

Abstract
To identify the upstream signals of neuronal apoptosis in patients with medically intractable temporal lobe epilepsy (TLE), we evaluated by immunohistochemistry and confocal microscopy brain tissues of 13 TLE patients and 5 control patients regarding expression of chemokines and cell-cycle proteins. The chemokine RANTES (CCR5) and other CC-chemokines and apoptotic markers (caspase-3, -8, -9) were expressed in lateral temporal cortical and hippocampal neurons of TLE patients, but not in neurons of control cases. The chemokine RANTES is usually found in cytoplasmic and extracellular locations. However, in TLE neurons, RANTES was displayed in an unusual location, the neuronal nuclei. In addition, the cell-cycle regulatory transcription factor E2F1 was found in an abnormal location in neuronal cytoplasm. The pro-inflammatory enzyme cyclooxygenase-2 and cytokine interleukin-1β were expressed both in neurons of patients suffering from temporal lobe epilepsy and from cerebral trauma. The vessels showed fibrin leakage, perivascular macrophages and expression of IL-6 on endothelial cells. In conclusion, the cytoplasmic effects of E2F1 and nuclear effects of RANTES might have novel roles in neuronal apoptosis of TLE neurons and indicate a need to develop new medical and/or surgical neuroprotective strategies against apoptotic signaling by these molecules. Both RANTES and E2F1 signaling are upstream from caspase activation, thus the antagonists of RANTES and/or E2F1 blockade might be neuroprotective for patients with medically intractable temporal lobe epilepsy. The results have implications for the development of new medical and surgical therapies based on inhibition of chemotactic and mitogenic stimuli of neuronal apoptosis in patients with medically intractable temporal lobe epilepsy.
Keywords: Medically intractable temporal lobe epilepsy, RANTES, interleukin-1β, cyclooxygenase-2, E2F-1, neuronal apoptosis
INTRODUCTION
The complex signaling pathways leading to neuronal apoptosis in epilepsy have not been completely elucidated. Seizures stimulate calcium influx, glutamate receptor activation and/or release of death receptor ligands [1] and may provoke neuronal death by mitochondrial [2] and death receptor pathways [3, 4]. The E2F family (E2F1–E2F6) is responsible for regulating cell cycle progression; however, E2F1 is also able to induce cell death through several mechanisms [5]. The transcription factor, E2F1, and the interacting pocket-binding protein, Rb, are implicated in the death of neurons in neurodegenerative disorders [6, 7]. E2F1 protein expression is increased in the neuronal cytoplasm of involved brain regions in HIV encephalitis, simian-immunodeficiency virus (SIV) encephalitis [8], Alzheimer disease [9], amyotrophic lateral sclerosis [10], and Parkinson’s disease [11]. Affected neurons in Parkinson’s disease display altered distribution of phosphorylated retinoblastoma protein (Rb) [12]. In amyotrophic lateral sclerosis, E2F1 is redistributed into the cytoplasm of motor neurons and the transcriptional regulator Rb is hyperphosphorylated [10]. The International League Against Epilepsy defines medically intractable epilepsy as the failure of two tolerated, appropriately chosen and used antiepileptic medications schedules (either monotherapy or combination therapy) to achieve sustained seizure freedom [12a]. The expression and distribution of E2F-1 and Rb have not been previously investigated in brain tissue of patients with medically intractable temporal lobe epilepsy.
Inflammation leads to apoptosis through induction of inflammatory cytokines and chemokines. Inflammatory cytokines including interleukin-1 β IL-1 β) are not constitutively expressed in normal brain [13] but are detected in a wide range of neurodegenerative disorders [14]. Cytokine expression has been noted in autopsy tissues of patients with temporal lobe epilepsy [15] and rodent brain tissues after seizure induction [16]. The cytokine IL-1 β may induce pro-apoptotic and excitatory signals, which lead to long-lasting changes in gene transcription [17]. In animal models of epileptogenesis, activation of the IL-1β system is associated with neurodegeneration and blood–brain barrier breakdown [18]. Anti-inflammatory drugs are considered for use in epilepsy both for their anticonvulsant activity and modulation of gene transcription [19]. Vagus nerve stimulation has been shown to have immune rebalancing functions which may be related to its antiseizure action [20]. However, expression of IL-1, TNF-alpha and IL-10 in the brain may be associated with cell injury other than that associated with seizures and these cytokines may in certain situations have neuroprotective effects [21, 22].
Chemokines are chemotactic proteins classified into four subfamilies known as CXC-, CC-, C-, and CXXXC-chemokines [23]. In the central nervous system, chemokines have additional functions, including control of neural plasticity by CCL5 [24], a role of CCL5-CCR5 in inflammation and apoptosis, and a dichotomous role of CXCR4, which has positive neurodevelopmental effects through its ligand SD F-1[25] and neuronotoxic effects mediated by the HIV-1 protein gp120 [26]. Chemokines may lead to neuronal death through E2F1 signaling [27].
In this study, we examine upstream signals for apoptosis, including inflammatory signals by chemokines, cytokines and cyclooxygenase-2 (COX-2) and the cell cycle transcription factor, E2F1. The results suggest that apoptosis in neurons of patients with medically intractable temporal lobe epilepsy (“TLE neurons”) may be induced by a combination of chemotactic and mitogenic stimuli. These findings have implications for tailoring the development of new medical and surgical therapies targeted towards specific chemotactic and mitogenic stimuli of neuronal apoptosis in patients with medically intractable temporal lobe epilepsy.
MATERIALS & METHODS
Surgical tissues
The Institutional Review Boards at the University of Arizona and UCLA approved the protocol of the study and the Human Consent. The study involved 13 TLE patients, 18-50 years old with the average preoperative seizure frequency ranging from 0.02 to 18 seizures per day (rare seizures, seizure frequency ≤ 0.1 seizures per day, in 6 patients; frequent seizures, seizure frequency > 0.1 seizure per day, in 7 patients), temporal lobe origin of seizures and clinical phenomenology of complex partial seizures. All patients in this study underwent en bloc anterior temporal lobectomy and amygdalohippocampectomy after preoperative planning and seizure focus localization to a single temporal lobe in accordance with previously described technique [28]. Briefly, the anterior temporal lobectomy technique involved en bloc resection of the lateral temporal cortex followed by dissection of superior temporal gyrus white matter using subpial technique, exposure of the temporal horn, and identification of the amygdala and hippocampus using the choroidal fissure as the superior-most landmark for en bloc resection of the hippocampus. The neurosurgical specimens were removed within 30-45 min from the beginning of the operation. Postoperatively, eight patients became seizure-free, 5 significantly improved and one improved in accordance with previously described seizure outcome criteria [28] (Table 1). Lateral temporal cortical tissues included surgical specimens of one trauma and one brain tumor (glioblastoma multiforme) case, and three postmortem cases of non-epileptic temporal lobe tissues. Samples were split with sterile scalpels in an aseptic environment for processing into 10% buffered formalin, which was kept at room temperature, and O.C.T. compound, which was placed into liquid nitrogen for immediate freezing. Formalin samples were transferred into 70% ETOH the next morning and sent for routine paraffin embedding. Samples were cut at 5μm slices for immunohistochemistry. Frozen blocks were transferred to -80°C until sectioned at 5μm for immunofluorescence.
Table 1.
Clinical Data for the Temporal Lobe Epilepsy Patients
| Pt. # | Age of Onset (years) | Seizure frequency | Epilepsy Duration (years) | Follow-up (months) | Surgical Outcome | Seizure Focus | Pathology | Neuroimaging | |
|---|---|---|---|---|---|---|---|---|---|
| MRI | PET | ||||||||
| 1 | 12 | 1.4/day | 6 | 0.1 | SF | Left TL | - | Left H atrophy | Left TL hypometabolism |
| 2 | 29 | 18/day | 4 | 11 | SI | Right TL | Focal cortical dysplasia & gliosis | Right hemisphere polygyria | Left TL hypometabolism |
| 3 | 44 | 0.02/day | 7 | 16 | SF | Right TL | - | Right medial temporal sclerosis | Right TL hypometabolism |
| 4 | 28 | 0.1/day | 8 | 1 | SF | Right TL | - | Right H atrophy | Right TL hypometabolism |
| 5 | 0.5 | 0.3/day | 31 | 13 | I | Left TL | - | Left medial temporal sclerosis | Left TL hypometabolism |
| 6 | 6 | 7/day | 32 | 12 | SI | Left TL | - | Normal | Normal |
| 7 | 30 | 0.1/day | 15 | 2 | SI | Left TL | - | Left H atrophy | Left lateral and right medial TL hypometabolism |
| 8 | 13 | 0.05/day | 26 | 6 | SI | Right TL | - | Right H atrophy | Right anterior TL hypometabolism |
| 9 | 11 | 0.1/day | 2 | 36 | SF | Left TL | Amygdalar low grade ependymoma (WHO grade II) | Left amygdalar minimally enhancing mass | |
| 10 | 30 | 0.1/day | 10 | 11 | SF | Right TL | - | Right medial temporal sclerosis | Right anterior/medial TL hypometabolism |
| 11 | 19 | 1/day | 22 | 4 | SF | Left TL | - | Left frontal lobe gliosis | Left anterior TL hypometabolism |
| 12 | 21 | 1.5/day | 34 | 7 | SF | Left TL | Cortical micro-calcifications | Cystic lesion left medial TL | |
| 13 | 31 | 0.3/day | 8 | 4 | SI | Left TL | - | Left medial temporal sclerosis | Left TL hypometabolism |
SF Seizure-free
SI Significantly
I Improved
U Unchanged
H Hippocampus
TL Temporal Lobe
No seizures
> 90% seizure frequency reduction
50-90% seizure frequency reduction
< 50% seizure frequency reduction
Immunohistochemistry (IHC)
IHC was performed using the single or double Envision technique (with rabbit and mouse antibodies) or the LSAB 2 technique (with goat antibody) (both from DAKO, Carpinteria, CA) as previously described [29]. Briefly, the tissue sections were deparaffinized, subjected to antigen retrieval by steam in DAKO antigen retrieval solution, blocked with dual endogenous block solution followed by serum-free protein block solution (both DAKO), stained in the Sequenza apparatus with the primary antibody in the single Envision technique (DAKO) and stained additionally with the second primary antibody in the double Envision technique. The primary antibodies (at 5 or 10 μg/ml) were rabbit anti-RANTES (Torrey Pines Biolabs, Inc.), mouse-anti-RANTES (BioSource), goat anti-interleukin-1β (Santa Cruz Biotechnology), mouse anti-CD68 (DAKO), mouse anti-COX-2 (Cayman), goat anti- MIP-1α (CCL3), goat anti-MIP-1β (CCL4), goat anti-IL-6, goat anti-CXCR4, goat anti-CCR5, goat anti-CCR1 (R&D), rabbit anti-fibrinogen (DAKO), rabbit Phospho-Rb (Cell Signaling Technology), rabbit E2F-1 (Santa Cruz Biotechnology), rabbit Caspase 3 (AR-55 Burnham Institute), rabbit Caspase 8 (AR-17 Burnham Institute), rabbit Caspase 9 (AR-57 Burnham Institute), mouse anti-NeuN (Chemicon, Temecula, CA), mouse anti-MAP-2 (Sigma), mouse anti-GFAP (DAKO), normal rabbit IgG and goat IgG (CalTag). When indicated, rabbit anti-RANTES was absorbed with recombinant RANTES (Torrey Pines Biolabs, Inc.). Stained sections were examined in Olympus Bmax microscope and photographed with DP11 camera. Positive staining was semi-quantitatively evaluated as 1+ to 4 +. Apoptotic cells were detected using Dead-End Colorimetric TUNEL system (Promega) according to manufacturer’s instructions.
Confocal microscopy
Cryoprotected sections were fixed in 100% acetone, washed with phosphate-buffered saline (PBS), permeabilized with 0.1% Triton in PBS for 10 minutes, washed again with PBS, blocked with 3% normal goat serum, incubated with primary antibodies (rabbit anti-RANTES and mouse anti-MAP2) followed by incubation with secondary antibodies (anti-mouse ALEXA488 and anti-rabbit ALEXA 594) and DAPI and examined by confocal microscopy as described [30].
RESULTS
CC- chemokines RANTES, MIP-1α and MIP-1β, and their receptors CCR1 and CCR5 are expressed in TLE neurons (Table 2)
Table 2.
Inflammatory and apoptotic markers in TLE and control tissues
| Pt. | Disease | RANTES | MIP1α | MIP1β | SDF-1 | CCR5 | CXCR4 | IL-1β | COX-2 | CD-68 | E2F-1 | P-Rb | Casp3 | Casp8 | Casp9 | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| # | TL | H | TL | H | TL | H | TL | H | TL | H | TL | H | TL | H | TL | H | TL | H | TL | H | TL | H | TL | H | TL | H | TL | H | |
| 1 | TLE | nucl+ | nucl+ | 2+ | 2+ | + | + | + | + | + | + | 2+ | + | + | + | + | - | + | + | + | |||||||||
| 2 | TLE | nucl+ | nucl+ | + | + | + | + | + | + | + | + | + | + | + | + | - | + | + | + | ||||||||||
| 3 | TLE | nucl+ | nucl+ | + | - | + | + | + | + | 2+ | + | + | + | + | + | - | - | ||||||||||||
| 4 | TLE | nucl+ | nucl+ | + | + | + | + | + | 2+ | + | + | + | + | ||||||||||||||||
| 5 | TLE | nucl+ | 2+ | + | + | - | + | + | + | - | |||||||||||||||||||
| 6 | TLE | nucl+ | + | 2+ | + | + | + | + | 4+ | + | + | + | + | + | |||||||||||||||
| 7 | TLE | cytopl+ | nucl+ | + | + | - | + | + | + | + | + | + | + | - | - | ||||||||||||||
| 8 | TLE | nucl 2+ | nucl+ | + | + | + | + | + | + | + | + | - | - | + | + | + | - | - | + | + | + | + | + | + | |||||
| 9 | TLE | nucl+ | 2+ | + | + | - | + | + | - | + | + | + | + | ||||||||||||||||
| 10 | TLE | nucl+ | nucl+ | + | - | + | + | + | + | + | - | + | + | - | + | + | |||||||||||||
| 11 | TLE | nucl 4+ | + | + | + | + | + | + | + | + | + | - | + | + | + | + | - | - | + | + | |||||||||
| 12 | TLE | - | + | 2+ | + | + | - | - | - | + | + | ||||||||||||||||||
| 13 | TLE | nucl+ | + | + | - | - | - | - | + | + | + | ||||||||||||||||||
| Control | trauma | - | + | + | - | + | + | + | + | - | - | - | - | - | - | - | - | - | - | ||||||||||
| Glioblastoma Multiforme | + | + | + | + | - | + | 2+ | ||||||||||||||||||||||
| angiosarcoma of the pleura | - | - | + | + | - | - | - | - | |||||||||||||||||||||
| congenital heart disease | - | - | + | + | - | - | - | - | |||||||||||||||||||||
| primary pulmonary hypertension | - | - | + | + | - | - | - | - | |||||||||||||||||||||
RANTES (CCL5) was expressed in patchy fashion in neuronal nuclei (identified by NeuN staining) (Fig. 1A-D) of all TLE tissues but one (Table 2). Nuclear localization of RANTES was confirmed by IHC (Fig.1B) and confocal microscopy, which showed colocalization of RANTES with the nucleus (Fig. 1C). Hippocampal TLE tissues were tested in 7 patients and all were positive for nuclear RANTES (Table 2). RANTES staining was absorbed by RANTES antigen (not shown). Astrocytes did not express RANTES (Fig. 1D). IP-1α (CCL3), MIP-1β (CCL4) (Figs. 1 E, F) and MCP-1 (CCL2) (not shown) were expressed in neuronal cytoplasm spreading into the neuropil. Neurons in the control tissues were negative for RANTES (Fig. 1G). The chemokine receptors CCR1 and CCR5 were expressed in lateral temporal cortical and hippocampal neurons of TLE cases (Figs.1 I, J) but not in the control tissues (Fig.1L).
Figure 1. Expression of CC and CXC chemokines and chemokine receptors in MIE (medically intractable temporal lobe epilepsy) and trauma.
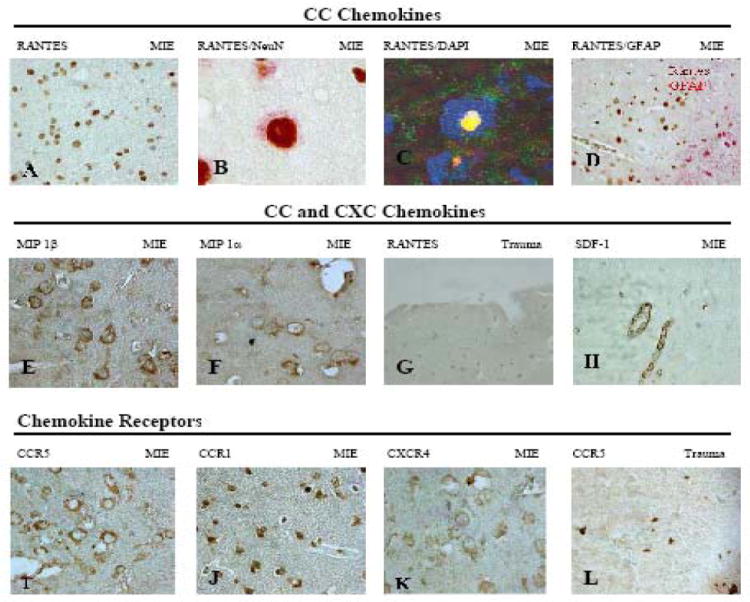
Immunohistochemistry or confocal microscopy of temporal lobe (TL) and hippocampus (H) using the following antibodies: (A) RANTES (TL, 40X); (B) RANTES/NeuN (TL, 100x); (C) RANTES green /DAPI (TL, confocal microscopy 100x) (D) RANTES/GFAP (H, 40x); (E) MIP -1 β (40x); (F) MIP-1α(40x); (G) RANTES (Trauma cortex, 20x); (H) SDF-1(H,40x); (I) CCR5 (20x); (J) CCR1 (40x); (K) CXCR4 (40x); (L) CCR5 (Trauma cortex, 40x). Note RANTES expression in neuronal nuclei (A-D) but cytoplasmic expression of MIP-1α and MIP-1β extending into the neuropil (E, F).
TLE Endothelial cells express SDF-1 and IL-6
The TLE neurons expressed the CXCR4 receptor (Fig. 1K) but not its ligand, the chemokine SDF-1 (CXCL12) (Fig. 1H). Endothelial cells in TLE tissues expressed SDF-1 (Fig. 1H) and IL-6 (Fig. 2D), a cytokine probably responsible for induction of SDF-1[31].
Figure 2. Expression of COX-2 and cytokines in MIE (medically intractable temporal lobe epilepsy) and trauma.
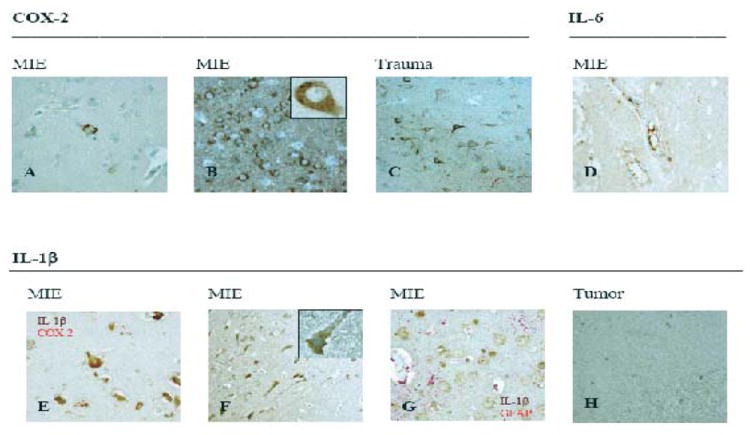
Immunohistochemistry of temporal lobe (TL) and hippocampus (H) using the following antibodies: (A) COX-2 (Case 2,TL, 40x); (B) COX-2 (Case 7,TL, 40x/100x); (C) COX-2 (cortex, trauma case, 20x); (D) IL-6 (40x); (E) IL-1β/COX 2 (40x); (F) IL-1β (40x/100x); (G) IL-1β/GFAP (H, 40x); (H) IL-1β (cortex, tumor case, 40x).
COX-2 and interleukin-1β expression is non-specific for TLE
In TLE neurons COX-2 expression was cytoplasmic and variable in its intensity, involving either rare neurons (Fig.2A) or all neurons in zonal fashion (Fig.2B), and while it did not consistently correlate with the frequency of seizures COX-2 expression was strong in the patients #3, #4 and # 7 with rare seizures (Table 1 and 2). Neuronal expression of COX-2 was also noted in the traumatic brain injury tissue (Fig. 2C). IL-1β was expressed by TLE neurons (Fig. 2F) but not by astrocytes (Fig. 2G) or neurons in peritumoral tissue (Fig. 2H). When double-stained, COX-2 and IL-1β co-localized in TLE neurons (Fig. 2E).
Cell cycle proteins and apoptotic markers are expressed in TLE neurons
E2F1 was expressed exclusively in the neuronal cytoplasm of all tested TLE specimens (Figs. 3E, F), but not in the trauma case (Fig. 3G) or control autopsy brain tissues (Table 2). Phosphorylated Rb was not expressed in any TLE tissue (Fig. 3H) (Table 2). The apoptotic markers, cleaved caspase 3, 8 and 9, were positive in neurons of all tested TLE specimens (Figs. 3 B, C, D) (Table 2). Only one TLE specimen showed widespread TUNEL staining of neurons; the remaining specimens were negative (Fig. 3A).
Figure 3. Expression of apoptotic markers and cell cycle proteins in MIE (medically intractable temporal lobe epilepsy) and trauma.
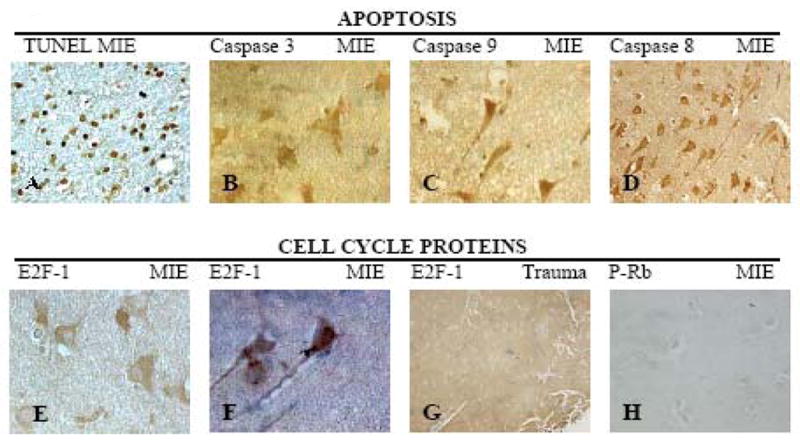
TUNEL reaction or immunohistochemistry of temporal lobe (TL) and hippocampus (H) using the following antibodies (A) TUNEL (TL, 40x); (B) Caspase 3 (AR-55) (TL,40x) (C) TL Caspase 9 (AR-57) (40x); (D) Caspase 8 (AR-17) (TL, 40x); (E) E2F-1 (TL, 100x); (F) E2F-1 (H,100x); (G) E2F-1 (Trauma cortex, 10x); (H) P-Rb (H, 20x).
Perivascular infiltration with macrophages and leakage of blood-brain barrier in TLE
Temporal lobe epilepsy tissues showed perivascular infiltration with macrophages and visible microglia (Fig. 4 A, B), whereas the trauma tissue showed only microglia but not perivascular macrophages (Fig. 4C). The TLE tissue showed perivascular fibrinogen leakage (Fig. 4D). No infiltration by CD3 T or CD20 B cells of TLE tissues was detected (not shown).
Figure 4. CD 68 and fibrinogen expression in MIE (medically intractable temporal lobe epilepsy) and trauma.
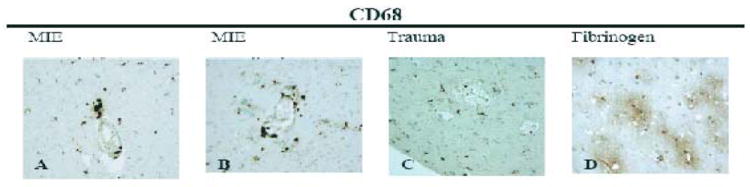
Perivascular infiltration and blood brain barrier leakage were demonstrated by immunohistochemical staining of the temporal lobe and hippocampus using anti-CD 68 in TL (A, 40X) and H (B, 40X) of MIE, TL of trauma (C,40X), as well as fibrinogen staining in TL (D, 20x).
DISCUSSION
This study identifies upstream chemokine and cell-cycle protein signals of neuronal apoptosis through immunohistochemistry and confocal microscopy of lateral cortical and hippocampal tissues in patients who have undergone surgical treatment of medically intractable temporal lobe epilepsy (TLE). In this study, we have examined surgical specimens of TLE patients for the expression of chemokines and the transcription factor, E2F1, and neuronal apoptosis. The neurons expressed CC-chemokines but not CXC-chemokines. The salient finding was RANTES expression in the nuclei of TLE neurons but not control neurons. The nuclear localization of RANTES contrasts with the typical cytoplasmic and extracellular distribution of chemokines. Chemokine induction is unlikely a result of surgery because the time from onset to completion of surgical removal of both lateral temporal cortex and hippocampus (30-45 minutes) was too short for protein expression. After a surgical insult to the mouse brain, MCP-1 induction required at least 3 h and that of RANTES at least 24h [32]. Furthermore, RANTES does not translocate to the nucleus [33].
Chemokines are chemotactic proteins classified within four sub-families (CXC-, CC-, C-, and CXXXC) [23] which, in the central nervous system, may exert control of neural plasticity (via CCL5) [24], inflammation and apoptosis (via CCL5-CCR5) and may have positive neurodevelopmental effects (via the SD F-1 ligand) [25] or neurootoxic effects (via the HIV-1 protein gp120) [26]. Chemokines may lead to neuronal death through the E2F1 signaling pathway [27]. CC-chemokines have been shown to be upregulated in the hippocampus in the pilocarpine model of status epilepticus and are implicated in pathological neuroplasticity and neuroinflammation following seizures [34]. The chemokine, RANTES has been implicated in the recruitment of inflammatory cells to the central nervous system in the kanic acid seizure model [35]. In the current study, the chemokine RANTES (CCR5) and other CC-chemokines were expressed in lateral temporal cortical and hippocampal neurons of TLE patients, but not in neurons of control cases. The chemokine RANTES is usually found in cytoplasmic and extracellular locations. However, in TLE neurons, RANTES was displayed in an unusual location, the neuronal nuclei. In surgical specimens of TLE patients in the current study, neurons expressed CC-chemokines, but not CXC-chemokines.
Induction of inflammatory cytokines and chemokines leads to apoptosis. Inflammatory cytokines, such as interleukin 1β (IL-1β), are not usually expressed in normal brain tissue but are demonstrated in a diverse range of neurodegenerative disorders [14]. Cytokine expression has been detected in temporal lobe epilepsy at autopsy [15] and in rodent brain tissues after seizure induction [16]. The cytokine IL-1β may induce pro-apoptotic and excitatory signals, which lead to long-lasting changes in gene transcription [17]. IL-1β is proconvulsive [36], has increased expression in the dentate gyrus following lithium-pilocarpine-induced seizures and has been implicated in the pathophysiology of status epilepticus and functional brain impairment after seizures [37]. In the current study, TLE and trauma case neurons strongly expressed IL-1β and COX-2. IL-1β is induced by pyrogenic signals in the anterior hypothalamus in febrile states. IL-1β induction in epilepsy may be due to febrile stimuli and/or ischemic, traumatic or excitotoxic stimuli [19]. IL-1β induces inflammatory activation of the brain, as shown in mixed glial/astrocyte cultures where bacterial lipopolysaccharide stimulates IL-1β followed by release of prostaglandin E2 and IL-6, and activation of NF-κB, p38, JNK and ERK1/2 [38].
In the central nervous system, chemokines and chemokine receptors play crucial developmental functions and modulate the central nervous system for neurorepair and neuroregeneration or inflammation and cell death. Examples of such neuroprotective and neuroregenerative activities include induction of RANTES-responsive genes involved in neuronal survival, neurite outgrowth and synaptogenesis [24] and attraction of neuronal progenitors towards inflammatory stimuli [39]. Chemokines are small molecules, which diffuse readily from the cytoplasm, thus the restricted nuclear location of RANTES in epileptic neurons belies its chemotactic role. However, another chemokine PESKY/CCL27 has a nuclear translocation residue and, upon nuclear translocation, induces cytoskeletal actin reorganization [35], which could lead to apoptosis, as is the case in HIV-1 infection [36]. As discussed below, in TLE brains, nuclear RANTES could play a role in neuronal apoptosis because all but one of the examined TLE tissues showed neurons positive for RANTES and activated caspases. RANTES, MIP-1α/β and MCP-1 may have chemotactic function leading to perivascular infiltration by macrophages.
The chemokine, SDF-1, is the ligand for the CXCR4 receptor. SDF-1 itself was expressed only by endothelial cells in the TLE cases but its receptor, CXCR4, was expressed in TLE neurons. Chemokine signaling by HIV-1 through CXCR4 induces neuronal apoptosis [41]. CXCR4 signaling may be pro-apoptotic through activation of E2F1 [27] or anti-apoptotic through activation of Akt [42]. Although the role of SDF-1 signaling in epilepsy is unresolved, we speculate that it could be important therapeutically using synthetically modified CXCR4 and CCR5 ligands modulating neuronal survival rather than apoptosis [43]
Apoptotic markers (caspase-3, -8, -9) were expressed in lateral temporal cortical and hippocampal neurons of TLE patients but not in neurons of control cases. The cell-cycle regulatory transcription factor E2F1 was found in an abnormal location, exclusively in TLE neuronal cytoplasm but phosphorylated Rb (pRb) was not detected. RANTES has been shown to cause subcellular translocation of E2F1 from neuronal nuclei to the cytoplasm potentially explaining the cytoplasmic localization of E2F1 in neurons of patients with medically intractable temporal lobe epilepsy [44]. However, in the current study, this cytoplasmic localization of E2F1 was not associated with the previously reported concurrent observation of increased phosphylorated Rb [44], suggesting a potentially alternate and as yet unidentified mechanism for the subcellular translocation of E2F1 in human temporal lobe epilepsy. The mechanism by which cytoplasmic E2F1 produces apoptosis in temporal lobe epilepsy is not known. However, E2F1 has been shown to induce apoptosis independent of its transcriptional activity [45]. Specifically, E2F1 can induce apoptosis by a death receptor-dependent mechanism through downregulating TRAF2 protein levels [45]. TRAF proteins are intracellular signal transducers for several superfamilies of immune receptors [45]. TRAF2 interacts with TNF receptors mediating the survival of the receptors [45]. TRAF2, which E2F1 downregulates, is required for protection against TNF-induced apoptosis [46]. While the transcription factor E2F1 is known for its interaction with pRb, particularly in controlling cell proliferation, E2F1 also appears to have a pivotal role in regulating apoptosis in the absence of phosphylorated Rb [47]. For instance, in certain breast cancer cell lines which lack pRb, E2F1 overexpression results in apoptosis [47]. E2F family transcription factors are crucial in the induction of aberrant cell cycle and neuronal apoptosis [48]. Phosphorylation of Rb releases E2F-1 from a complex with Rb and activates cell cycle programming [49]. E2F1 and molecules that either regulate E2F activity or that are transcriptional targets of E2F, in particular P-Rb and p53, are induced in diverse neurodegenerative diseases, such as SIV encephalitis [50] and Parkinson’s disease [11, 12]. E2F1 is involved in chemokine related neurotoxicity [27], through mechanisms which upregulate proteins Cdc2 and Puma [51]. For example, deletion of puma appears to protect hippocampal neurons in a status epilepticus model [52]. Specifically, neuronal death is reduced by approximately 50% in the hippocampus of puma-deficient mice [52]. Supporting the concept that apoptotic pathway activation is a trigger of epileptogenesis, data have been reported which show that genetic deletion of the proapoptotic protein, puma, acting acutely influences neuronal death and subsequently alters the phenotype of epilepsy in the long-term [53]. In a rodent model, in comparison to puma-expressing mice, puma-deficient mice had significantly smaller hippocampal lesions following status epilepticus [53]. Long-term EEG recording detected an approximately 60% reduction in the frequency of epileptic seizures in puma-deficient mice compared to puma-expressing mice [53].
RANTES stimulates increased cytoplasmic expression of E2F1 and nuclear localization of the hyperphosphorylated retinoblastoma susceptibility gene product (ppRb) [54]. The balance of cytoplasmic and nuclear location of E2F1 may play a role in pathological role of E2F1 in neurodegenerative disorders [55]. While RANTES alone has been shown to induce apoptosis through activation of caspase-9, caspase-3, release of cytochrome c and poly(ADP-ribose) polymerase cleavage, there is evidence that IFN-gamma stimulates RANTES which in turn produces apoptosis via stimulation of caspase-3 and caspase-9 [56, 57]. Production of IF-N gamma has been shown to result from Con A-stimulated splenocytes in left side kindled rats with temporal lobe seizures [58]. Therefore, future research is warranted to investigate the complex interactions involving IFN-gamma, RANTES, caspase-3, caspase-9 and their roles in neuronal apoptosis in temporal lobe epilepsy.
If E2F1 is involved in the pathogenesis of neuronal apoptosis in TLE, E2F1 blockade might be beneficial in TLE as has been shown in 1-methyl-4-phenyl 1,2,3,6 tetrahydropyridine model of Parkinson’s disease [11]. Based on these considerations, neuroprotective drugs for epileptic neurons should be sought among anti-inflammatory drugs, such as flavonoids [59], neurotrophic factors [60], small molecular inhibitors of RANTES signaling through Met-RANTES/CC chemokine ligand 5 (CCL5) [61], CCR5 antagonists, such as the Merck’s compound 167 (CMPD 167) [62], and caspase inhibitors [63, 64]. The role of cytoplasmic E2F1 in neurodegenerative diseases, such as HIV encephalitis, SIV encephalitis, Alzheimer Disease, Parkinson Disease, Huntington Disease, and amyotrophic lateral sclerosis, is not understood but, as suggested by Jordan-Sciutto [65], the insight into cytoplasmic effects of E2F1 could lead to novel and unique therapeutic opportunities in neurodegenerative diseases.
The results of this study suggest that chemotactic and mitogenic stimuli of neuronal apoptosis are expressed in both cortical and hippocampal tissues in patients with medically intractable temporal lobe epilepsy. These findings may have implications for the development of new medical and surgical therapies for TLE. Recent evidence suggests that increasing resection of temporal lobe tissue beyond selective hippocampectomy to include entorhinal and lateral temporal cortex may optimize seizure outcome in TLE [66, 67, 68]. Improved seizure outcome with more extensive resection of hippocampus with lateral temporal cortex may be a result of increased removal of diseased neuronal populations affected by inflammation and apoptosis. Anti-inflammatory drugs are considered for use in epilepsy both for their anticonvulsant activity and modulation of gene transcription [19]. Vagus nerve stimulation has been shown to have immune rebalancing functions which may be related to its antiseizure action [20]. Inhibition of neuronal inflammation and apoptosis may provide a new paradigm for development of medical and surgical strategies including neuromodulation of both the central and peripheral nervous system.
CONCLUSION
In this study, we have examined surgical specimens of TLE patients for the expression of chemokines and the transcription factor, E2F1, and neuronal apoptosis. The results of this study suggest that apoptosis in neurons of patients with medically intractable temporal lobe epilepsy may be induced by a combination of chemotactic and mitogenic stimuli. The cytoplasmic effects of E2F1 and nuclear effects of RANTES might have novel roles in apoptosis of TLE neurons and indicate a need to develop new neuroprotective strategies against apoptotic signaling by these molecules. Both RANTES and E2F1 signaling are upstream from caspase activation, thus the authors hypothesize that medical and/or surgical therapeutic antagonists of RANTES and/or E2F-1 blockade might be neuroprotective for patients with medically intractable temporal lobe epilepsy. Temporal cortical, hippocampal and vagus nerve stimulation should be evaluated for their neuromodulatory effects in blocking neuronal RANTES and E2F1 expression. The results offer a new paradigm for development of medical and surgical therapy, including resective surgery and neuromodulation, based on removal and/or inhibition of chemotactic and mitogenic stimuli of neuronal apoptosis in patients with medically intractable temporal lobe epilepsy.
Acknowledgments
We thank S. Krajewski and J. Reed, Burnham Institute, San Diego for antibodies to cleaved caspases, and P. Murphy, NIH, Bethesda for discussion of chemokine expression. Santosh Desai, Karolina Simonyan, and Rosalie Khakshoor provided excellent technical assistance with the study. Jane Lee assisted with the manuscript.
Footnotes
The authors report no financial interest in any product or service mentioned in this article.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Henshall DC, Simon RP. Epilepsy and apoptosis pathways. J Cereb Blood Flow Metab. 2005;25(12):1557–72. doi: 10.1038/sj.jcbfm.9600149. [DOI] [PubMed] [Google Scholar]
- 2.Henshall DC, et al. Formation of the Apaf-1/cytochrome c complex precedes activation of caspase-9 during seizure-induced neuronal death. Cell Death Differ. 2001;8(12):1169–81. doi: 10.1038/sj.cdd.4400921. [DOI] [PubMed] [Google Scholar]
- 3.Meller R, et al. Activation of the caspase 8 pathway mediates seizure-induced cell death in cultured hippocampal neurons. Epilepsy Res. 2006;70(1):3–14. doi: 10.1016/j.eplepsyres.2006.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yamamoto A, et al. Evidence of tumor necrosis factor receptor 1 signaling in human temporal lobe epilepsy. Exp Neurol. 2006;202(2):410–20. doi: 10.1016/j.expneurol.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 5.Hallstrom TC, Nevins JR. Specificity in the activation and control of transcription factor E2F-dependent apoptosis. Proc Natl Acad Sci U S A. 2003;100(19):10848–53. doi: 10.1073/pnas.1831408100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nguyen MD, Mushynski WE, Julien JP. Cycling at the interface between neurodevelopment and neurodegeneration. Cell Death Differ. 2002;9(12):1294–306. doi: 10.1038/sj.cdd.4401108. [DOI] [PubMed] [Google Scholar]
- 7.Herrup K, et al. Divide and die: cell cycle events as triggers of nerve cell death. J Neurosci. 2004;24(42):9232–9. doi: 10.1523/JNEUROSCI.3347-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jordan-Sciutto KL, et al. Cell cycle proteins exhibit altered expression patterns in lentiviral-associated encephalitis. J Neurosci. 2002;22(6):2185–95. doi: 10.1523/JNEUROSCI.22-06-02185.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jordan-Sciutto K, Rhodes J, Bowser R. Altered subcellular distribution of transcriptional regulators in response to Abeta peptide and during Alzheimer’s disease. Mech Ageing Dev. 2001;123(1):11–20. doi: 10.1016/s0047-6374(01)00334-7. [DOI] [PubMed] [Google Scholar]
- 10.Ranganathan S, Bowser R. Alterations in G(1) to S phase cell-cycle regulators during amyotrophic lateral sclerosis. Am J Pathol. 2003;162(3):823–35. doi: 10.1016/S0002-9440(10)63879-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hoglinger GU, et al. The pRb/E2F cell-cycle pathway mediates cell death in Parkinson’s disease. Proc Natl Acad Sci U S A. 2007;104(9):3585–90. doi: 10.1073/pnas.0611671104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jordan-Sciutto KL, et al. Expression patterns of retinoblastoma protein in Parkinson disease. J Neuropathol Exp Neurol. 2003;62(1):68–74. doi: 10.1093/jnen/62.1.68. [DOI] [PubMed] [Google Scholar]
- 12a.Xiao-Ting H, Patrick K. Update and overview of the International League Against Epilepsy consensus definition of drug-resistant epilepsy. Eur Neurol Rev. 2011;6(1):57–9. [Google Scholar]
- 13.Schobitz B, De Kloet ER, Holsboer F. Gene expression and function of interleukin 1, interleukin 6 and tumor necrosis factor in the brain. Prog Neurobiol. 1994;44(4):397–432. doi: 10.1016/0301-0082(94)90034-5. [DOI] [PubMed] [Google Scholar]
- 14.Zhao B, Schwartz JP. Involvement of cytokines in normal CNS development and neurological diseases: recent progress and perspectives. J Neurosci Res. 1998;52(1):7–16. doi: 10.1002/(SICI)1097-4547(19980401)52:1<7::AID-JNR2>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 15.Sheng JG, et al. Increased neuronal beta-amyloid precursor protein expression in human temporal lobe epilepsy: association with interleukin-1 alpha immunoreactivity. J Neurochem. 1994;63(5):1872–9. doi: 10.1046/j.1471-4159.1994.63051872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.De Simoni MG, et al. Inflammatory cytokines and related genes are induced in the rat hippocampus by limbic status epilepticus. Eur J Neurosci. 2000;12(7):2623–33. doi: 10.1046/j.1460-9568.2000.00140.x. [DOI] [PubMed] [Google Scholar]
- 17.Vezzani A, Baram TZ. New roles for interleukin-1 Beta in the mechanisms of epilepsy. Epilepsy Curr. 2007;7(2):45–50. doi: 10.1111/j.1535-7511.2007.00165.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ravizza T, et al. Innate and adaptive immunity during epileptogenesis and spontaneous seizures: evidence from experimental models and human temporal lobe epilepsy. Neurobiol Dis. 2008;29(1):142–60. doi: 10.1016/j.nbd.2007.08.012. [DOI] [PubMed] [Google Scholar]
- 19.Vezzani A, Granata T. Brain inflammation in epilepsy: experimental and clinical evidence. Epilepsia. 2005;46(11):1724–43. doi: 10.1111/j.1528-1167.2005.00298.x. [DOI] [PubMed] [Google Scholar]
- 20.Maijoie HJ, Rijkers K, Berfelo MW, Hulsman JA, Myint A, Schwarz M, Vies JS. Vagus nerve stimulation in refractory epilepsy: Effects of Pro- and Anti-inflammatory cytokines in peripheral blood. Neuroimmunomodulation. 2010;18(1):52–6. doi: 10.1159/000315530. [DOI] [PubMed] [Google Scholar]
- 21.Burkovetskaya ME, Levin SG, Godukhin OV. Neuroprotective effects of interleukin-10 and tumor necrosis factor-alpha against hypoxia-induced hyperexcitability in hippocampal slice neurons. Neurosci Lett. 2007;416(3):236–40. doi: 10.1016/j.neulet.2006.12.059. [DOI] [PubMed] [Google Scholar]
- 22.Jauneau AC, et al. Interleukin-1beta and anaphylatoxins exert a synergistic effect on NGF expression by astrocytes. J Neuroinflammation. 2006;3:8. doi: 10.1186/1742-2094-3-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Murphy PM, et al. International union of pharmacology. XXII. Nomenclature for chemokine receptors. Pharmacol Rev. 2000;52(1):145–76. [PubMed] [Google Scholar]
- 24.Valerio A, et al. Gene expression profile activated by the chemokine CCL5/RANTES in human neuronal cells. J Neurosci Res. 2004;78(3):371–82. doi: 10.1002/jnr.20250. [DOI] [PubMed] [Google Scholar]
- 25.Pujol F, Kitabgi P, Boudin H. The chemokine SDF-1 differentially regulates axonal elongation and branching in hippocampal neurons. J Cell Sci. 2005;118(Pt 5):1071–80. doi: 10.1242/jcs.01694. [DOI] [PubMed] [Google Scholar]
- 26.Brandimarti R, et al. Regulation of cell cycle proteins by chemokine receptors: A novel pathway in human immunodeficiency virus neuropathogenesis? J Neurovirol. 2004;10(Suppl 1):108–12. doi: 10.1080/753312761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shimizu S, et al. Role of the transcription factor E2F1 in CXCR4-mediated neurotoxicity and HIV neuropathology. Neurobiol Dis. 2007;25(1):17–26. doi: 10.1016/j.nbd.2006.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Weinand ME, Wyler AR, Richey ET, Phillips BB, Somes GW. Long-term ictal monitoring with subdural strip electrrodes: prognostic factors for selecting temporal lobectomy candidates. J Neurosurg. 1992;77(1):20–8. doi: 10.3171/jns.1992.77.1.0020. [DOI] [PubMed] [Google Scholar]
- 29.Fiala M, et al. Cyclooxygenase-2-positive macrophages infiltrate the Alzheimer’s disease brain and damage the blood-brain barrier. Eur J Clin Invest. 2002;32(5):360–71. doi: 10.1046/j.1365-2362.2002.00994.x. [DOI] [PubMed] [Google Scholar]
- 30.Fiala M, et al. Innate immunity and transcription of MGAT-III and Toll-like receptors in Alzheimer’s disease patients are improved by bisdemethoxycurcumin. Proc Natl Acad Sc USA. 2007;104:12849–12854. doi: 10.1073/pnas.0701267104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hideshima T, et al. Cytokines and signal transduction. Best Pract Res Clin Haematol. 2005;18(4):509–24. doi: 10.1016/j.beha.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 32.Babcock AA, et al. Chemokine expression by glial cells directs leukocytes to sites of axonal injury in the CNS. J Neurosci. 2003;23(21):7922–30. doi: 10.1523/JNEUROSCI.23-21-07922.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gortz A, et al. The chemokine ESkine/CCL27 displays novel modes of intracrine and paracrine function. J Immunol. 2002;169(3):1387–94. doi: 10.4049/jimmunol.169.3.1387. [DOI] [PubMed] [Google Scholar]
- 34.Foresti ML, Arisi GM, Katki K, Montafiez A, Sanchez RM, Shapiro LA. Chemokine CCL2 and its receptor CCR2 are increased in the hippocampus following pilocarpine-induced status epilepticus. J Neuroinflammation. 2009;6:40. doi: 10.1186/1742-2094-6-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Laing JM, Aurelian L. Delta RR vaccination protects from KA-induced seizures and neuronal loss through ICP10PK-mediated modulation of the neuronal-microglial axis. Genet Vaccines Ther. 2008;6:1. doi: 10.1186/1479-0556-6-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lehtmakl KA, Keranen T, Palmio J, Peltola J. Levels of IL-1 beta and IL-1ra in cerebrospinal fluid of human patients after single and prolonged seizures. Neuromodulation. 2010;17(1):19–22. doi: 10.1159/000243081. [DOI] [PubMed] [Google Scholar]
- 37.Zhang SJ, Li XW, Wei D, Jiang W. Expression of IL-1 mRNA in the dentate gyrus of adult rats following lithium-pilocarpine-induced seizures. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi. 2010;26(3):288–90. [PubMed] [Google Scholar]
- 38.Pinteaux E, et al. Expression of interleukin-1 receptors and their role in interleukin-1 actions in murine microglial cells. J Neurochem. 2002;83(4):754–63. doi: 10.1046/j.1471-4159.2002.01184.x. [DOI] [PubMed] [Google Scholar]
- 39.Belmadani A, et al. Chemokines regulate the migration of neural progenitors to sites of neuroinflammation. J Neurosci. 2006;26(12):3182–91. doi: 10.1523/JNEUROSCI.0156-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Matarrese P, Malorni W. Human immunodeficiency virus (HIV)-1 proteins and cytoskeleton: partners in viral life and host cell death. Cell Death Differ. 2005;12(Suppl 1):932–41. doi: 10.1038/sj.cdd.4401582. [DOI] [PubMed] [Google Scholar]
- 41.Hesselgesser J, et al. Neuronal apoptosis induced by HIV-1 gp120 and the chemokine SDF-1 alpha is mediated by the chemokine receptor CXCR4. Curr Biol. 1998;8(10):595–8. doi: 10.1016/s0960-9822(98)70230-1. [DOI] [PubMed] [Google Scholar]
- 42.Vlahakis SR, et al. G protein-coupled chemokine receptors induce both survival and apoptotic signaling pathways. J Immunol. 2002;169(10):5546–54. doi: 10.4049/jimmunol.169.10.5546. [DOI] [PubMed] [Google Scholar]
- 43.Choi WT, et al. Neuronal apoptotic signaling pathways probed and intervened by synthetically and modularly modified (SMM) chemokines. J Biol Chem. 2007;282(10):7154–63. doi: 10.1074/jbc.M611599200. [DOI] [PubMed] [Google Scholar]
- 44.Jordan-Sciutto KL, Murray Fenner BA, Wiley CA, Achim CL. Response of cell cycle proteins to neurotrophic factor and chemokine stimulation in human neuroglia. Exp Neurol. 2001;167(2):205–14. doi: 10.1006/exnr.2000.7594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Phillips AC, Ernst MK, Bates S, Rice NR, Vousden KH. E2F-1 potentiates cell death by blocking antiapoptotic signaling pathways. Mol Cell. 1999;4(5):771–81. doi: 10.1016/s1097-2765(00)80387-1. [DOI] [PubMed] [Google Scholar]
- 46.Vince JE, Pantaki D, Feltham R, Mace PD, Cordier SM, Schmukle AC, Davidson AJ, Callus BA, Wong WW, Gentle IE, Carter H, Lee EF, Walczak H, Day CL, Vaux DL, Silke J. TRAF2 must bind to cellular inhibitors of apoptosis for tumor necrosis factor (tnf) to efficiently activate nf-{kappa}b and to prevent tnf-induced apoptosis. J Biol Chem. 2009;284(51):35906–15. doi: 10.1074/jbc.M109.072256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sun B, Wingate H, Swisher SG, Keyomarsi K, Hunt KK. Absence of pRb facilitates E2F1-induced apoptosis in breast cancer cells. Cell Cycle. 2010 Mar 20;9(6) doi: 10.4161/cc.9.6.10990. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 48.Greene LA, Biswas SC, Liu DX. Cell cycle molecules and vertebrate neuron death: E2F at the hub. Cell Death Differ. 2004;11(1):49–60. doi: 10.1038/sj.cdd.4401341. [DOI] [PubMed] [Google Scholar]
- 49.Chellappan SP, et al. The E2F transcription factor is a cellular target for the RB protein. Cell. 1991;65(6):1053–61. doi: 10.1016/0092-8674(91)90557-f. [DOI] [PubMed] [Google Scholar]
- 50.Jordan-Sciutto KL, et al. Induction of cell-cycle regulators in simian immunodeficiency virus encephalitis. Am J Pathol. 2000;157(2):497–507. doi: 10.1016/S0002-9440(10)64561-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shimizu S, Khan MZ, Hippensteel RL, Parkar A, Raghupathi R, Meucci O. Role of the transcription factor E2F1 in CXCR4-mediated neurotoxicity and HIV neuropathology. Neurobiol Dis. 2007;25(1):17–26. doi: 10.1016/j.nbd.2006.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Engel T, Hatazaki S, Tanaka K, Prehn JH, Henshall DC. Deletion of Puma protects hippocampal neurons in a model of severe status epilepticus. Neuroscience. 2010;168(2):443–50. doi: 10.1016/j.neuroscience.2010.03.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Engel T, Murphy BM, Hatazaki S, Jimenez-Mateos EM, Concannon CG, Woods I, Prehn JH, Henshall DC. Reduced hippocampal damage and epileptic seizures after status epilepticus in mice lacking proapoptotic Puma. FASEB J. 2010;24(3):853–61. doi: 10.1096/fj.09-145870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Strachan GD, et al. Chemokine- and neurotrophic factor-induced changes in E2F1 localization and phosphorylation of the retinoblastoma susceptibility gene product (pRb) occur by distinct mechanisms in murine cortical cultures. Exp Neurol. 2005;193(2):455–68. doi: 10.1016/j.expneurol.2004.08.038. [DOI] [PubMed] [Google Scholar]
- 55.Chu CT, et al. Location, location, location: altered transcription factor trafficking in neurodegeneration. J Neuropathol Exp Neurol. 2007;66(10):873–83. doi: 10.1097/nen.0b013e318156a3d7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Murooka TT, Wong MM, Rahbar R, Majchrzak-Kita B, Proudfoot AE, Fish EN. CCL5-CCR5-mediated apoptosis in T cells: Requirement for glycosaminoglycan binding and CCL5 aggregation. J Biol Chem. 2006;281(35):25184–94. doi: 10.1074/jbc.M603912200. Epub 2006 Jun 28. [DOI] [PubMed] [Google Scholar]
- 57.Ryu HJ, Kim JE, Kim MJ, Kwon HJ, Suh SW, Song HK, Kang TC. The protective effects of interleukin-18 and interferon-γ on neuronal damages in the rat hippocampus following status epilepticus. Neuroscience. 2010;170(3):711–21. doi: 10.1016/j.neuroscience.2010.07.048. [DOI] [PubMed] [Google Scholar]
- 58.Goldstein KR, Bhatt R, Barton BE, Zalcman SS, Rameshwar P, Siegel A. Effects of hemispheric lateralization and site specificity on immune alterations induced by kindled temporal lobe seizures. Brain Behav Immun. 2002;16(6):706–19. doi: 10.1016/s0889-1591(02)00024-7. [DOI] [PubMed] [Google Scholar]
- 59.Sharma V, Mishra M, Gosh S, Tewari R, Basu A, Seth P, Sen E. Modulation of interleukin-1beta mediated inflammatory response in human astrocytes by flavonoids: implications in neuroprotection. Brain Res Bull. 2007;73(1-3):55–63. doi: 10.1016/j.brainresbull.2007.01.016. [DOI] [PubMed] [Google Scholar]
- 60.Mocchetti I, Bachis A, Masliah E. Chemokine receptors and neurotrophic factors: Potential therapy against aids dementia? J Neurosci Res. 2008;86(2):243–55. doi: 10.1002/jnr.21492. [DOI] [PubMed] [Google Scholar]
- 61.Chvatchko Y, et al. Inhibition of airway inflammation by amino-terminally modified RANTES/CC chemokine ligand 5 analogues is not mediated through CCR3. J Immunol. 2003;171(10):5498–506. doi: 10.4049/jimmunol.171.10.5498. [DOI] [PubMed] [Google Scholar]
- 62.Schroder C, et al. CCR5 blockade modulates inflammation and alloimmunity in primates. J Immunol. 2007;179(4):2289–99. doi: 10.4049/jimmunol.179.4.2289. [DOI] [PubMed] [Google Scholar]
- 63.Tompkins MM, et al. Apoptotic-like changes in Lewy-body-associated disorders and normal aging in substantia nigral neurons. Am J Pathol. 1997;150(1):119–31. [PMC free article] [PubMed] [Google Scholar]
- 64.Patil K, Sharma SC. Broad spectrum caspase inhibitor rescues retinal ganglion cells after ischemia. Neuroreport. 2004;15(6):981–4. doi: 10.1097/00001756-200404290-00010. [DOI] [PubMed] [Google Scholar]
- 65.Wang Y, et al. E2F1 localizes predominantly to neuronal cytoplasm and fails to induce expression of its transcriptional targets in human immunodeficiency virus-induced neuronal damage. Neurosci Lett. 2010;479(2):97–101. doi: 10.1016/j.neulet.2010.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Boniha L, Yasuda CL, Rorden C, Li LM, Tedeschi H, de Oliveira E, Cendes F. Does resection of the medial temporal lobe improve the outcome of temporal lobe epilepsy surgery? Epilepsia. 2007;48(3):571–8. doi: 10.1111/j.1528-1167.2006.00958.x. [DOI] [PubMed] [Google Scholar]
- 67.Cukiert A, Burattini JA, Mariani PP, Cukiert CM, Argentoni M, Baise-Zung C, Forster CR, Mello VA. Outcome after cortico-amygdalo-hippocampectomy in patients with temporal lobe epilepsy and normal MRI. Seizure. 2010;19(6):319–23. doi: 10.1016/j.seizure.2010.04.012. [DOI] [PubMed] [Google Scholar]
- 68.Murphy M, Smith PD, Wood M, Bowden S, O’Brien TJ, Bulluss KJ, Cook MJ. Surgery for temporal lobe epilepsy associated with mesial temporal sclerosis in the older patient: A long-term follow-up. Epilepsia. 2010;51(6):1024–9. doi: 10.1111/j.1528-1167.2009.02430.x. [DOI] [PubMed] [Google Scholar]