

Abstract
Cell-free protein synthesis has emerged as a powerful technology platform to help satisfy the growing demand for simple and efficient protein production. While used for decades as a foundational research tool for understanding transcription and translation, recent advances have made possible cost-effective microscale to manufacturing scale synthesis of complex proteins. Protein yields exceed grams protein produced per liter reaction volume, batch reactions last for multiple hours, costs have been reduced orders of magnitude, and reaction scale has reached the 100-liter milestone. These advances have inspired new applications in the synthesis of protein libraries for functional genomics and structural biology, the production of personalized medicines, and the expression of virus-like particles, among others. In the coming years, cell-free protein synthesis promises new industrial processes where short protein production timelines are crucial as well as innovative approaches to a wide range of applications.
Keywords: Cell-free protein synthesis, in vitro translation, recombinant DNA protein production, E. coli extract, wheat germ extract, personalized medicines, high-throughput, synthetic biology, cell-free biology
1. Introduction
Cell-free protein synthesis (CFPS) systems derived from crude cell extracts have been used for decades as a research tool in fundamental and applied biology (Fig. 1). They were used in the ground-breaking experiments of Nirenberg and Mattaei in 1961, playing an essential role in the discovery of the genetic code (Nirenberg and Matthaei, 1961). More recently, CFPS has shown remarkable utility as a protein synthesis technology (Katzen et al., 2005, Swartz, 2006), including the production of pharmaceutical proteins (Goerke and Swartz, 2008, Kanter et al., 2007, Yang et al., 2005), and high-throughput production of protein libraries for protein evolution and structural genomics (Madin et al., 2000, Takai et al., 2010).
Figure 1.
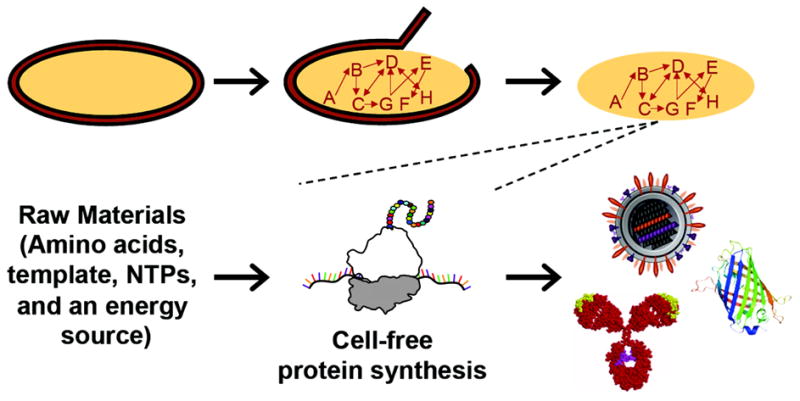
Cell-free protein synthesis systems exploit crude cell extracts to produce valuable therapeutics and vaccines, among other products.
The driving force behind the development of this technology has been its potential to rapidly express bioactive recombinant DNA (rDNA) proteins. In particular, cell-free systems have distinct advantages over in vivo methods for recombinant protein production (Katzen et al., 2005, Swartz, 2006, Zawada et al., 2011). Without the need to support ancillary processes required for cell viability and growth, CFPS allows optimization of the cell extract towards the exclusive production of a single protein product. The absence of a cell wall enables an open and versatile environment for active monitoring, rapid sampling, and direct manipulation of the protein synthesis process. Finally, the cell-free format allows for screening without requiring a gene-cloning step (Fig. 2), enabling rapid process/product development pipelines (Kanter et al., 2007, Zawada et al., 2011).
Figure 2.
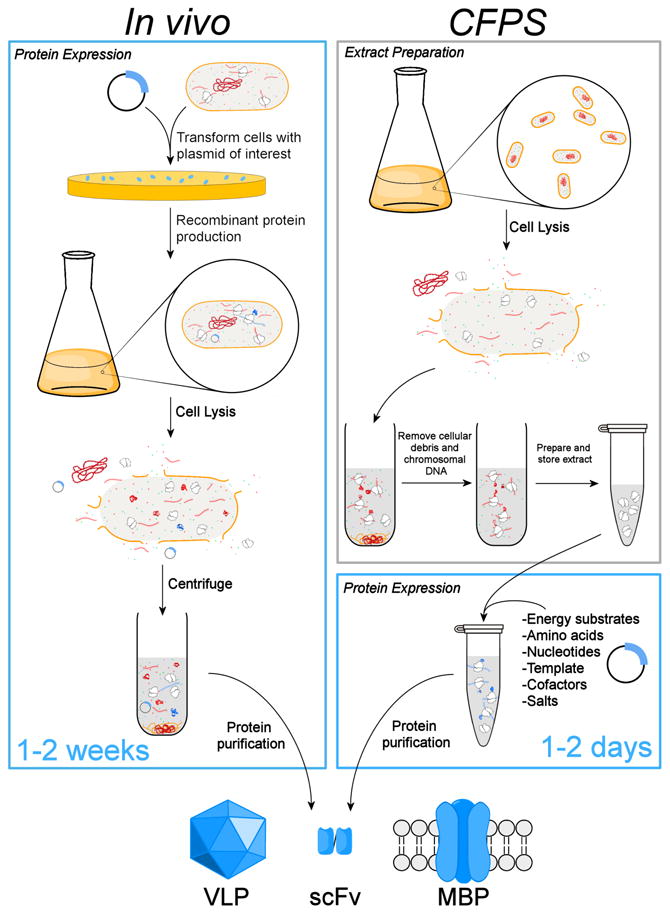
Cartoon comparison of in vivo recombinant DNA protein expression with cell-free protein synthesis (CFPS). CFPS systems provide a more rapid process/product development timeline. Example proteins shown include a virus-like particle (VLP), single-chain antibody variable fragment (scFv), and a membrane bound protein (MBP).
Despite many promising aspects of cell-free systems, several obstacles have previously limited their use as a protein production technology. These obstacles have included short reaction durations of active protein synthesis, low protein production rates, and difficulty in supplying the intense energy and substrate needs of protein synthesis without deleterious concomitant changes in the chemical environment. Furthermore, expensive reagent costs (particularly high energy phosphate chemicals in the form of nucleotides and secondary energy sources), small reaction scales, a limited ability to correctly fold proteins containing multiple disulfide bonds, and its initial development as a “black-box” science were limitations (Swartz, 2006). However, technical advances in the last decade have addressed these limitations and revitalized CFPS systems to meet the increasing demands for protein synthesis (Katzen et al., 2005). Moreover, a recent demonstration of cost-effective cell-free protein synthesis in a 100-liter reaction by Sutro Biopharma, Inc. (Zawada et al., 2011) shows the potential of CFPS systems to become a powerful recombinant DNA protein production platform at the industrial scale.
In this review, we focus on developments that have transformed crude extract CFPS systems into a platform technology for industrial and high-throughput protein production. With due respect to the many advances in purified translation systems, such as the PURE system developed by Ueda and colleagues (Ohashi et al., 2010) as well as New England Biolabs (Asahara and Chong, 2010, Hillebrecht and Chong, 2008), we concentrate on crude extract based systems because the expense of the PURE system currently restricts large-scale commercial applicability. In addition, a review on the PURE system was recently published (Ohashi et al., 2010). Here, we begin with a brief introduction describing the technological capabilities of the field. In the next section, we discuss historical trends in protein yields, cost, reaction duration, and scale of CFPS systems. Finally, we examine frontier applications made possible by the recent technical renaissance.
2. Cell-free protein synthesis primer
To produce proteins of interest, CFPS systems harness an ensemble of catalytic components necessary for energy generation and protein synthesis from crude lysates of microbial, plant, or animal cells. Crude lysates contain the necessary elements for transcription, translation, protein folding, and energy metabolism (e.g., ribosomes, aminoacyl-tRNA synthetases, translation initiation and elongation factors, ribosome release factors, nucleotide recycling enzymes, metabolic enzymes, chaperones, foldases, etc.). Activated catalysts within the cell lysate act as a chemical factory to synthesize and fold desired protein products upon incubation with essential substrates, which include amino acids, nucleotides, DNA or mRNA template encoding the target protein, energy substrates, cofactors, and salts. After initiation of cell-free protein synthesis, production typically continues until one of the substrates (e.g., ATP, cysteine, etc.) is depleted or byproduct accumulation (e.g., inorganic phosphate) reaches an inhibitory concentration.
Although any organism can potentially provide a source of crude lysate, the most common cell-free translation systems consist of extracts from Escherichia coli (ECE), rabbit reticulocytes (RRL), wheat germ (WGE), and insect cells (ICE). Since these cells behave very differently, the extracts derived from them do as well. Thus, the first decision when attempting to produce biologically active proteins using CFPS is choosing the source of extract. Typically this decision begins by considering the availability of materials and convenience of extract preparation, yield of protein needed, protein origin and complexity, downstream processing needs, and cost. In the remainder of this section we highlight the most commonly used CFPS systems (Table 1).
Table 1. Comparison of various cell-free protein synthesis systems.
| Type | Advantages | Disadvantages |
|---|---|---|
| E.coli S30 extract |
|
|
| Wheat-germ extract |
|
|
| Rabbit reticulocyte |
|
|
| Insect cell extract |
|
|
| Yeast |
|
|
Batch system
The prokaryotic E. coli CFPS system is the most popular and is also commercially available. The adoption of the E. coli system is due to several factors. First, E. coli is easily fermented in large quantities using low-cost media and easily ruptured using high-pressure homogenizers. Thus, extract preparation is simple and inexpensive. Second, E. coli based systems generally achieve the highest protein yields, from hundreds of micrograms per milliliter to milligrams per milliliter in a batch reaction, depending on the protein of interest (e.g., 1.7 mg mL-1 chloramphenicol acetyl transferase (Kim et al., 2011), 0.7 mg mL-1 human granulocyte-macrophage colony-stimulating factor (Zawada et al., 2011), and 0.022 mg mL-1 FeFe hydrogenase (Boyer et al., 2008)). Third, the reaction cost of the E. coli system is the lowest. This is due in large part to the ability to activate metabolic reactions in the extract that fuel high-level protein synthesis, which has obviated the need for using expensive energy substrates such as phosphoenolpyruvate (Swartz, 2006).
WGE, RRL and ICE systems are the most widely used eukaryotic CFPS systems. They are also commercially available. Compared to the E. coli system, these methods have advantages for producing some types of complex proteins and can achieve post-translational modifications not found in bacteria (Chang et al., 2005). However, eukaryotic CFPS systems generally have more laborious extract preparation procedures, are more costly, and have lower protein yields in batch reactions.
In terms of protein yields, WGE, pioneered by Endo and colleagues, is the most productive. WGE is prepared from isolated wheat seed embryos (Madin et al., 2000, Takai et al., 2010), typically producing between several hundred micrograms to milligrams of recombinant protein per milliliter reaction, depending on the protein and format (Madin et al., 2000). RRL reactions are approximately 2 orders of magnitude lower, with typical reported protein yields of several to tens of micrograms protein per milligram reaction (Jackson and Hunt, 1983, Shields and Blobel, 1978, Tarui et al., 2001). Reported protein yields from ICE, usually prepared from Spodoptera frugiperda cells (Ezure et al., 2010, Tarui et al., 2001), are several tens of micrograms per milliliter reaction.
While WGE is the most efficient at making proteins, it is not readily suitable for some post-translational processing like glycosylation (Tarui et al., 2001). On this front, RRL and ICE have shown the most versatility. Isoprenylation (Hancock, 1995, Suzuki et al., 2007), acetylation (Gibbs et al., 1985, Suzuki et al., 2006b), N-myristoylation (Suzuki et al., 2006b), phosphorylation (Safer and Jagus, 1979), ubiquitin-conjugation (Suzuki et al., 2010), signal peptide processing (Shields and Blobel, 1978), and core glycosylation (Shields and Blobel, 1978, Tarui et al., 2001) have been achieved. With respect to glycosylation, ICE has the advantage over RRL that core glycosylation does not require the addition of microsomal membranes, which have been shown to provide the compartmentalization and enzymes needed for proper post-translational modifications in RRL. These microsomal membranes must be separately purified and added into the CFPS reaction. This extra processing step is not desirable. Thus, ICE is emerging as the fastest growing CFPS platform. Beyond those platforms listed above, eukaryotic CFPS systems based on yeast (Iizuka et al., 1994), cancer cells (Weber et al., 1975), and hybridoma (Mikami et al., 2006), among others, have also been developed.
Although each of the CFPS systems developed to date has their merits, the trade-off between yield, cost, and post-translational modification requirements must be carefully considered. In the next section, we examine benchmarks in the capacity to synthesize proteins at high concentrations, reduced costs, increased scale, and improved protein folding.
3. Technological advances in CFPS
Guided by the pioneering work of Spirin and co-workers (Spirin et al., 1988), the last ∼25 years have illuminated general rules for achieving high protein yields in vitro. To briefly summarize, requirements for optimal cell-free expression include: adequate substrate supply, a homeostatic environment, and the removal or avoidance of inhibitory byproducts. Not surprisingly, these requirements are characteristic properties of the in vivo state of a rapidly growing cell. Thus, a guiding principle that has emerged in the development of CFPS systems is that activating authentic biological processes in vitro through cytoplasmic mimicry enables highly productive systems (Jewett et al., 2008, Jewett and Swartz, 2004a).
3.1 Benchmark trends in CFPS
Figure 3 shows the progression of the CFPS field from sampled publications, highlighting protein synthesis yield (batch and fed-batch/continuous exchange formats), batch reaction duration, protein synthesis rates, protein yield per dollar energy substrates and nucleotides, and batch reaction volume. Strikingly, the observed trends increase dramatically with time, providing a clear picture of the growth of the field. Figure 3a, for example, demonstrates an increasing trend for batch E. coli CFPS reactions (∼100 μg mL-1 protein produced per year). To date, the biggest improvements in protein yields have been observed for the E. coli and WGE systems. These systems now routinely produce proteins in batch (Fig. 3a; E. coli) and fed-batch or continuous exchange (Fig. 3b; E. coli & WGE) formats in the milligram per milliliter range for a variety of proteins. The fed-batch or continuous exchange yields presented in Figure 3b are normalized for reaction plus feed solution volumes, to take into account the costs of feed solution chemicals.
Figure 3.

Historical trends of cell-free protein synthesis systems. Blue squares = E. coli extract (ECE), red circles = wheat germ extract (WGE), purple triangles = insect cell extract (ICE), and green diamonds = rabbit reticulocyte lysate (RRL). (A) Cell-free protein synthesis yields for a batch reaction. (B) “Effective” cell-free protein synthesis yields for a fed-batch or continuous exchange cell-free (CECF) reaction based on the total volume of reaction and feeding solutions. (C) Reaction length for active protein synthesis in a batch cell-free reaction. (D) Rate of protein synthesis during a cell-free reaction. (E) Protein yield per dollar of NXPs (e.g., ATP, ADP, AMP, GTP, etc.) and energy source, which are the dominant substrate costs of CFPS reactions. (F) Scale of cell-free reaction volumes. Cited references can be found in Supplementary Table 1.
What has driven the transformational increases in protein yields? From the observed trends, it is clear that increases in protein yield in the batch format are intimately tied to increases in batch reaction duration (Fig 3c; ∼40 minutes per year) and increases in protein synthesis rate (Fig. 3d; ∼30 μg mL-1 h-1 per year). Thus, longer reaction duration and increased protein synthesis rates result in higher protein yields. What then has enabled longer reactions and increased rates? Philosophically, the linear growth of these process parameters has been enabled by a new way of thinking. Now more than ten years ago, a series of elegant experiments by Kim and Swartz first revealed that metabolic networks, not just simple one-step phosphorylation reactions, could be harnessed in vitro to supply energy for protein biosynthesis (Kim and Swartz, 1999, 2000, 2001). Equally important, their results also demonstrated that deleterious activities, which direct resources away from protein production, could be specifically identified and controlled. In sum, these landmark experiments transformed cell-free systems into sets of biochemical reactions that could be analyzed and controlled (i.e., not a “black box”) in order to improve cell-free system performance. Moreover, it enabled the realization that cytoplasmic mimicry was crucial for enabling highly active cell-free systems (Jewett and Swartz, 2004a). Such a frame of reference shift enabled substrate limitations to be assessed and alleviated and extract quality to be improved.
In recent years, for example, changes in extract preparation procedures have led to more robust extracts, the ability to activate central metabolism for fueling CFPS, and the ability to increase reaction scale. Because CFPS systems exploit an ensemble of catalytic proteins prepared from the crude lysates of cells, cell extract (whose composition is sensitive to growth media, lysis method, and processing conditions) is the most critical component in the CFPS reaction. It is therefore somewhat surprising that only a few studies have focused on alterations to extract preparation procedures for improved productivity. Let's consider the E. coli CFPS system. Up until about 7 years ago, the extract preparation procedure in E. coli systems had remained relatively constant since its original inception in the early 1960s (Liu et al., 2005). Recently, however, systematic optimization of each step in extract preparation has been carried out. As a result of these new reports, a defined medium has been developed for consistent growth of source cells (Zawada et al., 2003), active extracts can be produced from high-density fermentations (Zawada and Swartz, 2005), simplifications to the original protocol have significantly reduced the time and cost associated with extract preparation (Liu et al., 2005), and the extract preparation procedure has been modified for manufacturing scale-up of CFPS reactions (Zawada et al., 2011). A key improvement has been the inclusion of excess glucose in the growth media, which has enabled activation of low-cost energy regenerating pathways and more productive extracts (Jewett and Swartz, 2004a, b, Kim and Choi, 2000, Zawada et al., 2011). In the WGE system, the discovery of a method for preventing contamination by a protein synthesis inhibitor originating from the endosperm opened the way to its emergence as a powerful technology in high-throughput protein production (Madin et al., 2000).
Beyond extract preparation, stabilizing reaction substrates without the concomitant accumulation of harmful side products has underpinned the growth of the CFPS field. Indeed, focusing on substrate availability, rather than protein production, has led to the major transitions in technology development. These transitions are highlighted by the non-linear trends in Figure 3 (panels b, e, and f). One of these disruptive technologies is the use of continuous exchange or bilayer systems, where passive diffusion enables substrates to be replenished and byproducts to be removed (Fig. 3b). Although a closed batch system provides reproducibility, efficient use of energy substrates, ease of scale-up, and operational convenience for parallel expression of numerous proteins, continuously feeding substrates greatly lengthens reaction lifetime and protein yields per reaction volume (Endo and Sawasaki, 2006).
A second key transition for the field has been the ability to activate pathways in the cell extract that enhance protein synthesis. Much attention has been given towards stimulating central metabolism to fuel high-level CFPS, rather than costly one-step phosphorylation reactions driven by phosphoenolpyruvate (PEP) or similar compounds. In one approach, Jewett et al. (2008) co-activated central metabolism, oxidative phosphorylation, and protein synthesis in a single reaction to fuel high-level, cost-effective protein synthesis (up to 1.2 mg mL-1 in 2 h) (Jewett et al., 2008). In another approach, Calhoun and Swartz (2005a, b) demonstrated that glucose could fuel protein synthesis (Calhoun and Swartz, 2005a, b). Substituting nucleoside monophosphates (NMPs) for nucleoside triphosphates (NTPs) further reduced energy costs while maintaining high protein yields (Calhoun and Swartz, 2005a, Jewett et al., 2008). Complementary efforts have more recently utilized polymeric carbohydrates such as maltodextran (Wang and Zhang, 2009) and soluble starch or glycogen (Kim et al., 2011) as energy substrates because they are slowly metabolized. A key advantage of these energy substrates is that environmental factors like pH and inorganic phosphate concentration are more stable, which can lead to higher protein expression. Indeed, the recent work from Kim and colleagues reported the synthesis of 1.7 mg mL-1 protein in an E. coli CFPS system, the highest known reported batch yield to our knowledge (Fig. 3a) (Kim et al., 2011). In addition to activating beneficial pathways, removal of harmful pathways has also paid dividends. For example, stabilization of amino acid substrates by deleting genes encoding deleterious enzymes (e.g., those that deplete substrates) has also been shown to enable high-level CFPS (Calhoun and Swartz, 2006, Michel-Reydellet et al., 2004, Swartz, 2006).
A third technological breakthrough is the recent demonstration of CFPS at the manufacturing scale (Fig. 3f). Combining advances in activating cost-effective energy metabolism that support long-lived protein production with new robust extract preparation procedures, Zawada et al. (2011) of Sutro Biopharma, Inc. developed a cost-competitive large-scale E. coli based CFPS system. Their open cell-free synthesis (OCFS) system was able to produce 700 mg L-1 of human granulocyte-macrophage colony-stimulating factor (rhGM-CSF) in 10 hours at the 100 liter scale (Zawada et al., 2011). The linear scalability of the system over a 106 range in volume is essential for rapid and effective optimization of reaction parameters for a given recombinant protein. Future extension of such technologies will make possible the commercial production of protein pharmaceuticals that are inaccessible to cells, because they are either toxic or difficult to express (i.e., not soluble).
3.2 Template preparation
Outside of the three technological milestones highlighted above, key advances in template preparation have also been realized in recent years. A particular focus has been on lowering the cost of DNA template preparation. Since the lowering of energy substrate costs, preparation of large quantities of highly purified DNA now represents one of the most expensive substrates for CFPS reactions. In addition, this time-consuming step can be a bottleneck when expressing a large number of proteins. DNA rolling circle amplification may provide one solution. In a recent report, DNA rolling circle amplification was used to amplify 5 μg of circular DNA from 100 ng starting material, which served as a high-quality template for protein synthesis (Kumar and Chernaya, 2009). Another focus has been directed towards enabling high-throughput gene construction. The Gateway vector system, available from Invitrogen (Hartley et al., 2000), utilizes integrase enzymes for one-step insertion of a desired gene into a vector while avoiding excess restriction digest and ligation reactions. This platform was used for high-throughput production of 33,275 entry clones that were subsequently used for CFPS of a portion of the human proteome (Goshima et al., 2008).
Other major developments in template preparation have included a “universal” sequence for translation initiation for eukaryotic CFPS systems (Swartz, 2009). In vivo, capped and poly-adenylated mRNA is required for efficient translation initiation. Early on, most eukaryotic CFPS systems used capped mRNA as a template, which aids in ribosome binding to the mRNA. However, the capping reaction is costly and has a low efficiency. Moreover, any free m7GpppG cap analogue is a strong inhibitor of the initiation factor eIF-4E, which greatly lowers translation efficiency if not properly removed. As a result most of eukaryotic cell-free systems utilize internal ribosome entry site (IRES) sequences to initiate translation (Fitzgerald and Semler, 2009). IRES sequences are small RNA fragments ranging from several tens to hundreds of nucleotides. These IRES sequences are particular for each organism normally derived from viruses. For example, in WGE, the 5′-UTR fragment of Ω gene from tobacco mosaic virus is able to efficiently initiate translation (Gallie, 2002); ICE systems use the 5′-UTR fragment of polyhedrin gene from baculovirus (Suzuki et al., 2006a); RRL uses an RNA fragment from encephalomyocarditis virus (Craig et al., 1992, Kozak, 1986). Some IRES sequences function through an unstructured region at the 5′ end of the message that binds to the ribosome and initiates translation. In an exemplary report, Mureev et al. (2009) exploited this phenomenon to generate an artificial IRES sequence made from polyA or AT-rich sequences at the 5′ end of mRNA. Termed species-independent translational sequences (SITS), these templates were able to initiate cap-independent translation in almost all known cell-free expression systems, including E. coli (Mureev et al., 2009).
Increasing the effective template concentration through localization is another technology that has increased CFPS productivity. In one example, Park et al. (2009) cross-linked linear template DNA molecules with X-shaped DNA adapters to generate a DNA hydrogel for use in combined cell-free transcription and translation systems. This method improved protein production in WGE 300-fold as compared to the soluble DNA template control. This improvement is mainly attributed to gene protection from endogenous DNase digestion, higher overall gene concentration by removing DNA solubility limitations, and faster enzyme turnover rates due to confined localization of the genes (Park et al., 2009). Combined, a variety of improvements in template design have led to higher protein synthesis yields, which have concurrently enabled the field to synthesize more complex proteins.
3.3 Protein Folding
Over the last decade, efforts to synthesize complex proteins, such as those containing multiple disulfide bonds has intensified. Figure 4 shows a timeline highlighting milestones in both the E. coli and wheat germ CFPS systems. To fold complex proteins, the cell-free system must shield hydrophobic regions of the target protein from one another, provide the proper natural chemical environment, incorporate cofactors such as iron-sulfur clusters, encourage disulfide bond formation, and promote disulfide bond isomerization. One main challenge for CFPS systems arises in reproducing in vivo oxidative folding pathways to allow for formation and isomerization of disulfide bonds. Whereas organisms have evolved to use different regions in space to separate protein biosynthesis from oxidative folding, cell-free systems seek to accomplish both tasks in the same compartment. In spite of this contrast, considerable progress has been made towards enhancing the folding of eukaryotic proteins with multiple disulfide bonds. In several exemplary examples, Swartz and colleagues have shown it possible to establish an oxidizing environment in the CFPS reaction that promotes disulfide bond formation through balancing the redox potential reaction. By pre-treating the cell extract with iodoacetamide (IAM), an alkylating agent that covalently blocks the free sulfhydryl groups of cellular enzymes, using a glutathione buffer to provide an oxidizing environment, and providing the disulfide bond forming enzyme DsbC, they demonstrated the synthesis of active urokinase (Kim and Swartz, 2004) and a truncated form of tissue plasminogen activator (Yin and Swartz, 2004).
Figure 4.

Timeline: Synthetic biology milestones in the production of complex therapeutic proteins. Abbreviations: scFv: single-chain antibody variable fragment, vtPA: variant of human tissue-type plasminogen activator, GM-CSF: granulocyte macrophage colony stimulating factor, IGF-I: insulin-like growth factor I, cIFN-α: human consensus interferon-alpha, rhGM-CSF: human granulocyte macrophage colony-stimulating factor. Cited references can be found in Supplementary Table 2.
To form and isomerize disulfide bonds and to help nascent polypeptides attain their active conformation without aggregation, nature also exploits a variety of enzymes (e.g., the Dsb system in E. coli and other chaperones). Simple addition of these molecules has been important for production of complex proteins in vitro (Katzen et al., 2005). Beyond addition of natural foldases, synthetic approaches have also been used. In one approach, Welsh et al. (2011) tethered the eukaryotic Hsp70 chaperone BiP to trigger factor. This method was meant to mimic chaperone-assisted folding in the ER because trigger factor is a ribosome-associating E. coli chaperone. The result was an improvement in soluble protein yields for secreted eukaryotic proteins (Welsh et al., 2011). In another approach, Sasaki et al. (2011) improved proper bond formation and protein folding by incorporating amphiphilic polysaccharide nanogels into the cell-free reaction, allowing for the binding and then controlled release of peptide chains, preventing aggregation and misfolding for some proteins. Together, these examples, and others (Zawada et al., 2011), showcase the freedom of design in adjusting cell-free system components by direct addition of new components (in this case folding aids and chaperones).
4. Applications
Marked advancements in productivity, cost, scale, and complexity of recombinant protein synthesized have rapidly expanded the utility and now industrialization of CFPS systems (Swartz, 2006). In this section, we highlight several emerging applications made possible by these advances. These include the production of protein libraries, personalized medicines, evolved proteins, membrane proteins, and virus-like particles.
4.1 High Throughput Production
In this post-genomic era, high-throughput protein expression platforms are becoming increasingly important. Cell-free systems have many advantages for meeting this need. First, direct use of PCR templates avoids time-intensive molecular cloning steps (Fig. 2). Second, improvements in cost-effective high-yield batch reactions make multi-well (96 or 384) protein production feasible. Third, there is tremendous potential for miniaturization and automation using microchips. Fourth, the lack of a cell wall barrier allows for easy manipulation of reaction conditions, including the incorporation of isotope-labeled amino acids.
Stable-isotope labeling of proteins for NMR structure assignment or X-ray crystallography using CFPS is playing a critical role in structural biology projects. A key advantage of cell-free systems is that the efficiency of labeled amino acid incorporation, high-protein expression yields, and purity of expressed products in cell-free systems can allow for direct heteronuclear NMR analysis without purification (Morita et al., 2003, Ozawa et al., 2005, Takai et al., 2008). Already, several thousands of protein structures have been determined using cell-free systems (Endo and Sawasaki, 2003).
CFPS synthesis platforms also serve as a foundational technology platform for the large-scale synthesis of protein libraries for functional genomics. Protein in situ arrays (PISA), for example, have been quickly and efficiently generated using CFPS to comprehensively study protein interaction networks on microchips (He et al., 2008, He and Taussig, 2007). In another illustrative example, a WGE system was used as a “human protein factory” in an attempt to synthesize 13,364 human proteins (Goshima et al., 2008). Of the synthesized proteins (12,996 or 97.2%), many of those tested demonstrated function (e.g., 58 of 75 tested phosphatases) and 99.86% were successfully printed onto glass slides to build a protein microarray (Goshima et al., 2008). Because the cell-free approach obviates the need to synthesize, purify and immobilize proteins separately, it seems poised to offer an improved toolbox and faster process for probing different aspects of protein function. Quantitative improvements in lowering the binding detection limit, such as a recent report that functionalized carbon nanotubes with cell-free synthesized proteins to go from the 100 nM to the 10 pM scale, are helping to pave the way (Ahn et al., 2011). Beyond protein arrays, other functional genomics approaches, like sequential protein expression (Woodrow et al., 2006, Woodrow and Swartz, 2007), promise to help unravel the function of each and every gene product.
4.2 Therapeutics
As the issues of cost, scale, and protein folding are no longer insurmountable barriers to the adoption of cell-free technology, efforts to exploit CFPS for commercial production of therapeutics will be intensified (Zawada et al., 2011). A unique and exciting development is the potential to enable the production of personalized medicines. In one example, Kanter et al. (2007) synthesized a cytokine-fused single chain antibody fragment (scFv) of immunoglobulin (Ig) idiotype found on the surface of specific B-cell lymphoma using an E. coli CFPS system (Kanter et al., 2007). This “personalized” scFv fusion that was specific for a particular lymphoma successfully elicited an immune response against the native Ig protein. Strikingly, this purified vaccine for treatment of lymphoma was produced in a matter of days as compared to months in traditional mammalian cell expression. The ability for quick, flexible, and high-yield expression of therapeutics, combined with simple downstream processing demonstrates exciting new possibilities for protein based patient specific medicines.
Outside of patient specific medicines, CFPS could also help identify new drug candidates for existing and emerging threats in cancer, hepatitis, and malaria. Already, the quick and rapid expression platform has a growing role in screening pipelines. Endo's group, for example, has recently utilized WGE to express 124 genes from the malaria genome as possible vaccine candidates. The majority of these products, 93 (75%), were expressed in soluble form. Notably, genes with native codon usage have as high a yield as optimized codon usage (Tsuboi et al., 2010, Tsuboi et al., 2008). In another example, CFPS was used to synthesize vaccine candidates for botulinum toxins at more than 1 mg mL-1 concentrations (Zichel et al., 2010).
4.3 Protein evolution
CFPS systems provide a versatile platform for protein, or enzyme, engineering. Since the 1990s, several extraordinary methods in directed evolution have been developed based in cell-free protein synthesis systems such as ribosome display (Mattheakis et al., 1994, Zahnd et al., 2007), mRNA display (Roberts and Szostak, 1997), and in vitro compartmentalization (Tawfik and Griffiths, 1998). Broadly, these technologies have shown many advantages over in vivo-based display methods, which include a broader library size range, phenotype-genotype coupling efficiency, and high-throughput screening methods. So far, CFPS protein evolution technologies have been used successfully in selecting scFvs antibody fragments (Fukuda et al., 2006), DNA-binding factors (Ihara et al., 2006), and drug molecules used as cancer therapeutics (Yan and Xu, 2006). In a recent example, Stapleton and Swartz (2010) developed a high-throughput method to display hydrogenases based on microbead display, in vitro compartmentalization, and fluorescence-activated cell sorting (FACS) (Stapleton and Swartz, 2010). Since hydrogenases catalyze the formation of hydrogen 2H++ 2e- → H2, which make them potentially key biocatalysts for hydrogen fuel production, but are sensitive to oxygen, this approach could be a potentially powerful tool for engineering oxygen tolerant hydrogenases for compelling applications in energy production.
4.4 Membrane Proteins
Membrane protein production is another application that has received considerable attention. It is reported that membrane proteins account for three-quarters of all potential drug targets (Khnouf et al., 2010). However, their overexpression in vivo remains a bottleneck due to their complex structure, hydrophobic transmembrane region, host toxicity, and the time consuming and low efficiency refolding steps required. Evidence now suggests the possibility of high-level membrane protein expression for biochemical or structural studies using CFPS systems. The key idea is to synthesize membrane proteins in the presence of natural or synthetic lipids and/or detergents that help solubilize the membrane protein. For example, direct addition of surfactants or purified lipids can prevent aggregation of membrane protein polypeptides (Klammt et al., 2005). In lieu of detergents, the addition of purified E. coli phospholipid bilayer vesicles has also been used. Using this approach, two membrane proteins, the tetracycline pump (TetA) and mannitol permease (MtlA), were expressed and achieved the high yield of 570 and 130 μg mL-1 respectively, up to 400 times as previous methods (Wuu and Swartz, 2008). Nanolipoprotein particles, which are lipid bilayers confined within a ring of amphipathic protein of defined diameter (Cappuccio et al., 2009), as well as unilamellar liposomes (Goren et al., 2009) have also shown tremendous promise.
4.5 Virus-Like Particles
Virus-like particles (VLPs) are 25-100 nm complexes self-assembled from one or more structural proteins (Johnson and Chiu, 2000). Being structurally similar to viruses, they elicit an immunogenic response, but have the potential to be used as safe vaccines because they do not contain genetic material (Jennings and Bachmann, 2008). Furthermore, their self-assembled and hollow structure gives interest in using VLPs as drug delivery and gene therapy agents (Bundy et al., 2008). As defined and regular structures are important for the immunogenic response of VLPs, key design considerations in their recombinant production are the composition and consistency of VLP subunits and the purity and distribution of the final product. Producing VLPs recombinantly in vivo is challenging because of the structural inconsistencies involved in the scale up, protein impurities from in vivo production (Pattenden et al., 2005), and the high costs associated with recombinant strain development (Rothengass, 2007). E. coli based CFPS platforms have been developed to greatly improve the manufacturability of VLPs. Bundy et al., for example, efficiently synthesized the MS2 coat protein in batch CFPS reactions (Bundy et al., 2008). Furthermore, CFPS systems allow for fast reaction and assembly optimization at the bench top level for new VLP targets, which may be scaled up to industrial production levels.
In addition to the rapid process and product development pipelines that are enabled by CFPS, the ability to functionalize VLPs could greatly expand their applications. In one example, Patel and Swartz (2011) used E. coli CFPS to incorporate click-chemistry functionalizable non-natural amino acids into VLPs at a yield of 300 μg mL-1 (Patel and Swartz, 2011). These functionalized VLPs were decorated with antibody fragments, GM-CSF, DNA, and poly(ethylene glycol). In fact, multiple ligands can be added to these VLPs at once, with the surface composition depending on the ligand ratios introduced. For improved VLP stability, Bundy and Swartz (2011) controlled the redox potential of the E. coli CFPS system, and were able to control disulfide bond formation between the capsid monomers (Bundy and Swartz, 2011). These advances demonstrate the merits of CFPS systems as a potentially powerful VLP production platform for drug delivery and vaccines applications.
5. Summary
In the coming years, we anticipate that the utility of CFPS will only expand. This is due to their potential for high-throughput, cost-effective, and high-level protein production. Immediate challenges for the field include the gap in our ability to reliably synthesize any biologically active protein in a universal platform, the lack of a cost-effective and scalable eukaryotic CFPS platform, and the inability to carry out humanized glycosylation patterns. By addressing such challenges, we will be limited not by the technicalities in facilitating synthesis of proteins, but by the number of growing applications that cell-free protein synthesis can resolve. Given the exquisite capability to modify and control CFPS systems and the emergence of cell-free systems on the industrial scale, cell-free applications have now come of age, but are only beginning to reach their full potential.
Supplementary Material
Acknowledgments
The authors gratefully acknowledge funding from the National Institutes of Health (Grant Number R00GM081450), the National Academies Keck Futures Initiative (Grant Number NAFKI-SB5), the National Science Foundation (Grant Number MCB-0943393), the Office of Naval Research (Grant Number N00014-11-1-0363), DARPA (Grant Number N66001-11-1-4137), and the ARMY Research Office (Grant Number W911NF-11-1-044).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahn JH, Kim JH, Reuel NF, Barone PW, Boghossian AA, Zhang J, et al. Label-free, single protein detection on a near-infrared fluorescent single-walled carbon nanotube/protein microarray fabricated by cell-free synthesis. Nano Lett. 2011;11:2743–52. doi: 10.1021/nl201033d. [DOI] [PubMed] [Google Scholar]
- Asahara H, Chong S. In vitro genetic reconstruction of bacterial transcription initiation by coupled synthesis and detection of RNA polymerase holoenzyme. Nucleic Acids Res. 2010;38:e141. doi: 10.1093/nar/gkq377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer ME, Stapleton JA, Kuchenreuther JM, Wang Cw, Swartz JR. Cell-free synthesis and maturation of [FeFe] hydrogenases. Biotechnol Bioeng. 2008;99:59–67. doi: 10.1002/bit.21511. [DOI] [PubMed] [Google Scholar]
- Bundy BC, Franciszkowicz MJ, Swartz JR. Escherichia coli-based cell-free synthesis of virus-like particles. Biotechnol Bioeng. 2008;100:28–37. doi: 10.1002/bit.21716. [DOI] [PubMed] [Google Scholar]
- Bundy BC, Swartz JR. Efficient disulfide bond formation in virus-like particles. J Biotechnol. 2011;154:230–9. doi: 10.1016/j.jbiotec.2011.04.011. [DOI] [PubMed] [Google Scholar]
- Calhoun KA, Swartz JR. An economical method for cell-free protein synthesis using glucose and nucleoside monophosphates. Biotechnol Prog. 2005a;21:1146–53. doi: 10.1021/bp050052y. [DOI] [PubMed] [Google Scholar]
- Calhoun KA, Swartz JR. Energizing cell-free protein synthesis with glucose metabolism. Biotechnol Bioeng. 2005b;90:606–13. doi: 10.1002/bit.20449. [DOI] [PubMed] [Google Scholar]
- Calhoun KA, Swartz JR. Total amino acid stabilization during cell-free protein synthesis reactions. J Biotech. 2006;123:193–203. doi: 10.1016/j.jbiotec.2005.11.011. [DOI] [PubMed] [Google Scholar]
- Cappuccio JA, Hinz AK, Kuhn EA, Fletcher JE, Arroyo ES, Henderson PT, et al. Cell-free expression for nanolipoprotein particles: Building a high-throughput membrane protein solubility platform. In: Doyle SA, editor. Methods Mol Biol: High Throughput Protein Expression and Purification. Totowa, NJ: Humana Press; 2009. pp. 273–95. [DOI] [PubMed] [Google Scholar]
- Chang HC, Kaiser CM, Hartl FU, Barral JM. De novo folding of GFP fusion proteins: high efficiency in eukaryotes but not in bacteria. J Mol Biol. 2005;353:397–409. doi: 10.1016/j.jmb.2005.08.052. [DOI] [PubMed] [Google Scholar]
- Craig D, Howell MT, Gibbs CL, Hunt T, Jackson RJ. Plasmid cDNA-directed protein synthesis in a coupled eukaryotic in vitro transcription-translation system. Nucleic Acids Res. 1992;20:4987–95. doi: 10.1093/nar/20.19.4987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endo Y, Sawasaki T. Cell-free expression systems for eukaryotic protein production. Curr Opin Biotechnol. 2006;17(4):373–380. doi: 10.1016/j.copbio.2006.06.009. [DOI] [PubMed] [Google Scholar]
- Endo Y, Sawasaki T. High-throughput, genome-scale protein production method based on the wheat germ cell-free expression system. Biotechnol Adv. 2003;21:695–713. doi: 10.1016/s0734-9750(03)00105-8. [DOI] [PubMed] [Google Scholar]
- Ezure T, Suzuki T, Shikata M, Ito M, Ando E. A cell-free protein synthesis system from insect cells. Methods Mol Biol. 2010;607:31–42. doi: 10.1007/978-1-60327-331-2_4. [DOI] [PubMed] [Google Scholar]
- Fitzgerald KD, Semler BL. Bridging IRES elements in mRNAs to the eukaryotic translation apparatus. Biochim Biophys Acta Gene Regulatory Mechanisms. 2009;1789:518–28. doi: 10.1016/j.bbagrm.2009.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda I, Kojoh K, Tabata N, Doi N, Takashima H, Miyamoto-Sato E, et al. In vitro evolution of single-chain antibodies using mRNA display. Nucleic Acids Res. 2006;34:e127. doi: 10.1093/nar/gkl618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallie DR. The 5′-leader of tobacco mosaic virus promotes translation through enhanced recruitment of eIF4F. Nucleic Acids Res. 2002;30:3401–11. doi: 10.1093/nar/gkf457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs PEM, Zouzias DC, Freedberg IM. Differential post-translational modification of human type-I deratins synthesized in a rabbit reticulocyte cell-free system. Biochim Biophys Acta. 1985;824:247–55. doi: 10.1016/0167-4781(85)90055-7. [DOI] [PubMed] [Google Scholar]
- Goerke AR, Swartz JR. Development of cell-free protein synthesis platforms for disulfide bonded proteins. Biotechnol Bioeng. 2008;99:351–67. doi: 10.1002/bit.21567. [DOI] [PubMed] [Google Scholar]
- Goren MA, Nozawa A, Makino Si, Wrobel RL, Fox BG. Cell-free translation of integral membrane proteins into unilamelar liposomes. In: Richard RB, Murray PD, editors. Methods Enzymol. Academic Press; 2009. pp. 647–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goshima N, Kawamura Y, Fukumoto A, Miura A, Honma R, Satoh R, et al. Human protein factory for converting the transcriptome into an in vitro-expressed proteome. Nat Methods. 2008;5:1011–7. doi: 10.1038/nmeth.1273. [DOI] [PubMed] [Google Scholar]
- Hancock JF. Reticulocyte lysate assay for in vitro translation and posttranslational modification of Ras proteins. In: Balch WE, CJDAH, editors. Methods Enzymol. Academic Press; 1995. pp. 60–5. [DOI] [PubMed] [Google Scholar]
- Hartley JL, Temple GF, Brash MA. DNA cloning using in vitro site-specific recombination. Genome Res. 2000;10:1788–98. doi: 10.1101/gr.143000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He M, Stoevesandt O, Taussig MJ. In situ synthesis of protein arrays. Curr Opin Biotechnol. 2008;19:4–9. doi: 10.1016/j.copbio.2007.11.009. [DOI] [PubMed] [Google Scholar]
- He M, Taussig MJ. Rapid discovery of protein interactions by cell-free protein technologies. Biochem Soc Trans. 2007;35:962–5. doi: 10.1042/BST0350962. [DOI] [PubMed] [Google Scholar]
- Hillebrecht J, Chong S. A comparative study of protein synthesis in in vitro systems: from the prokaryotic reconstituted to the eukaryotic extract-based. BMC Biotechnol. 2008;8:58. doi: 10.1186/1472-6750-8-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ihara H, Mie M, Funabashi H, Takahashi F, Sawasaki T, Endo Y, et al. In vitro selection of zinc finger DNA-binding proteins through ribosome display. Biochem Bioph Res Co. 2006;345:1149–54. doi: 10.1016/j.bbrc.2006.05.029. [DOI] [PubMed] [Google Scholar]
- Iizuka N, Najita L, Franzusoff A, Sarnow P. Cap-dependent and cap-independent translation by internal initiation of mRNAs in cell extracts prepared from Saccharomyces cerevisiae. Mol Cell Biol. 1994;14:7322–30. doi: 10.1128/mcb.14.11.7322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson RJ, Hunt T. Preparation and use of nuclease-treated rabbit reticulocyte lysates for the translation of eukaryotic messenger RNA. In: Sidney F, Becca F, editors. Methods Enzymol. Academic Press; 1983. pp. 50–74. [DOI] [PubMed] [Google Scholar]
- Jennings GT, Bachmann MF. The coming of age of virus-like particle vaccines. Biol Chem. 2008;389:521–36. doi: 10.1515/bc.2008.064. [DOI] [PubMed] [Google Scholar]
- Jewett MC, Calhoun KA, Voloshin A, Wuu JJ, Swartz JR. An integrated cell-free metabolic platform for protein production and synthetic biology. Mol Syst Biol. 2008;4 doi: 10.1038/msb.2008.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jewett MC, Swartz JR. Mimicking the Escherichia coli cytoplasmic environment activates long-lived and efficient cell-free protein synthesis. Biotechnol Bioeng. 2004a;86:19–26. doi: 10.1002/bit.20026. [DOI] [PubMed] [Google Scholar]
- Jewett MC, Swartz JR. Substrate replenishment extends protein synthesis with an in vitro translation system designed to mimic the cytoplasm. Biotechnol Bioeng. 2004b;87:465–71. doi: 10.1002/bit.20139. [DOI] [PubMed] [Google Scholar]
- Johnson JE, Chiu W. Structures of virus and virus-like particles. Curr Opin Struct Biol. 2000;10:229–35. doi: 10.1016/s0959-440x(00)00073-7. [DOI] [PubMed] [Google Scholar]
- Kanter G, Yang J, Voloshin A, Levy S, Swartz JR, Levy R. Cell-free production of scFv fusion proteins: an efficient approach for personalized lymphoma vaccines. Blood. 2007;109:3393–9. doi: 10.1182/blood-2006-07-030593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katzen F, Chang G, Kudlicki W. The past, present and future of cell-free protein synthesis. Trends Biotechnol. 2005;23:150–6. doi: 10.1016/j.tibtech.2005.01.003. [DOI] [PubMed] [Google Scholar]
- Khnouf R, Olivero D, Jin S, Coleman MA, Fan ZH. Cell-free expression of soluble and membrane proteins in an array device for drug screening. Anal Chem. 2010;82:7021–6. doi: 10.1021/ac1015479. [DOI] [PubMed] [Google Scholar]
- Kim DM, Swartz JR. Prolonging cell-free protein synthesis with a novel ATP regeneration system. Biotechnol Bioeng. 1999;66:180–8. [PubMed] [Google Scholar]
- Kim DM, Swartz JR. Prolonging cell-free protein synthesis by selective reagent additions. Biotechnol Prog. 2000;16:385–90. doi: 10.1021/bp000031y. [DOI] [PubMed] [Google Scholar]
- Kim DM, Swartz JR. Regeneration of adenosine triphosphate from glycolytic intermediates for cell-free protein synthesis. Biotechnol Bioeng. 2001;74:309–16. [PubMed] [Google Scholar]
- Kim DM, Swartz JR. Efficient production of a bioactive, multiple disulfide-bonded protein using modified extracts of Escherichia coli. Biotechnol Bioeng. 2004;85:122–9. doi: 10.1002/bit.10865. [DOI] [PubMed] [Google Scholar]
- Kim HC, Kim TW, Kim DM. Prolonged production of proteins in a cell-free protein synthesis system using polymeric carbohydrates as an energy source. Process Biochem. 2011;46:1366–9. [Google Scholar]
- Kim RG, Choi CY. Expression-independent consumption of substrates in cell-free expression system from Escherichia coli. J Biotech. 2000;84:27–32. doi: 10.1016/s0168-1656(00)00326-6. [DOI] [PubMed] [Google Scholar]
- Klammt C, Schwarz D, Fendler K, Haase W, Dötsch V, Bernhard F. Evaluation of detergents for the soluble expression of α -helical and β -barrel-type integral membrane proteins by a preparative scale individual cell-free expression system. FEBS J. 2005;272:6024–38. doi: 10.1111/j.1742-4658.2005.05002.x. [DOI] [PubMed] [Google Scholar]
- Kozak M. Point mutations define a sequence flanking the AUG initiator codon that modulates translation by eukaryotic ribosomes. Cell. 1986;44:283–92. doi: 10.1016/0092-8674(86)90762-2. [DOI] [PubMed] [Google Scholar]
- Kumar G, Chernaya G. Cell-free protein synthesis using multiply-primed rolling circle amplification products. Biotechniques. 2009;47:637–9. doi: 10.2144/000113171. [DOI] [PubMed] [Google Scholar]
- Liu DV, Zawada JF, Swartz JR. Streamlining Escherichia coli S30 Extract preparation for economical cell-free protein synthesis. Biotechnol Prog. 2005;21:460–5. doi: 10.1021/bp049789y. [DOI] [PubMed] [Google Scholar]
- Madin K, Sawasaki T, Ogasawara T, Endo Y. A highly efficient and robust cell-free protein synthesis system prepared from wheat embryos: Plants apparently contain a suicide system directed at ribosomes. Proc Natl Acad Sci U S A. 2000;97:559–64. doi: 10.1073/pnas.97.2.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattheakis LC, Bhatt RR, Dower WJ. An in vitro polysome display system for identifying ligands from very large peptide libraries. Proc Natl Acad Sci U S A. 1994;91:9022–6. doi: 10.1073/pnas.91.19.9022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel-Reydellet N, Calhoun K, Swartz J. Amino acid stabilization for cell-free protein synthesis by modification of the Escherichia coli genome. Metab Eng. 2004;6:197–203. doi: 10.1016/j.ymben.2004.01.003. [DOI] [PubMed] [Google Scholar]
- Mikami S, Kobayashi T, Yokoyama S, Imataka H. A hybridoma-based in vitro translation system that efficiently synthesizes glycoproteins. J Biotechnol. 2006;127:65–78. doi: 10.1016/j.jbiotec.2006.06.018. [DOI] [PubMed] [Google Scholar]
- Morita EH, Sawasaki T, Tanaka R, Endo Y, Kohno T. A wheat germ cell-free system is a novel way to screen protein folding and function. Protein Sci. 2003;12:1216–21. doi: 10.1110/ps.0241203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mureev S, Kovtun O, Nguyen UT, Alexandrov K. Species-independent translational leaders facilitate cell-free expression. Nat Biotechnol. 2009;27:747–52. doi: 10.1038/nbt.1556. [DOI] [PubMed] [Google Scholar]
- Nirenberg MW, Matthaei JH. The dependence of cell-free protein synthesis in E. coli upon naturally occurring or synthetic polyribonucleotides. Proc Natl Acad Sci U S A. 1961;47:1588–602. doi: 10.1073/pnas.47.10.1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohashi H, Kanamori T, Shimizu Y, Ueda T. A highly controllable reconstituted cell-free system -a breakthrough in protein synthesis research. Curr Pharm Biotechnol. 2010;11:267–71. doi: 10.2174/138920110791111889. [DOI] [PubMed] [Google Scholar]
- Ozawa K, Dixon NE, Otting G. Cell-free synthesis of 15N-labeled proteins for NMR studies. IUBMB Life. 2005;57:615–22. doi: 10.1080/15216540500217859. [DOI] [PubMed] [Google Scholar]
- Park N, Um SH, Funabashi H, Xu J, Luo D. A cell-free protein-producing gel. Nat Mater. 2009;8:432–7. doi: 10.1038/nmat2419. [DOI] [PubMed] [Google Scholar]
- Patel KG, Swartz JR. Surface functionalization of virus-like particles by direct conjugation using azide–alkyne click chemistry. Bioconjug Chem. 2011;22:376–87. doi: 10.1021/bc100367u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattenden LK, Middelberg APJ, Niebert M, Lipin DI. Towards the preparative and large-scale precision manufacture of virus-like particles. Trends Biotechnol. 2005;23:523–9. doi: 10.1016/j.tibtech.2005.07.011. [DOI] [PubMed] [Google Scholar]
- Roberts RW, Szostak JW. RNA-peptide fusions for the in vitro selection of peptides and proteins. Proc Natl Acad Sci U S A. 1997;94:12297–302. doi: 10.1073/pnas.94.23.12297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothengass BE. Advocating for the quadravalent HPV vaccination, Gardasil, by Merck. Int J Pediatr Otorhinolaryngol. 2007;71:671–2. doi: 10.1016/j.ijporl.2006.12.007. [DOI] [PubMed] [Google Scholar]
- Safer B, Jagus R. Control of eIF-2 phosphatase activity in rabbit reticulocyte lysate. Proc Natl Acad Sci U S A. 1979;76:1094–8. doi: 10.1073/pnas.76.3.1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki Y, Asayama W, Niwa T, Sawada Si, Ueda T, Taguchi H, et al. Amphiphilic polysaccharide nanogels as artificial chaperones in cell-free protein synthesis. Macromol Biosci. 2011;11:814–20. doi: 10.1002/mabi.201000457. [DOI] [PubMed] [Google Scholar]
- Shields D, Blobel G. Efficient cleavage and segregation of nascent presecretory proteins in a reticulocyte lysate supplemented with microsomal membranes. J Biol Chem. 1978;253:3753–6. [PubMed] [Google Scholar]
- Spirin A, Baranov V, Ryabova L, Ovodov S, Alakhov Y. A continuous cell-free translation system capable of producing polypeptides in high yield. Science. 1988;242:1162–4. doi: 10.1126/science.3055301. [DOI] [PubMed] [Google Scholar]
- Stapleton JA, Swartz JR. Development of an in vitro compartmentalization screen for high-throughput directed evolution of [FeFe] hydrogenases. PLoS ONE. 2010;5:e15275. doi: 10.1371/journal.pone.0015275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T, Ezure T, Ando E, Nishimura O, Utsumi T, Tsunasawa S. Preparation of ubiquitin-conjugated proteins using an insect cell-free protein synthesis system. J Biotechnol. 2010;145:73–8. doi: 10.1016/j.jbiotec.2009.10.009. [DOI] [PubMed] [Google Scholar]
- Suzuki T, Ito M, Ezure T, Kobayashi S, Shikata M, Tanimizu K, et al. Performance of expression vector, pTD1, in insect cell-free translation system. J Biosci Bioeng. 2006a;102:69–71. doi: 10.1263/jbb.102.69. [DOI] [PubMed] [Google Scholar]
- Suzuki T, Ito M, Ezure T, Shikata M, Ando E, Utsumi T, et al. N-terminal protein modifications in an insect cell-free protein synthesis system and their identification by mass spectrometry. Proteomics. 2006b;6:4486–95. doi: 10.1002/pmic.200600126. [DOI] [PubMed] [Google Scholar]
- Suzuki T, Ito M, Ezure T, Shikata M, Ando E, Utsumi T, et al. Protein prenylation in an insect cell-free protein synthesis system and identification of products by mass spectrometry. Proteomics. 2007;7:1942–50. doi: 10.1002/pmic.200700237. [DOI] [PubMed] [Google Scholar]
- Swartz J. Developing cell-free biology for industrial applications. J Ind Microbiol Biotechnol. 2006;33:476–85. doi: 10.1007/s10295-006-0127-y. [DOI] [PubMed] [Google Scholar]
- Swartz JR. Universal cell-free protein synthesis. Nat Biotechnol. 2009;27:731–2. doi: 10.1038/nbt0809-731. [DOI] [PubMed] [Google Scholar]
- Takai K, Sawasaki T, Endo Y. Development of key technologies for high-throughput cell-free protein production with the extract from wheat embryos. In: Andrzej J, editor. Adv Protein Chem Struct Biol. Academic Press; 2008. pp. 53–84. [DOI] [PubMed] [Google Scholar]
- Takai K, Sawasaki T, Endo Y. Practical cell-free protein synthesis system using purified wheat embryos. Nat Protoc. 2010;5:227–38. doi: 10.1038/nprot.2009.207. [DOI] [PubMed] [Google Scholar]
- Tarui H, Murata M, Tani I, Imanishi S, Nishikawa S, Hara T. Establishment and characterization of cell-free translation/glycosylation in insect cell (Spodoptera frugiperda 21) extract prepared with high pressure treatment. Appl Microbiol Biotechnol. 2001;55:446–53. doi: 10.1007/s002530000534. [DOI] [PubMed] [Google Scholar]
- Tawfik DS, Griffiths AD. Man-made cell-like compartments for molecular evolution. Nat Biotechnol. 1998;16:652–6. doi: 10.1038/nbt0798-652. [DOI] [PubMed] [Google Scholar]
- Tsuboi T, Takeo S, Arumugam TU, Otsuki H, Torii M. The wheat germ cell-free protein synthesis system: A key tool for novel malaria vaccine candidate discovery. Acta Trop. 2010;114:171–6. doi: 10.1016/j.actatropica.2009.10.024. [DOI] [PubMed] [Google Scholar]
- Tsuboi T, Takeo S, Iriko H, Jin L, Tsuchimochi M, Matsuda S, et al. Wheat germ cell-free system-based production of malaria proteins for discovery of novel vaccine candidates. Infect Immun. 2008;76:1702–8. doi: 10.1128/IAI.01539-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Zhang YHP. Cell-free protein synthesis energized by slowly-metabolized maltodextrin. BMC Biotechnol. 2009;9:58. doi: 10.1186/1472-6750-9-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber LA, Feman ER, Baglioni C. A cell free system from HeLa cells active in initiation of protein synthesis. Biochemistry. 1975;14:5315–21. doi: 10.1021/bi00695a015. [DOI] [PubMed] [Google Scholar]
- Welsh JP, Bonomo J, Swartz JR. Localization of BiP to translating ribosomes increases soluble accumulation of secreted eukaryotic proteins in an Escherichia coli cell-free system. Biotechnol Bioeng. 2011;108:1739–48. doi: 10.1002/bit.23111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodrow KA, Airen IO, Swartz JR. Rapid expression of functional genomic libraries. J Proteome Res. 2006;5:3288–300. doi: 10.1021/pr050459y. [DOI] [PubMed] [Google Scholar]
- Woodrow KA, Swartz JR. A sequential expression system for high-throughput functional genomic analysis. Proteomics. 2007;7:3870–9. doi: 10.1002/pmic.200700471. [DOI] [PubMed] [Google Scholar]
- Wuu J, Swartz J. High yield cell-free production of integral membrane proteins without refolding or detergents. Biochim Biophys Acta Biomembranes. 2008;1778:1237–50. doi: 10.1016/j.bbamem.2008.01.023. [DOI] [PubMed] [Google Scholar]
- Yan X, Xu Z. Ribosome-display technology: Applications for directed evolution of functional proteins. Drug Discov Today. 2006;11:911–6. doi: 10.1016/j.drudis.2006.08.012. [DOI] [PubMed] [Google Scholar]
- Yang J, Kanter G, Voloshin A, Michel-Reydellet N, Velkeen H, Levy R, et al. Rapid expression of vaccine proteins for B-cell lymphoma in a cell-free system. Biotechnol Bioeng. 2005;89:503–11. doi: 10.1002/bit.20283. [DOI] [PubMed] [Google Scholar]
- Yin G, Swartz JR. Enhancing multiple disulfide bonded protein folding in a cell-free system. Biotechnol Bioeng. 2004;86:188–95. doi: 10.1002/bit.10827. [DOI] [PubMed] [Google Scholar]
- Zahnd C, Amstutz P, Pluckthun A. Ribosome display: selecting and evolving proteins in vitro that specifically bind to a target. Nat Methods. 2007;4:269–79. doi: 10.1038/nmeth1003. [DOI] [PubMed] [Google Scholar]
- Zawada J, Swartz J. Maintaining rapid growth in moderate-density Escherichia coli fermentations. Biotechnol Bioeng. 2005;89:407–15. doi: 10.1002/bit.20369. [DOI] [PubMed] [Google Scholar]
- Zawada JF, Richter B, Huang E, Loades E, Shah A, Swartz JR. High density, defined media culture for production of Escherichia coli extracts. Fermentation Biotechnology. 2003;862:142–56. [Google Scholar]
- Zawada JF, Yin G, Steiner AR, Yang J, Naresh A, Roy SM, et al. Microscale to manufacturing scale-up of cell-free cytokine production—a new approach for shortening protein production development timelines. Biotechnol Bioeng. 2011;108:1570–8. doi: 10.1002/bit.23103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zichel R, Mimran A, Keren A, Barnea A, Steinberger-Levy I, Marcus D, et al. Efficacy of a potential trivalent vaccine based on Hc fragments of botulinum toxins A, B, and E produced in a cell-free expression system. Clin Vaccine Immunol. 2010;17:784–92. doi: 10.1128/CVI.00496-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.