

Abstract
We present a novel methodology to determine the phase of single-nucleotide polymorphisms (SNPs) on a chromosome, which we term clone-based systematic haplotyping (CSH). The CSH procedure is based on separating the allelic chromosomes of a diploid genome by fosmid/cosmid cloning, and subsequent SNP typing of 96 clone pools, each representing ∼10% of the genome. The pools are screened by PCR for the sequence of interest, followed by SNP typing on the PCR products using the GOOD assay. We demonstrate that by CSH, the haplotype of SNPs separated by more than 50 kilobases can definitely be assigned. We propose this method as being suitable for constructing maps of ancestral haplotypes, analysis of complex diseases, and for diagnosis of rare defects in which the molecular haplotype is crucial. In addition, by amplifying the initial DNA by many orders of magnitude, the original DNA resource is effectively immortalized, enabling the haplotyping of hundreds of thousands of SNPs per individual.
Genetic variation and allelic complexity in the human population seem to play an enormous role in the genetics of complex traits, as well as in individual drug responses. The most common variations in the human genome, comprising ∼90% of all differences, are single-nucleotide polymorphisms (SNPs). Following the recent deciphering of the human genome by sequencing (Lander et al. 2001; Venter et al. 2001), the next step for gaining additional insight into the genetic contribution to the risk for complex diseases should be the analysis of genetic variations, such as SNPs associated with a special phenotype or disease. However, frequent failure of complex disease studies as well as the great body of controversial results in single SNP association studies indicate the importance of haplotype analysis, that is, the determination of the phase of SNPs on a single chromosome, over simply SNP genotyping (Davidson 2000; Drysdale et al. 2000; Hoehe et al. 2000; Maat-Kievit et al. 2001; Boldt and Petzl-Erler 2002). Furthermore, the finding that haplotypes occur in blocks of limited diversity (Daly et al. 2001) will considerably cut down the workload involved in large-scale haplotyping of individuals in the future.
Current procedures for haplotyping, and thus gaining the maximal possible information from the data, are very labor-intensive and expensive. The most frequently used methods require either somatic cell hybrid technology, which converts a diploid cell into haploid cell lines (Douglas et al. 2001) or excessive statistical analysis (Rohde and Fuerst 2001; Sachidanandam et al. 2001). In a different approach, parental genotyping can be used to infer haplotypes in a family study (Tapadar et al. 2000). An interesting proof-of-principle for direct haplotyping of kilobase-sized DNA using carbon nanotube probes and multiplex detection of labeled probes by atomic force microscopy was presented recently (Woolley et al. 2000). Direct haplotyping can also be achieved by the newly published polony-technique (Mitra et al. 2003). This technique relies on embedding intact chromosomal DNA in polyacrylamide gels on a glass slide and subsequent PCR. Another method includes diluting the DNA sample to the degree that quasi-single molecule PCRs are performed (Ruano et al. 1990). A more recent development simply exploits the fact that in a good DNA preparation the average DNA is about 100–150 kb in length and that several short-range PCRs can be performed on a single molecule (Ding and Cantor 2003). A further procedure employs allele-specific PCR and relies on subsequent gel-based separation and fluorescence detection (Fugger et al. 1990). The combination of allele-specific PCR and mass spectrometry analysis of SNPs was achieved by Gut and coworkers (Tost et al. 2002).
Here we present a method for fast and efficient molecular haplotyping that we have termed clone-based systematic haplotyping (CSH). In this method, large-insert cloning (insert size ∼40 kb) and subsequent screening of clone pools (each representing ∼10% of the genome) by PCR are coupled with a mass spectrometric procedure for SNP typing that we have termed the GOOD assay (Sauer et al. 2000a,b, Sauer and Gut 2003).
RESULTS
Reconstruction of the Haplotypes
As an example, we analyzed the phase of 20 SNPs in the region of the opioid receptor mu1 gene (OPRM1) that are distributed over ∼61 kb (see Fig. 2 below). These SNPs were derived from a recent article (Hoehe et al. 2000) and the NCBI-SNP-database (http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=snp). We designed primer pairs that amplify a small fragment containing one to three of these SNPs, and primers for subsequent discrimination of alleles by the GOOD assay. Oligonucleotide sequences are available as online Supplemental data at www.genome.org (Table 1). Two DNAs (DNA11 and DNA28) out of a cohort of 35 samples were found to be heterozygous in several positions. We constructed an ∼10x coverage fosmid/cosmid library of each of these DNAs. The libraries were allotted into 96 pools, each representing ∼10% of the genome (Fig. 1). The clone-pool DNA served as template for PCRs that amplify the regions containing the SNPs. The PCRs were performed in a 96-well format and scored by agarose gel electrophoreses. In the case of a heterozygous SNP, the PCR products of the positive pools were further used for the GOOD assay to determine the haplotype. Otherwise, the scoring of positive PCRs served to estimate the length of the fosmids/cosmids contained within the pools, for internal validation and to judge whether two markers analyzed are on the same clone. Since the pool size is ∼10%–12% of the genome, the chance that two different clones of the same region are contained within the same pool and would thus hinder proper analysis is only ∼1%–2%. As can be seen in Figure 2A, the distances between the heterozygous and therefore informative SNPs are not evenly spaced and differ slightly for the two DNAs. For DNA11, an SNP on PCR fragments 5, 6, 10, 11, and 3 is heterozygous, and for DNA28 on fragments 2, 5, 12, and 3. The largest distance of heterozygous SNPs for DNA11 is between rs3778150 and Mu_Opiat_IVS2+31 on fragments PCR11 and PCR3, respectively, and approximates 33.3 kb. For DNA28, the largest distance between informative SNPs is 31.8 kb, and is between rs510769 on fragment 5 and rs3778152 on fragment 12.
Figure 2.

(A) Schematic drawing of the OPRM1 genomic region. SNPs are indicated and tagged with the published names (NCBI dbSNP-database; Hoehe et al. 2000). The genotypes of DNA11 and DNA28 are shown in the top lines. (B) Results for the haplotyping of DNA11 and DNA28. The different haplotypes are represented in red and green, respectively. A“+” indicates positive a PCR of SNPs that were previously found to be homozygous for DNA11 and DNA28, respectively. The letters A, C, G, and T denote the result of the haplotyping by the GOOD assay of the pools analyzed. Blue bars: pools where only homozygous SNPs are detected. Brown indicates the presence of two clones of different haplotypes in one pool.
Figure 1.

(Left) A schematic representation of the CSH method; (right) a corresponding example of “real data.” (A) Intact high-molecular-weight DNA is prepared, and the quality is checked by PFG electrophoresis. (B) DNA is sheared into pieces of ∼40–100 kb, as can be seen by PFG. (C) After end-repair, a cosmid/fosmid library is constructed by ligation of the fragments to a suitable vector, in vitro packaging, and transfection of bacteria. The quality of the library is checked by restriction analysis of a few randomly chosen clones. (D) The titer of the library is determined, and ∼10,000 clones are plated onto one plate. (E) The region of interest is amplified by PCR, and the positive pools are selected for subsequent analysis by the GOOD assay; as an example PCR3 of DNA11 is shown. (F) The genotypes of the SNPs are analyzed using the GOOD assay (Fig. 3).
The PCR screening and haplotyping results for DNA11 and DNA28 are shown in Figure 2B. On average we had 12.14 positive pools per locus for DNA28 and 15.21 for DNA11, ranging from 9 to 18 positive pools per locus. These results are summarized in Figure 2B; positive PCRs are marked by a “+”, or in the case of heterozygous SNPs, the genotype is indicated. The green and orange bars reflect the different phases of the haplotype, and the light blue bars depict those clones for which the phase cannot yet be detected because they contain no heterozygous SNP. The brown color indicates the presence of two clones of different haplotype in one pool.
In the case of DNA11, we found five clones bridging the distance between rs3778150 and Mu_Opiat_IVS2+31 unambiguously; two with T(rs3778150)-C(Mu_Opiat_IVS2+31) in phase and three with C(rs3778150)-T(Mu_Opiat_IVS2+31). In pool B5 ofDNA11, the situation appears to be a bit more complex, as it contains two clones of different phases and therefore would not allow the reconstruction of the haplotype. In the case of DNA28 there are only three clones bridging the largest gap between the two informative SNPs (rs510769 on fragment 5 and rs3778152 on fragment 12). All three are of the same phase A(rs510769)-G(rs3778152), and the corresponding haplotype must be deduced from the other data. Because it is crucial for CSH to find at least one clone that bridges the distance between two informative SNPs, we summarized the interrelation of distances between the heterozygous SNPs and the number of clones bridging the distance in Table 2 which is available as online Supplemental data at www.genome.org. It becomes clear that SNPs lying closer together appear more frequently together on the same clone than SNPs lying further apart.
For both DNAs, it is possible to reconstruct the haplotype by using overlapping clones. In the case of DNA11, the pools C1 and H9 would suffice to reconstruct the following haplotype: rs510769(A) – rs607759(T) – rs3778148(T) – rs3778150(C) – Mu_Op31(T). Pools C5, E2 (and A2) would suffice to reconstruct the allelic haplotype: rs510769(G) – rs607759(C) – rs3778148(G) – rs3778150(T) – Mu_Op31(C). For DNA28, the haplotype can be inferred; for example, from pools E12 and E2 to be Mu_Opiat_-172(T) – rs510769(A) – rs3778152(G) – Mu_Op691(C), and the other haplotype must be deduced from the other data to be Mu_Opiat_-172(G) – rs510769(G) – rs3778152(A) – Mu_Op691(G).
For whole-genome haplotype analysis, it is essential that sufficient material is generated by the DNA preparation of the clone pools to enable 1–2 million PCRs. In a dilution series, we determined that the pool DNA (initially 300 μL) could be diluted up to 6000-fold and still obtain a PCR product. For one PCR, 0.5 μL DNA was required. Thus 3,400,000 PCRs could be set up in 25-μL reactions and used as template for genotyping by the GOOD assay in singleplex reactions. Because only 3-μL of the PCRs was used, enough template was generated to type up to 6 × 3,400,000 SNPs. This implies that 4 μL for gel scoring of the PCR products and 3 μL for the GOOD assays are required.
One approach to increase the throughput of this method is to multiplex the PCRs; that is, to include the primers for two or more PCR products in one reaction, and GOOD assays. In Figure 3, an example of a duplex GOOD assay can be seen where two SNPs that are about 10 kb apart were typed in the same reaction. Thus the haplotype of these two SNPs could be determined in one reaction. The phase determination of SNPs, lying 10 kb apart, is—thus far—not possible with other mass spectrometric procedures.
Figure 3.

A duplex haplotype analysis of two SNPs lying 10 kb apart. For SNP rs510769, primer TATGGCATTTCACATTCACATGptTA was used; for SNP rs607759 of the OPRM1 gene primer, AATTGAATGGCTCTAGGptAC was used. Respective products for rs510769 were Gpt-TApt(G/A) with masses 1256 Da and 1240 Da, and respective products for rs607759 were GptAC(T/C) with masses 1216 and 1201. Top trace: The mass spectrometry analysis of SNPs rs510769 and rs607759 of the OPRM1 gene in a specific fosmid pool. As can be deduced from the masses, the A allele of SNP rs510769 and the T allele of SNP rs607759 were in phase. Bottom trace: The analysis of the complementary fosmid pool, where the G allele of SNP rs510769 and the C allele of SNP rs607759 were in phase.
One might wonder whether it is more efficient to analyze fewer and larger pools. To simulate larger pools, we `electronically' combined two or three neighboring pools to result in one larger pool. The results are illustrated in Figure 4. For DNA28, the two pools combined resulted in super-pools containing ∼24.3% of the genome (Fig. 4A). The three clones that were bridging the gap between rs510769 and rs3779152 are still in pools that do not contain an allelic clone of different phase. However, in three other pools (C3/D3, E9/F9, E12/F12), clones of different phases can be detected. For DNA11, which initially had slightly larger pools, the combination of two pools resulted in pools bearing on average 30.4% of the genome (Fig. 4A). This results in one of originally seven clones that were bridging the gap between rs3779150 and rs3779162 being combined with a clone of different phase (C4/D4).
Figure 4.

The effect of larger pools was simulated `electronically'. (A) Two pools were combined; (B) three pools.
For DNA28, the combination of three pools results in apparently four clones bridging the gap between rs510769 and rs3779152, and three pools contain clones of detectably different phases (D1-F1, E2-G2, D10-F10, and F9-H9). However, from the original data (Fig. 2), we know that two clones are included within pool F9-H9. For DNA11, the combination of three pools each has a much more severe effect: One pool now consists on average of 45% of the genome, resulting in only three clones unambiguously bridging the gap between rs3779150 and rs3779162, and six pools contain clones of detectably different phase, of which five are at the same locus. When combining two pools, only two to three pools contain clones of detectably different phase, as expected.
DISCUSSION
We have shown the proof-of-concept of an efficient procedure to directly determine the haplotype of SNPs. This involved separating allelic DNA fragments adopting fosmid- or cosmid-cloning and analyzing 96 pools that represent less than 20% of the genome. Using this approach we were able to experimentally reconstruct the haplotype of a region spanning more than 60 kb.
In contrast to methods which rely on separating the allelic chromosomes by establishing somatic cell hybrid lines, in our method the complexity of the DNA template for subsequent PCR is much lower. This can be attributed to the following: First, we have only cosmid DNA of one-tenth to one-fifth of the human genome in our pools, and secondly we do not have an additional mammalian genome in the background. The initial step, namely cloning the DNA in a cosmid vector, raises the question of whether this method can be applied to regions of unclonable DNA. However, most SNPs currently available from SNP databases are derived from sequencing of cloned DNAs. Furthermore, the libraries were constructed by random shearing of the DNA, instead of using restriction endonucleases. This allows even cloning of regions that do not contain a restriction site for the chosen endonuclease, and thus only a very small proportion of the genome may be underrepresented in our libraries.
As shown here, the CSH method works reliably if the heterozygous SNPs are up to 30 kb apart, and if a 10-fold coverage fosmid/cosmid library is provided. The initial choice to plate the library in pools of 10,000 clones (∼10% of the genome) proved to efficiently ensure that each clone could unambiguously be classified as one or the other haplotype. As expected, in 10%–15% of the pools, a PCR product could be amplified. Gel-based detection of successful amplification was therefore reasonable to select the positive pools prior to performing the GOOD assay. To reduce times spent on PCR work and analysis, one might consider making pools as large as 0.3–0.4 genome equivalents. In this case, time and money for the preselection of positive pools might be saved, because the overall cost of the subsequent analysis even of negative pools by the GOOD assay is about the same as for the gel analysis. However, when choosing the pool size, one should consider that the probability of two (or more) clones of the opposite haplotype being within one pool increases with the size of the pools, based on the following equation:T(Mu_Opiat_IVS2+31). In pool B5 of
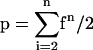 |
where f is the fraction of the genome per pool, and p is the probability of two or more clones of different alleles in one pool.
As described in the Results section, scaling up the pool size results in a reduced workload for DNA preparation and PCR setup, but scoring fewer but larger pools may compromise the ease of interpretation of the results, because pools containing clones of different phases cannot be used to infer the haplotypes. The limited size of cosmids combined with the scoring of PCR results, which we used as internal validation, enables an estimation of whether two clones are contained within the same pool. Pool B5 of DNA11 (Fig. 2) is the only pool out of over 30 (15 “orange” pools and 16 “green” pools) which cannot be assigned unambiguously to one or the other haplotype. In DNA28 (Fig. 2), all pools can definitely be assigned. This clearly validates that CSH is a straightforward and accurate method.
Furthermore, in the “electronically” combined pool E2/F2 of DNA11 (Fig. 4A), markers of more than 55 kb are covered, although all of the same phase, and it is clear that this bar must be the result of two clones, because a single cosmid covers a region of maximally 45–48 kb. Obviously, two clones of the same phase do not interfere with the subsequent analysis, pools containing two clones of different phases must be excluded from further analysis. For instance, it can be seen in Figure 4A that the combined pool C4/D4 of DNA11 contains two clones of different phases that cover the same region. The examples of Figure 4 clearly demonstrate the effect of pool size and number of pools that should be excluded from further analysis.
For the CSH methodology, we see in principle two major applications. First is the analysis of ancestral haplotypes in a rather small number of DNA samples, thus suggesting CSH as an efficient alternative to cell-hybrid technologies and an experimental complement to statistical analysis. Although the construction of fosmid/cosmid libraries still requires days of work, it offers several advantages. Most importantly, constructing a fosmid or cosmid library is significantly easier and cheaper than using somatic cell hybrids (Patil et al. 2001). Another advantage of fosmids/cosmids over almost any other type of library is that they render lengthy characterization of the library unnecessary. It is clear that the size of cosmid/fosmid clones is limited by the lambda-phage-head used for the in vitro packaging, and including the vector between 35 and 53 kb, with most clones being between 44 and 48 kb; therefore the size of the pools can quite easily be determined by simple colony counts.
A second application of the CSH procedure is the determination of the molecular haplotype in case control studies, where statistical analyses yielded ambiguous results. Ultimately, the physical haplotypes of a limited number of DNAs must be determined to back up statistical analyses. A third, rather minor application of CSH might be the haplotype reconstruction of duplicated regions, especially those containing disease-associated genes. However, in this case not two, but four phases must be reconstructed.
To apply CSH in a haplotyping project, some steps in the CSH procedure will have to be automated. This includes the initial DNA preparation, PCR set-up, scoring by 384 gels, set-up of the GOOD assay, and bioinformatics (a laboratory information management system). We will apply CSH as an experimental complement to large-scale statistical analysis of (ancestral) haplotypes in the future.
METHODS
Materials
Oligonucleotides for PCR were synthesized and HPSF-purified by MWG. dNTPs and ddNTPs were purchased from Roche Diagnostics, and phosphodiesterase II (calf spleen) was supplied from Cell Systems. α-S-ddNTPs were synthesized by Biolog. Thermosequenase and shrimp alkaline phosphatase were obtained from Amersham Buchler. Chemical reagents were purchased from Aldrich. The MALDI matrix (α-cyano-4-hydroxy-cinnamic acid methyl ester) is available from Bruker Saxonia.
Genomic DNA Isolation
Anonymous DNAs were prepared using leukocyte filters that were obtained from a blood donation center in Berlin. Filters were reverse-flushed with 80 mL of PBS, and the cells were subsequently collected by centrifugation. The erythrocytes were lysed by resuspending the cells in erythrocyte lysis buffer (155 mM NH4Cl, 5 mM NH4HCO3, 0.1 mM EDTA, 40 μM KCl, pH 7.4) and incubation on ice for 15 min. The remaining leucocytes were collected again by centrifugation. This procedure was repeated if necessary. The volume of the cell pellet was determined and about the same volume of PBS was added. The cells were gently resuspended, and then mixed with an equal volume of 1.5% low-melting-point agarose in PBS. The suspension was immediately distributed into block molds of ∼100 μL. After solidifying, the blocks were pushed into NDS (500 mM EDTA, 1% N-Lauroyl Sarcosinat, 10 mM Tris-HCl, pH 9.0) containing 1 mg/mL ProteinaseK, and incubated overnight at 50°C. The next day, the blocks were washed five times for 30 min in TE, incubated for 2 h in TE with 1 mM PMSF, followed by 4–5 further washes in TE.
Construction and Pooling of Fosmid and Cosmid Libraries
Library constructions employed protocols recommended by Epicentre. The fosmid libraries were constructed using the EpiFos5 cloning kit and the cosmid library with the pWEB kit, respectively. Alternatively, if only a small amount of initial material is available, the procedure described by Nizetic et al. (1994), which relies on partially digesting the DNA with a mixture of dammethylase and MboI, could be applied.
Fragmentation of DNA
In brief, three agarose blocks each containing ∼5–7 μg DNA were liquified at 65°C with 300 μL TE. The DNA was sheared by passing 12 times through a 23G-11/4 needle attached to a 1-mL syringe. Gelase buffer and 1 μL Gelase (Epicentre) were added. After incubation for 30 min at 42°C, ammonium acetate was added to a final concentration of 2.5 M, carefully mixed, and centrifuged for 10 min at 13,000 rpm to remove residual undigested agarose. The supernatant was transferred to a fresh tube, and the DNA precipitated by adding 2 vol of ethanol.
End Repair
The DNA was carefully resuspended in a small volume and subjected to end repair as directed in the manual from Epicentre. In brief, 40 μg DNA was incubated for 45 min at room temperature in a total volume of 80 μL containing 66 mM K acetate, 33 Tris-acetate, 10 mM Mg acetate, 0.5 mM DTT, 1 mM ATP, 0.25 mM of each dNTP, and 4 μL of the supplied end-repair enzyme mix, which includes T4 polynucleotidekinase and T4 DNA polymerase.
Ligation
Approx. 900 ng of DNA fragments was ligated to 1.5 μg of the supplied blunt-end Epifos-5 or the pWEB vector, respectively, for 2 h in a total volume of 60 μL, using the supplied fast-link ligation buffer, according to the Epicentre manual.
Packaging and Transfection
The ligation reaction was packaged in vitro using six MAX-Plax Packaging extracts (Epicentre) according to the manufacturer's manual. XL1 blue cells, grown in LB supplemented with 10 mM MgSO4 and 0.2 % maltose to the mid-log phase, were used for transfection with the fosmids/cosmids. Pools of the library comprising ∼10,000 single colonies were plated onto LB plates (150 mm dia. plates, LB-Medium with 1.2% agarose, because in previous experiments we found that agar might contain substances that inhibit the subsequent PCR), supplemented with the appropriate antibiotic.
DNA Preparation of the Pools
After incubation at 37°C for 15–16 h, the bacteria were washed from the plates with 9 mL LB. The DNAs from the pools were prepared according to standard alkaline lysis protocol (Sambrook et al. 1989). In brief, the bacteria were pelleted by centrifugation, resuspended in 2.5 mL AL1 (50 mM Tris-HCl, pH 8.0, 10 mM EDTA), and then 2.5 mL AL2 (0.2 NNaOH, 1% SDS) was added, gently mixed, and incubated for 5 min at room temperature. Lastly, the solution was neutralized with 2.5 mL AL3 (3 M KAcetate, pH 5.5). The cell debris was removed by centrifugation, and the supernatant transferred into a fresh tube. The DNA was precipitated by adding 0.6 vol iso-propanol and spun down. After washing with 70% ethanol, the DNA was finally resuspended in 300 μL TE. Usually, 0.5 μL of a 1:40 dilution was used for subsequent PCRs.
The GOOD Assay
PCR
PCR was performed in a volume of 25 μL containing 1 × PCR buffer (20 mM ammonium sulfate, 75 mM Tris-HCl, pH 9.0, 0.01% Tween 20, 1.5 mm MgCl2), 0.5 M Betaine, 50 μM dNTPs, 0.5 μM each primer (see Supplemental Table 1), and 1 U Taq DNA polymerase. The PCR regime was a touch-down protocol: After an initial denaturation step of 4 min at 95°C, 14 cycles of 15 sec at 95°C, 45 sec at 70°C, -1°C/cycle, and 1 min at 72°C followed. Afterwards, 21 cycles of 15-sec denaturation at 95°C, annealing at 56°C for 30 sec, and extension at 72°C for 1 min were performed, followed by a final extension of 5 min at 72°C. Three μL of the PCR were taken for subsequent reactions.
SAP Digest
Here, 0.5 μL (1 U/μL) of shrimp alkaline phosphatase (SAP) and 1.5 μL 50 mM Tris-HCl (pH 8.0) were added to the PCR and incubated for 1 h at 37°C, followed by denaturation at 90°C for 10 min.
Primer Extension Reaction
The primers used (see Supplemental Table 1) contained two phosphorothioate (pt) bridges. 7.5 pmol of the modified primers, 2 mM MgCl2, 100 μM respective α-S-ddNTPs, and 1 U Thermosequenase were added to the SAP-digested PCR. The reaction volume was increased to 5 μL by the addition of water. The reaction mix was cycled 35 times for 10 sec at 96°C, 30 sec at the corresponding annealing temperature, and 30 sec at 72°C. The annealing temperatures are shown in Supplemental Table 1.
Phosphodiesterase II Digestion
Here, 0.5 μL of a 0.5 M acetic acid solution and 1.5 μL of phosphodiesterase (5 U/ml) diluted in ammonium citrate (0.1 M, pH 6.0) were added. The reaction was incubated for 1.5 h at 37°C.
Alkylation Reaction and Preparation for MALDI Analysis
Twelve μL of acetonitrile, 3 μL bidistilled water, and 6 μL of methyliodide were added. The reaction was incubated for 35 min at 40°C. Upon cooling, a biphasic system was obtained. Five μL of the upper layer was sampled and diluted in 10 μL of 40% acetonitrile. From this solution, 0.5 μL was transferred onto the matrix-prepared MALDI target plates. For the preparation of the MALDI target, 0.5 μL of a 1.5% solution of α-cyano-4-hydroxycinnamic acid methyl ester matrix in acetone was spotted.
Mass Spectrometric Analysis
Spectra were recorded on a Bruker Biflex III time-of-flight mass spectrometer. This mass spectrometer is equipped with a Scout MTP ion source with delayed extraction. Spectra were recorded in a negative ion linear time-of-flight mode. Typical acceleration potentials were 18 kV. For delayed extraction, the extraction delay was 200 nsec.
Acknowledgments
We thank Roman Pawlik for providing us with Taq polymerase and James Adjaye for critical reading of the manuscript. This work was supported by the German National Genome Research Network (NGFN grant 01GR0155) and by the Max-Planck-Society.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.1442303.
Footnotes
[Supplemental material is available online at www.genome.org. The following individuals kindly provided reagents, samples, or unpublished information as indicated in the paper: R. Pawlik.]
References
- Boldt, A.B. and Petzl-Erler, M.L. 2002. A new strategy for mannose-binding lectin gene haplotyping. Hum. Mutat. 19: 296-306. [DOI] [PubMed] [Google Scholar]
- Daly, M.J., Rioux, J.D., Schaffner, S.F., Hudson, T.J., and Lander, E.S. 2001. High-resolution haplotype structure in the human genome. Nat. Genet. 29: 229-232. [DOI] [PubMed] [Google Scholar]
- Davidson, S. 2000. Research suggests importance of haplotypes over SNPs. Nat. Biotechnol. 18: 1134-1135. [DOI] [PubMed] [Google Scholar]
- Ding, C. and Cantor, C.R. 2003. Direct molecular haplotyping of long-range genomic DNA with M1-PCR. Proc. Natl. Acad. Sci. 100: 7449-7453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas, J.A., Boehnke, M., Gillanders, E., Trent, J.M., and Gruber, S.B. 2001. Experimentally-derived haplotypes substantially increase the efficiency of linkage disequilibrium studies. Nat. Genet. 28: 361-364. [DOI] [PubMed] [Google Scholar]
- Drysdale, C.M., McGraw, D.W., Stack, C.B, Stephens, J.C., Judson, R.S., Nandabalan, K., Arnold, K., Ruano, G., and Liggett, S.B. 2000. Complex promoter and coding region β2-adrenergic receptor haplotypes alter receptor expression and predict in vivo responsiveness. Proc. Natl. Acad. Sci. 97: 10483-10488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fugger, L., Morling, N., Ryder, L.P., Odum, N., and Svejgaard, A. 1990. Technical aspects of typing for HLA-DP alleles using allele-specific DNA in vitro amplification and sequence-specific oligonucleotide probes. Detection of single base mismatches. J. Immunol. Methods 129: 175-185. [DOI] [PubMed] [Google Scholar]
- Hoehe, M.R., Kopke, K., Wendel, B., Rohde, K., Flachmeier, C., Kidd, K.K., Berrettini, W.H., and Church, G.M. 2000. Sequence variability and candidate gene analysis in complex disease: Association of μ opioid receptor gene variation with substance dependence. Hum. Mol. Genet. 9: 2895-2908. [DOI] [PubMed] [Google Scholar]
- Lander, E.S., Linton, L.M., Birren, B., Nusbaum, C., Zody, M.C., Baldwin, J., Devon, K., Dewar, K., Doyle, M., FitzHugh, W., et al. 2001. Initial sequencing and analysis of the human genome. Nature 409: 860-921. [DOI] [PubMed] [Google Scholar]
- Maat-Kievit, A., Helderman-van den Enden, P., Losekoot, M., de Knijff, P., Belfroid, R., Vegter-van der Vlis, M., Roos, R., and Breuning, M. 2001. Using a roster and haplotyping is useful in risk assessment for persons with intermediate and reduced penetrance alleles in Huntington disease. Am. J. Med. Genet. 105: 737-744. [DOI] [PubMed] [Google Scholar]
- Mitra, R.D., Butty, V.L., Shendure, J., Williams, B.R., Housman, D.E., and Church, G.M. 2003. Digital genotyping and haplotyping with polymerase colonies. Proc. Natl. Acad. Sci. 100: 5926-5931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nizetic, D., Monard, S., Young, B., Cotter, F., Zehetner, G., and Lehrach, H. 1994. Construction of cosmid libraries from flow-sorted human chromosomes 1, 6, 7, 11, 13, and 18 for reference library resources. Mamm. Genome 5: 801-802. [DOI] [PubMed] [Google Scholar]
- Patil, N., Berno, A.J., Hinds, D.A., Barrett, W.A., Doshi, J.M., Hacker, C.R., Kautzer, C.R., Lee, D.H., Marjoribanks, C., McDonough, D.P., et al. 2001. Blocks of limited haplotype diversity revealed by high-resolution scanning of human chromosome 21. Science 294: 1719-1723. [DOI] [PubMed] [Google Scholar]
- Rohde, K. and Fuerst, R. 2001. Haplotyping and estimation of haplotype frequencies for closely linked biallelic multilocus genetic phenotypes including nuclear family information. Hum. Mutat. 17: 289-295. [DOI] [PubMed] [Google Scholar]
- Ruano, G., Kidd, K.K., and Stephens, J.C. 1990. Haplotype of multiple polymorphisms resolved by enzymatic amplification of single DNA molecules. Proc. Natl. Acad. Sci. 87: 6296-6300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachidanandam, R., Weissman, D., Schmidt, S.C., Kakol, J.M., Stein, L.D., Marth, G., Sherry, S., Mullikin, J.C., Mortimore, B.J., Willey, D.L., et al. 2001. A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature 409: 928-933. [DOI] [PubMed] [Google Scholar]
- Sambrook, J., Fritsch, E.F., and Maniatis, T. 1989. Molecular cloning: A laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Sauer, S. and Gut, I.G. 2003. Extension of the GOOD assay for genotyping single nucleotide polymorphisms by matrix-assisted laser desorption/ionization mass spectrometry. Rapid Commun. Mass Spectrom. 17: 1265-1272. [DOI] [PubMed] [Google Scholar]
- Sauer, S., Lechner, D., Berlin, K., Lehrach, H., Escary, J.L., Fox, N., and Gut, I.G. 2000a. A novel procedure for efficient genotyping of single nucleotide polymorphisms. Nucleic Acids Res. 28: e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauer, S., Lechner, D., Berlin, K., Plancon, C., Heuermann, A., Lehrach, H., and Gut, I.G. 2000b. Full flexibility genotyping of single nucleotide polymorphisms by the GOOD assay. Nucleic Acids Res. 28: e100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapadar, P., Ghosh, S., and Majumder, P.P. 2000. Haplotyping in pedigrees via a genetic algorithm. Hum. Hered. 50: 43-56. [DOI] [PubMed] [Google Scholar]
- Tost, J., Brandt, O., Derbala, D., Caloustian, C., Lechner, D., and Gut, I.G. 2002. Molecular haplotyping at high throughput. Nucleic Acids Res. 30: e96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venter, J.C., Adams, M.D., Myers, E.W., Li, P.W., Mural, R.J., Sutton, G.G., Smith, H.O., Yandell, M., Evans, C.A., Holt, R.A., et al. 2001. The sequence of the human genome. Science 291: 1304-1351. [DOI] [PubMed] [Google Scholar]
- Woolley, A.T., Guillemette, C., Li Cheung, C., Housman, D.E., and Lieber, C.M. 2000. Direct haplotyping of kilobase-size DNA using carbon nanotube probes. Nat. Biotechnol. 18: 760-763. [DOI] [PubMed] [Google Scholar]