpain proteolytic pathway. J Immunol. 1999;163:50–56. [PubMed] [Google Scholar]
- 34.Marquardt DL, Walker LL. Dependence of mast cell IgE-mediated cytokine production on nuclear factor-kappaB activity. J Allergy Clin Immunol. 2000;105:500–505. doi: 10.1067/mai.2000.104942. [DOI] [PubMed] [Google Scholar]
- 35.Jahn R, Scheller RH. SNAREs--engines for membrane fusion. Nat Rev Mol Cell Biol. 2006;7:631–643. doi: 10.1038/nrm2002. [DOI] [PubMed] [Google Scholar]
- 36.Guo Z, Turner C, Castle D. Relocation of the t-SNARE SNAP-23 from lamellipodia-like cell surface projections regulates compound exocytosis in mast cells. Cell. 1998;94:537–548. doi: 10.1016/s0092-8674(00)81594-9. [DOI] [PubMed] [Google Scholar]
- 37.Salinas E, Ventura J, Cordova LE, Luis Quintanar J. Presence of SNAP-25 in rat mast cells. Immunol Lett. 2004;95:105–108. doi: 10.1016/j.imlet.2004.05.018. [DOI] [PubMed] [Google Scholar]
- 38.Grumelli C, Berghuis P, Pozzi D, Caleo M, Antonucci F, Bonanno G, Carmignoto G, Dobszay MB, Harkany T, Matteoli M, Verderio C. Calpain activity contributes to the control of SNAP-25 levels in neurons. Mol Cell Neurosci. 2008;39:314–323. doi: 10.1016/j.mcn.2008.07.011. [DOI] [PubMed] [Google Scholar]
- 39.Tremblay R, Chakravarthy B, Hewitt K, Tauskela J, Morley P, Atkinson T, Durkin JP. Transient NMDA receptor inactivation provides long-term protection to cultured cortical neurons from a variety of death signals. J Neurosci. 2000;20:7183–7192. doi: 10.1523/JNEUROSCI.20-19-07183.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schoenwaelder SM, Yuan Y, Jackson SP. Calpain regulation of integrin alpha IIb beta 3 signaling in human platelets. Platelets. 2000;11:189–198. doi: 10.1080/09537100050057620. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.