

Abstract
Among human birth defect syndromes, malformations affecting the face are perhaps the most striking due to cultural and psychological expectations of facial shape. One such syndrome is auriculocondylar syndrome (ACS), in which patients present with defects in ear and mandible development. Affected structures arise from cranial neural crest cells, a population of cells in the embryo that reside in the pharyngeal arches and give rise to most of the bone, cartilage and connective tissue of the face. Recent studies have found that most cases of ACS arise from defects in signaling molecules associated with the endothelin signaling pathway. Disruption of this signaling pathway in both mouse and zebrafish results in loss of identity of neural crest cells of the mandibular portion of the first pharyngeal arch and the subsequent repatterning of these cells, leading to homeosis of lower jaw structures into more maxillary-like structures. These findings illustrate the importance of endothelin signaling in normal human craniofacial development and illustrate how clinical and basic science approaches can coalesce to improve our understanding of the genetic basis of human birth syndromes. Further, understanding the genetic basis for ACS that lies outside of known endothelin signaling components may help elucidate unknown aspects critical to the establishment of neural crest cell patterning during facial morphogenesis.
Keywords: endothelin, craniofacial development, neural crest cell, zebrafish, morpholino, knockout mice, transgenic mice, auriculocondylar syndrome, question mark ear, micrognathia, homeotic transformation, PLCB4, GNAI3, EDNRA
Introduction
Auriculocondylar syndrome (ACS) is a craniofacial birth defect characterized by outer ear and mandible malformations. Defects in these structures resemble those observed in animal mutant models in which endothelin 1 (EDN1)-endothelin receptor type A (EDNRA) signaling is disrupted. This information was recently used to identify the genetic basis for the majority of ACS cases. These findings illustrate the conservation and significance of endothelin signaling in jawed vertebrates for normal facial morphogenesis. In this review, we will discuss both the clinical appearance of ACS and the genetic studies that identified the basis for ACS. We will also review how animal models of endothelin signaling mutants have helped define the cellular and molecular events required for lower jaw development.
Clinical Characteristics of Auriculocondylar Syndrome
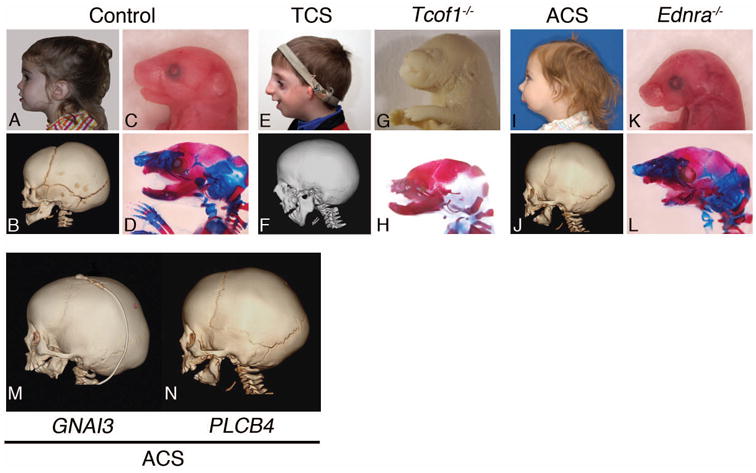
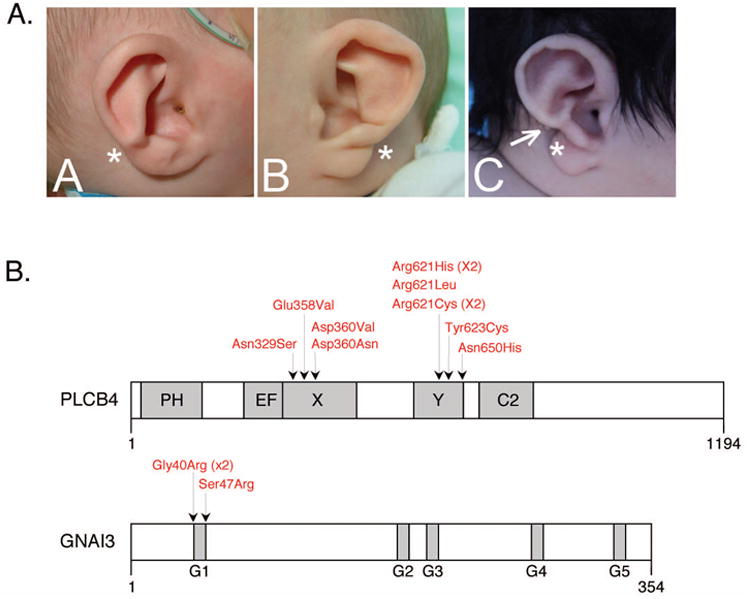
ACS (OMIM 602483 and 614669), also referred to as “question mark ear syndrome” or “dysgnathia complex”, is a craniofacial disorder affecting neural crest cell (NCC) development within the first and second pharyngeal arches. It is an apparently rare condition whose prevalence is still unknown. The first report appeared in 1978 [Uuspaa, 1978], which was followed by reports of several other isolated cases and a few familial cases [Baker et al., 2004; Divizia et al., 2002; Erlich et al., 2000; Gerkes et al., 2008; Gordon et al., 2013; Greig et al., 2012; Guion-Almeida et al., 1999; Guion-Almeida et al., 2002; Jampol et al., 1998; Kokitsu-Nakata et al., 2011; Masotti et al., 2008; Ozturk et al., 2005; Priolo et al., 2000; Propst et al., 2013; Rieder et al., 2012; Storm et al., 2005; Stuffken and Tuinzing, 2008]. Vertical disease segregation through several generations in a few families suggested an autosomal dominant pattern of inheritance for ACS [Guion-Almeida et al., 2002; Jampol et al., 1998; Masotti et al., 2008; Storm et al., 2005]. The triad of core features are micrognathia, mandibular condyle hypoplasia, and a unique ear malformation (Fig. 1A and Fig. 2I, J, M, N), which has been named as question mark ear (QME) or Cosman ear [Cosman et al., 1970]; the latter is a highly stereotypical malformation of the pinna, affecting the junction between the lobe and helix. The severity of the malformation varies and can range from a minor indentation to complete clefting between the lobe and helix (Figure 1). Other features of variable frequency include microstomia, full cheeks, palatal anomalies, glossoptosis, crowded teeth, facial asymmetry, postauricular tags and hearing loss (Table I) [Gordon et al., 2013; Kokitsu-Nakata et al., 2011]. Among ACS patients, there is a high degree of inter- and intra-familial phenotypic variation, including non-penetrance [Gordon et al., 2013; Ozturk et al., 2005; Rieder et al., 2012]. Isolated QME (IQME, OMIM 612798), where the range of ear dysmorphism is very similar to that in ACS, has also been reported in isolated and familial cases with an autosomal dominant mode of inheritance, and might represent a milder form of ACS [Al-Qattan, 1998; Brodovsky and Westreich, 1997; Cosman et al., 1970; Fumiiri and Hyakusoku, 1983; Pan et al., 2010; Park, 1998; Posso et al., 2011; Shkalim et al., 2008; Takato et al., 1989; Vayvada et al., 2005; Vincent et al., 1961].
Figure 1.
A-C. The phenotypic spectrum of question mark ear (QME). Panels A and B display the right and left ears, respectively, of the same individual. In A, the ear is mildly affected, with a raised fold between the helix and lobe (*). In B, the ear is more severely affected, with a crease at the lobe-helix junction (*). In C this characteristic crease extends further, almost separating the lobe and helix (*). A post-auricular tag is also present (arrow). Panels B and C are reproduced from the Journal of Medical Genetics, Gordon et al., 50(3):174-86, 2013, with permission from BMJ Publishing Group Ltd. D. Summary of missense mutations identified in PLCB4 and GNAI3 in ACS patients by Rieder et al. [2012] and Gordon et al. [2013]. Domains of each protein are depicted in grey. All PLCB4 mutations fall with the X and Y domains, which together comprise the catalytic domain. All GNAI3 mutations are within the G1 box, one of five motifs (G1-G5) that are involved in GDP/GTP binding. PH, pleckstrin homology; EF, EF hand-like.
Figure 2.
Comparison of human craniofacial syndromes caused by mutations in the EDNRA (ACS, GNAI3 and PLCB4) and non-EDNRA (TCOF1) pathways to mouse models. A-L. Human patients are shown in both lateral gross view (A, E, I) and lateral computed tomography (CT) scans (B, F and J), while embryonic day (E) 18.5 mouse embryos are shown in both lateral gross view (C, G, K) and laterally after skeleton staining (D, H and L). Skeletons were stained with alizarin red and alcian blue to visualize bone and cartilage staining, respectively. M. Comparison of skull malformation between patients with mutations in either GNAI3 or PLCB4. The mandible in both patients has taken on a maxilla-like (triangular) shape compared to the normal angular shape of the mandible. ACS, auriculocondylar syndrome; TCS, Treacher-Collins syndrome; Ednra-/-, endothelin receptor type A mutant mice.
Table 1. Main clinical features in ACS as compared to other conditions of the first and second pharyngeal arches and to mouse endothelin pathway mutants (AD: autosomal dominant; AR: autosomal recessive; + present; - not reported; ND, not determined).
| Clinical features | ACS cases reviewed in Kokitsu- Nakata et al. (2011), ie, prior to gene identification. N (%) |
ACS cases known to harbour mutations in PLCB4 or GNAI34 |
Clinical features in other syndromes affecting the 1st and 2nd pharyngeal arches |
Mice with null mutations in Edn1, Ece1 or Ednra8 |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||||
| ACS AD PLCB4 |
ACS AR PLCB4 |
ACS AD GNAI3 |
OAVS 1 | Isolated Pierre Robin sequence2 |
Treacher Collins syndrome AD TCOF13 |
Nager syndrome AD SF3B45 |
Miller syndrome AR DHODH6 |
MFDGA7 AD EFTUD2 |
|||
|
| |||||||||||
| FACE | |||||||||||
|
| |||||||||||
| • Facial asymmetry | 10/23 (44) | 0/11 | 0/1 | 1/3 | + | - | + | + | - | + | - |
|
| |||||||||||
| • Prominent cheeks | 17/23 (74) | 10/11 | 1/1 | 3/3 | - | - | - | _ | - | - | + |
|
| |||||||||||
| MOUTH | |||||||||||
|
| |||||||||||
| • Small mouth | 18/23 (78) | 10/11 | 1/1 | 3/3 | - | - | - | - | - | - | + |
|
| |||||||||||
| • Abnormal palate | 11/20 (55) | 1/10 | 0/1 | 1/2 | - | + | + | + | + | + | +/- |
|
| |||||||||||
| • Excess soft tissue / atypical uvula | 2/9 (22) | 1/10 | 0/1 | 1/2 | - | - | - | - | - | - | + |
|
| |||||||||||
| MANDIBLE | |||||||||||
|
| |||||||||||
| • Micro- and/or retrognathia | 18/23 (78) | 10/11 | 1/1 | 3/3 | + | + | + | + | + | + | + |
|
| |||||||||||
| • Mandibular condyle abnormality | 13/14 (93) | 6/6 | 1/1 | 1/1 | + | ND | + | + | ND | - | + |
|
| |||||||||||
| EARS | |||||||||||
|
| |||||||||||
| • Question mark ear | 23/23 (100) | 11/11 | 1/1 | 3/3 | - | - | - | - | - | - | ND |
| • Microtia | ND | 0/11 | 0/1 | 0/2 | + | - | + | + | + | + | + |
|
| |||||||||||
| • Pre-auricular tags | 4/23 (17) | 0/10 | 0/1 | 0/2 | + | - | +/- | +/- | +/- | + | ND |
| • post-auricular tags | ND | 3/10 | 1/1 | 0/2 | - | - | - | - | - | - | ND |
|
| |||||||||||
| • Hearing loss | 9/16 (56) | 2/10 | 0/1 | 1/1 | + | - | + | + | + | + | ND |
|
| |||||||||||
| EYES | |||||||||||
| • Downslanted palpebral fissures | ND | 0/11 | 0/1 | 0/3 | - | - | + | + | - | - | ND |
| • Epibulbar dermoids | ND | 0/10 | 0/1 | 0/2 | + | - | - | - | - | + | ND |
|
| |||||||||||
| Respiratory distress / apnoea / tracheotomy | 11/23 (48) | 6/10 | 1/1 | 2/3 | ND | + | + | + | ND | + | + |
|
| |||||||||||
| OTHER ALTERATIONS | |||||||||||
| • Vertebral defects | ND | ND | 0/1 | 0/2 | + | - | - | - | - | + | - |
| • Limb defects | ND | 0/10 | 0/1 | 0/2 | - | - | - | + | + | + | - |
–OAVS: oculoauriculovertebral spectrum. Highly heterogeneous disorder, most likely multifactorial and no genetic mechanism has yet been clearly established;
–Isolated Pierre Robin sequence (PRS) is likely to be genetically heterogeneous. The etiology of most cases is largely unknown, except for cases harboring deletions or translocations in the SOX9 regulatory region (reviewed in Gordon et al., 2009).
– TCOF1 is the major gene associated with autosomal dominant (AD) Treacher Collins syndrome (TCS); heterozygous mutations in POLR1D or in both alleles of POLR1C (autosomal recessive inheritance) account for a small proportion of TCS patients and were not included in this table due to the small number of reported cases.
- cases described in Rieder et al. (2012) and Gordon et al. (2013).
- cases described in Bernier et al. (2012) and Czeschik et al. (2013).
- cases described in Ng et al. (2010) and Rainger et al. (2012).
- MFDGA: mandibulofacial dysostosis, Guion-Almeida type. Cases described in Lines et al. (2012), Need et al. (2012), Gordon et al. (2012) and Luquetti et al. (2013).
– see text for references.
ACS clinically overlaps with other craniofacial syndromes with altered structures derived from the first and second pharyngeal arches, particularly oculoauriculovertebral spectrum (OAVS, OMIM 164210), though the ear malformations in OAVS are usually distinct from those observed in ACS [Gordon et al., 2013] (Table I). Similarly, in Treacher Collins syndrome (TCS, OMIM 154500), the ear defects (ie, microtia) are distinct from ACS (Fig. 1 and Table I), and TCS can also be distinguished from ACS by zygomatic arch hypoplasia and eyelid colobomas. In several familial cases of ACS, mildly affected individuals may present with isolated micrognathia, suggesting that some sporadic cases of more frequent mandibular dysplasias such as Pierre Robin sequence (PRS, OMIM 261800) may actually have an underlying genetic cause in common with that of ACS.
Loci and Mutation Types in Auriculocondylar Syndrome
The first disease locus for ACS was mapped to chromosome 1p21.1–q23.3 based on the study of a large ACS family [Guion-Almeida et al., 2002; Masotti et al., 2008]. However, the existence of at least a second locus was supported by the failure to find linkage to the chromosome 1 interval in a second ACS family [Masotti et al., 2008]. In 2012, the two major loci for ACS were identified by exome sequencing of several ACS kindreds [Rieder et al., 2012]. This group demonstrated that heterozygous missense mutations in phospholipase C, beta 4 (PLCB4, at 20p) and in guanine nucleotide-binding protein (G protein), alpha inhibiting activity polypeptide 3 (GNAI3, falling within the previously mapped interval on chromosome 1) led to the ACS phenotype. Subsequently, PLCB4 and GNAI3 were screened for mutations in a series of 11 ACS or IQME patients, with heterozygous missense mutations identified in PLCB4 in six cases and in GNAI3 in one case, while in another case a homozygous intragenic deletion within PLCB4 was identified [Gordon et al., 2013]. The latter finding was unexpected and provided the first evidence that ACS might also follow an autosomal recessive mode of inheritance while potentially shedding light on a previously reported case associated with consanguinity [Guion-Almeida et al., 2002]. Current data suggest that mutations in PLCB4 and GNAI3 account for about 80% of the ACS/IQME cases (15/19 families studied). Of the 15 solved cases, the majority (80%) are due to mutations in PLCB4 (11/15 PLCB4 heterozygous missense mutations, 1/15 homozygous PLCB4 deletion and 3/15 GNAI3 missense heterozygous mutations) (Fig. 1B). It is expected that genome/exome sequencing will bring new insights into the still unsolved ACS cases.
Incomplete penetrance and a high degree of clinical variability were found in ACS caused by PLCB4 or GNAI3 mutations [Gordon et al., 2013; Rieder et al., 2012], consistent with previous indications of variable penetrance and expressivity in ACS families [Guion-Almeida et al., 2002; Masotti et al., 2008; Ozturk et al., 2005]. Four of the 11 heterozygous PLCB4 mutations following an autosomal dominant inheritance pattern were de novo, while the three heterozygous GNAI3 mutations were inherited [Gordon et al., 2013; Rieder et al., 2012]. Molecular characterization of a larger number of families is still necessary to better evaluate the proportion of de novo mutations in PLCB4 and GNAI3 in ACS cases.
Mutational Mechanisms Causing Acs
The missense PLCB4 mutations so far identified are clustered within the catalytic domain of the protein, with recurrent mutations at Arg621 and Asp360. Structural protein modeling of PLCB4 missense mutations predicts that they act as dominant negatives, with the residues affected forming bonds with inositol triphosphate or calcium in the active site, or other amino acids participating in catalysis [Gordon et al., 2013; Rieder et al., 2012]. Interestingly, no heterozygous PLCB4 deletions, nonsense or frame shift mutations have been identified in ACS patients [Gordon et al., 2013; Rieder et al., 2012]. However, one patient with a homozygous deletion within PLCB4 (presumed to result in complete absence of functional PLCB4 protein) has been observed. The consanguineous parents of the homozygous patient each harbored the deletion in the heterozygous state but were phenotypically normal. In addition, other individuals identified via the copy number variant (CNV) database DECIPHER (http://decipher.sanger.ac.uk/) or from published literature, harboring deletions of varying sizes affecting PLCB4 and sometimes neighboring genes, presented varying phenotypes, but not ACS [Gordon et al., 2013]. These cases argue against haploinsufficiency of PLCB4 as a cause of ACS. Rather, it is plausible that ACS PLCB4 mutations result in dominant negative proteins that interfere with the function of the wild type version and/or other proteins. Evidence for this comes from the schmerle (she) mutation found in zebrafish, which, as described below, results from missense mutations in the catalytic domain of plcb3, a PLCB4 homologue [Walker et al., 2007]. Knocking down plcb3 function in zebrafish embryos using an antisense morpholino (in which the function of Plcb3 is blocked) results in a mild phenotype with low penetrance compared to she mutants, arguing strongly for a dominant negative effect of the schmerle mutations on other Plcb family members (there are four PLCB genes in each of zebrafish, mice and humans). Supporting this idea, PLCD4 produces a naturally occurring splice variant coding for an isoform that can dominantly interfere with various PLC proteins [Nagano et al., 1999]. An informative in vivo test of this dominant negative theory will require overexpression in zebrafish embryos of wild type human PLCB4/GNAI3 or versions of these proteins harbouring ACS mutations. The latter should dominantly interfere with the endogenous zebrafish version of each protein, resulting in pharyngeal arch phenotypes (and gene expression changes) similar to those observed for the germ line plcb3 mutants.
Only two different GNAI3 substitution mutations resulting in ACS in 3 familial cases have been reported so far, one at amino acid Gly40 and one at Ser47. These amino acids are the first and last amino acids of the G1 box, one of five highly conserved motifs of rat sarcoma (RAS) superfamily and G alpha proteins that play a role in guanosine diphosphate (GDP)/guanosine triphosphate (GTP) binding [Wennerberg et al., 2005]. Like the mutations in PLCB4, the fact that all reported mutations in GNAI3 fall within one structural domain supports the idea that they are not simply haploinsufficient alleles, but rather that they modify the activity of the protein in specific ways, given that mutations causing haploinsufficiency such as stops and frame shifts would be expected to be randomly distributed. Protein structural modeling of the GNAI3 missense substitution at Gly40 (p.Gly40Arg) suggested that it acts as a gain of function molecule [Rieder et al., 2012], while the modeling of the amino acid substitution at Ser47 (p.Ser47Arg), along with the effects of mutation of the equivalent residue in other G alpha and Ras proteins, suggested a dominant negative mechanism [Gordon et al., 2013]. Further functional analysis will be necessary in order to elucidate how these mutations result in ACS.
PLCB4, GNAI3 and EDNRA Signaling Pathway
Both PLCB4 and GNAI3 are predicted to function downstream of the EDNRA, a G protein-coupled receptor known to play a crucial role in pharyngeal arch patterning [Clouthier et al., 1998a]. As described above, this is supported by missense mutations in zebrafish plcb3, which result in fusion and hypoplasia of pharyngeal arch cartilage elements [Walker et al., 2007]. Interestingly, zebrafish with combined heterozygous mutations in plcb3 and edn1 (the ligand of the EDNRA) demonstrated a more severe phenotype than either mutant alone. As discussed in detail below, EDN1 signalling via the EDNRA regulates expression of Distalless homeobox (DLX) transcription factors in the pharyngeal arches and thereby plays a major role in specification of the lower jaw in vertebrates [Clouthier et al., 2000; Miller et al., 2003; Ozeki et al., 2004; Ruest et al., 2004]. Given that EDN1 signalling is thought to be mediated by phospholipase C enzymes in various cellular contexts [Ho et al., 2012] [Schiekel et al., 2013] [Kelley et al., 2006] and loss of zebrafish plcb3 disrupts pharyngeal arch development, it is plausible that disruption of PLCB4 activity in ACS patients has a similar negative effect on EDNRA signaling during human development. Likewise, as EDNRA is a G protein-coupled receptor, it is possible that mutations in GNAI3 repress signaling from EDNRA. That mandibular osteoblasts derived from PLCB4- or GNAI3-mutated ACS patients showed a significant reduction in the expression of DLX5 and DLX6 [Rieder et al., 2012] supports this view, as induction of Dlx5/Dlx6 expression in mouse pharyngeal arches is almost solely dependent on Ednra signaling [Ruest and Clouthier, 2009; Ruest et al., 2004]. While this further supports the disruption of the EDNRA signalling pathway in ACS, not all events downstream of EDNRA signalling in the pharyngeal arches are likely conserved in postnatal osteoblasts. Further functional experiments in animal models or induced pluripotent cells from ACS patients will be required to firmly establish the role played by PLCB4 and GNAI3 in the EDN1-EDNRA-DLX pathway.
Neither Plcb4 nor Gnai3 mutant mice develop an overt ACS phenotype. Plcb4 knockout mice develop a range of central nervous system defects, including ataxia, absence seizures and defects in visual responses [Cheong et al., 2009; Jiang et al., 1996; Kim et al., 1997], while Gnai3 null mice display rib and vertebral defects, a phenotype exacerbated in Gnai1/Gnai3 and Gnai2/Gnai3 mutants [Plummer et al., 2012]. It should be noted that the identification of a subtle craniofacial phenotype in the Plcb4 or Gnai3 null mice might require careful measurement of the size and shape of bones – such an investigation has not been reported. However, since these models do not actually represent the predicted dominant negative or gain-of-function mutations observed in human patients, a mouse model of ACS may require generation of mice harboring a missense mutation equivalent to that identified in humans. This assumes such a mutation would produce a more severe and representative model of the disease than complete loss of function, as appears to be the case for the plcb3 mutation in zebrafish [Walker et al., 2007].
As discussed in the second part of this review, mice that are homozygous null for Edn1, Ednra, endothelin converting enzyme 1 (Ece1) or Dlx5/Dlx6 display a homeotic transformation of the lower jaw into an upper jaw, highlighting the importance of the Ednra/Dlx pathway for specifying the mandibular portion of the first pharyngeal arch [Clouthier et al., 2010]. Rieder et al. (2012) drew on this information and the implied function of PLCB4 and GNAI3 in this pathway to support the argument that the lower jaw phenotype observed in ACS patients with PLCB4/GNAI3 mutations also represents a homeotic transformation of the mandible into a maxilla-like structure [Rieder et al., 2012]. This was based on alterations in skeletal morphology of the mandible in a small number of ACS patients, in which drastic reductions of the condyle, ramus and alveolar bone led to increased symmetry around the axis of the jaw joint. Soft tissue changes on the floor of the mouth were also presented that were reported to be palate-like. Interestingly, other reports have noted ectopic soft tissue masses in the posterior oral cavity in ACS patients [Gordon et al., 2013; Guion-Almeida et al., 2002] and may be equivalent to ectopic tissue found in the lower jaw of mouse endothelin pathway mutants [Barron et al., 2011; Ruest et al., 2004]. Clearly, detailed information about skeletal and soft tissue changes from a larger number of ACS patients is warranted to understand the mandibular to maxillary transformation. Considering the role of Dlx proteins in cranial myogenesis [Heude et al., 2010], additional imaging is also warranted to examine potential alterations in the morphology or identity of teeth and craniofacial muscles in ACS patients.
Phenotypic Variation, Other Loci
Among the patients harbouring PLCB4/GNAI3 missense mutations reported thus far [Gordon et al., 2013; Rieder et al., 2012], there is little evidence for genotype-phenotype correlations, even when comparing the PLCB4-mutant cohort with the GNAI3-mutant cohort. Indeed, there appears to be as much intrafamilial variation as there is between cases. However, one of the key outcomes from the series of patients tested by Gordon et al. [2013] was the extent to which the mutation-positive cases displayed a restricted phenotypic spectrum relative to (mutation-negative) patients presenting with a range of other first pharyngeal arch phenotypes (Table I). Amongst these atypical cases (negative for PLCB4/GNAI3 mutations) was a series of patients that had been previously described as ACS, but which displayed features of OAVS and condyle dysplasia, and which lacked the characteristic constriction between the lobe and helix [McGowan et al., 2011]. A small number of cases with non-syndromic auricular dysplasias distinct from QME were also tested but were negative for PLCB4 or GNAI3 mutations [Gordon et al., 2013]. It should be noted that condyle hypoplasia is seen in a number of craniofacial syndromes and therefore perhaps a less specific feature of ACS [Johnson et al., 2011; Travieso et al., 2013]. These findings highlight the fact that the presence of a QME is the most useful discriminating factor for a clinical diagnosis of ACS. Another distinctive feature of several mutation-positive cases was the presence of a post-auricular tag in a highly stereotypic location, above the lobe-helix junction [Gordon et al., 2013]. Key discriminating aspects of the ACS facial phenotype also include microstomia and prominent cheeks.
An important question is the nature of the genetic defect for PLCB4/GNAI3 mutation-negative cases with a convincing ACS phenotype. Rieder et al. (2012) and Gordon et al. (2013) each presented one such case. Case A001 in Rieder et al. (2012) was described only as “classic ACS”, though a detailed description of the pinna was not provided. Exome data for A001 revealed missense mutations in the DOCK1 and DOCK6 genes, which code for guanine nucleotide exchange factors; these factors activate G proteins by promoting the exchange of GDP for GTP [Cote and Vuori, 2007]. Information about the inheritance of these mutations in A001 was not available [Rieder et al., 2012]. As discussed in Gordon et al. (2013), the role of DOCK genes in ACS is not yet convincing. As noted above, there is evidence that PLCD4 can be alternatively spliced to generate a dominant negative isoform – therefore, the presence of intronic mutations in PLCB4 that lead to aberrant, dominantly-acting transcripts is a plausible disease mechanism that would not necessarily be detected by exome sequencing. Case 10 in Gordon et al. (2013) involved two PLCB4/GNAI3 mutation-negative siblings with features of ACS, including QMEs, born to consanguineous and unaffected parents. This case may involve a recessive or germline mosaic mutation in a gene other than the two candidate loci. It is also possible that some IQME cases may actually represent variable expressivity of PLCB4/GNAI3 mutations, given that in some familial ACS cases there can be individuals with a typical ear phenotype but no obvious mandibular dysplasia [Guion-Almeida et al., 2002; Rieder et al., 2012; Storm et al., 2005]. In addition, Gordon et al. (2013) identified two mutation-negative familial cases of IQME, suggesting that IQME may occasionally be a genetically distinct subgroup of ACS. Future investigation of such cases may provide the opportunity to identify novel regulators of the PLCB4/GNAI3 pathway.
In the ACS patient reported with homozygous loss of PLCB4, central apneas were recorded in multiple sleep studies [Gordon et al., 2013]. This finding was unexpected because although apneas and respiratory distress had been reported in several ACS patients, these events were typically in the context of oropharyngeal obstruction [Erlich et al., 2000; Guion-Almeida et al., 2002; Masotti et al., 2008]. Analysis of sleep studies that have been performed in PLCB4/GNAI3 missense mutation-positive ACS patients have suggested that central apneas can persist after mandibular distraction surgery [Gordon et al., 2013]. Comparison of standardized polysomnographic recordings from a larger number of ACS patients (ideally multiple recordings for each patient, before and after distraction surgery), would help clarify the possible disruption of central control of respiration in ACS. It is worth noting that endothelin signaling has previously been implicated in the regulation of respiration [Gaultier et al., 2004]. One previously published case, born to consanguineous parents, displayed ACS and some features suggestive of perturbed central nervous system development (learning, neuropsychomotor and language deficits), but genetic testing has not been reported for this patient [Guion-Almeida et al., 2002]. Whether extra-craniofacial phenotypes are more frequent in patients with homozygous loss of function of PLCB4 will require analysis of a larger number of such cases.
EDNRA Signaling and Neural Crest Cell Patterning
As described above, the phenotype associated with ACS likely results from disruption of the EDNRA pathway due to interruption of intracellular G protein-coupled signaling mediated by PLCB4 or GNAI3. This raises multiple questions, including how loss of this signal can lead to the endothelin mutant phenotype, which affects a relatively restricted portion of the craniofacial complex. To examine this aspect, one must first understand the basis of Ednra signaling and the types of defects observed following its loss.
Most craniofacial defects associated with ACS occur in cranial NCC derivatives. Migrating from the posterior midbrain and hindbrain to the pharyngeal arches, NCCs form bone, cartilage and connective tissue of the jaw (first pharyngeal arch), middle ear ossicles and ear pinnae (arches 1 and 2) and bone and cartilage structures of the throat (arches 2-4 in mammals, up to 7 arches in other vertebrates) [Couly et al., 1993; Fraser et al., 1990; Le Douarin et al., 1993; Lumsden et al., 1991]. In addition, cranial NCCs contribute to the cranial ganglia (all arches) [Le Douarin et al., 1993]. Ednra signaling is the earliest known event that establishes dorsal-ventral (D-V) identity of NCCs in the pharyngeal arches. Ednra (found on all cranial NCCs) is stimulated by Edn1, a 21 amino acid peptide [Clouthier et al., 2010; Clouthier et al., 1998a; Kurihara et al., 1994; Medeiros and Crump, 2012]. Edn1 is initially produced as a preproendothelin molecule from the overlying pharyngeal arch ectoderm [Yanagisawa et al., 1998; Yanagisawa, 1994], which is then processed by a furin protease to generate big-endothelin, a 38 amino acid inactive protein [Yanagisawa, 1994]. Big-endothelin is then cleaved by one of two Ece metalloproteases (Ece1 and Ece2) into mature, active Edn1 [Emoto and Yanagisawa, 1995; Xu et al., 1994]. In mammals, there are three ligands (Edn1, 2 and 3), which bind to the two known G-protein coupled receptors (Ednra and endothelin receptor type B (Ednrb)) with differential affinity [Yanagisawa, 1994]. Specific ligand/receptor combinations have specific physiological roles in adults and NCC patterning in embryos [Baynash et al., 1994; Clouthier et al., 1998a; Hosoda et al., 1994; Kurihara et al., 1994; Sakamoto et al., 1993; Yanagisawa et al., 1998; Yanagisawa, 1994], though only disruption of Edn1, Ece1 or Ednra leads to facial defects.
Mice lacking Edn1, Ece1 or Ednra die at birth, with multiple defects in craniofacial and cardiovascular structures [Clouthier et al., 1998b; Kurihara et al., 1994; Yanagisawa et al., 1998]. The observed craniofacial defects occur in structures derived from the mandibular portion of arch 1 and arches 2-4 and closely match those observed in ACS patients (Table I; Fig 2). The most evident change observed is an apparent homeotic transformation of the mandible into a more maxilla-like structure [Ozeki et al., 2004; Ruest et al., 2004]. This transformed maxilla-like structure articulates with the jugal bone of the zygomatic arch through a fibrous joint found between the jugal bone and a duplicated jugal bone in the lower jaw. Other first arch-derived elements are duplicated, including the palatine, pterygoid, jugal and lamina obturans bones and the ala temporalis cartilages, while the tympanic rings, malleus and incus are absent. The duplication of the palatine bones in the lower jaw is part of a mirror image duplication of the palate [Ruest et al., 2004]. This includes a large piece of soft tissue in the lower portion of the oral cavity that resembles the palate, complete with rugae, the raised epithelial ridges normally observed on the roof of the mouth. This tissue may be similar to the excessive soft tissue found in the oral cavity of ACS patients described above [Gordon et al., 2013; Guion-Almeida et al., 2002; Rieder et al., 2012]. Duplication of the mystacial vibrissae is also observed in Edn1, Ece1 and Ednra mutants, with these whisker-like sensory organs arrayed on the lower jaw in a similar pattern to that observed on the snout, suggesting that Ednra patterning can affect the identity of the overlying ectoderm. Additional changes include fusion of the hyoid bone with the pterygoid bones and low set/under-developed pinnae. Due to the different shape of pinna in rodents compared with human, it is not possible to define lobular changes as observed in ACS patients.
ENU-induced mutations in endothelin family members have also been found in zebrafish, including mutations in edn1 (sucker; suc/edn1) [Kimmel et al., 2003; Miller and Kimmel, 2001; Miller et al., 2000a], furinA (sturgeon, stu) [Walker et al., 2006], mef2c (hoover, hoo) [Miller et al., 2007] and plcb3 (she) [Walker et al., 2007]. In general, edn1-/- mutants have severe facial defects including malformed or hypoplastic ventral cartilages (Meckel's cartilage and the ceratohyal) and bones (branchiostegal rays and opercle) derived from arches 1 and 2. furinA, mef2c and plcb3 mutants have similar defects, though the severity and penetrance of defects differs between mutants and is generally less severe than that observed in edn1 mutants. In contrast, all four mutants have defects in the joints of the first and second arches resulting in fusions between dorsal and ventral cartilages, the significance of which is discussed below. Fusions also exist between the dentary (mandibular) and maxillary bones derived from the first arch, with the rudimentary dentary bone resembling a maxilla, suggestive of a homeotic transformation similar to that observed in Ednra mouse mutants [Kimmel et al., 2003]. As discussed above and below, mutations in plcb3 have been particularly useful in understanding the genetic basis of ACS.
EDNRA Signaling in the Distal and Intermediate Domains
Defects observed in ACS are limited to specific facial domains, a fact reflective of both the role of Ednra signaling in organizing the arch and the timing of its action. The pharyngeal arches can be divided into three domains [Miller et al., 2003; Talbot et al., 2010; Tavares et al., 2012; Zuniga et al., 2010]. The distal (ventral in fish) domain gives rise to Meckel's cartilage and the mandibular bone and some of the malleus in mice [Ruest et al., 2003]. The middle portion of the arches is referred to as the intermediate domain, which in zebrafish gives rise to a small portion of Meckel's cartilage and the joint regions between the Meckel's cartilage and the palatoquadrate (the first arch dorsal derivative in fish). In mice, the intermediate domain of the first arch gives rise to a small portion of the proximal mandible and a portion of the malleus, likely all of the incus and the fibrous tissue around the malleus/incus junction [Ruest et al., 2003; Tavares et al., 2012; Tucker et al., 2004]. The proximal (dorsal in fish) domain gives rise to dorsal elements that in zebrafish include the palatoquadrate (first arch) and hyosymplectic (second arch) cartilages. In mammals, the fate of the dorsal domain has not been as finely mapped as in zebrafish, though based on duplications observed in endothelin and Dlx mutants, likely gives rise to more proximal jaw structures that include the pterygoid and alisphenoid bones [Depew et al., 2005; Ozeki et al., 2004; Ruest et al., 2004].
The defects in endothelin pathway mutants reflect the role that Ednra signaling plays in patterning the distal and intermediate domains (Fig. 3). The first apparent action of Ednra signaling is induction of Dlx5/Dlx6 expression in distal and intermediate NCC-derived mesenchyme throughout the pharyngeal arches [Charité et al., 2001; Ozeki et al., 2004; Ruest et al., 2004]. Combined loss of both Dlx5 and Dlx6 leads to homeosis of lower jaw structures almost identical to that observed in Ednra, Edn1 or Ece1 mouse mutants, illustrating the close temporal relationship between Ednra signaling and Dlx5/Dlx6 expression. Dlx5/Dlx6 then work in combination with Mef2c [Miller et al., 2007; Verzi et al., 2007] to induce distal arch expression of the gene encoding the basic helix-loop-helix transcription factor Hand2, in part through direct binding of Dlx6 to the known arch-specific enhancer of Hand2 [Charité et al., 2000]. This enhancer is highly conserved among gnathostomes, as the zebrafish and mouse enhancers work interchangeably in both species with only minor differences in spatiotemporal expression [Ikle et al., 2012]. However, since expression of Hand2 is not affected in either Dlx5-/- or Dlx6-/- embryos [Jeong et al., 2008], Dlx5 and Dlx6 likely act interchangeably in inducing Hand2 expression. This is supported by the fact that mandibular hypoplasia is more severe in mice with progressive reduction of the total number of Dlx5 and Dlx6 alleles [Depew et al., 2005]. A similar compensation by DLX6 could explain why a patient with a homozygous mutation in DLX5 did not have an ACS-like phenotype [Shamseldin et al., 2012]. Hand2 subsequently establishes the distal arch domain described above through repression of at least Dlx5 and Dlx6 [Barron et al., 2011; Talbot et al., 2010]. This repression is necessary for tongue morphogenesis, as loss of Hand2 leads to both aglossia and duplication of the secondary palate as observed in Ednra/Edn1/Ece1 mutants. Ednra signaling is also crucial for gene expression within the intermediate domain, inducing both Dlx3 and Nkx3.2 (Fig. 3) [Clouthier et al., 2000; Miller et al., 2003; Talbot et al., 2010; Tavares et al., 2012]. Taken together, the primary event driving normal lower jaw patterning is an Ednra-Dlx5/Dlx6-Hand2 network, with disruption of this network leading to ACS-like phenotypes in the lower jaw.
Figure 3.
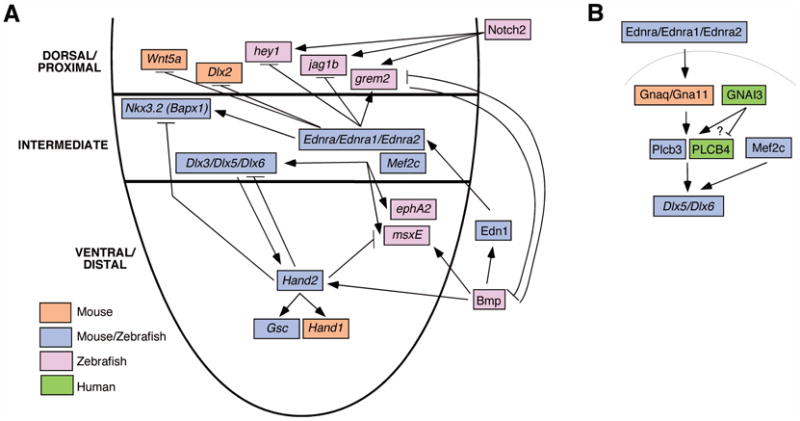
Signaling pathway initiated by Edn1-induced Ednra signaling. A. A schematized depiction of the first pharyngeal arch, with dorsal (proximal), intermediate and ventral (distal) aspects of the arch labeled on the left. Edn1 from the overlying ectoderm binds to the Ednra on the underlying neural crest-derived mesenchyme, initiating a signaling cascade that includes both positive and negative components. Part of this pathway includes additional activation potential for Bmp signaling in the ventral arch and repression of this activity by Grem2, found in the dorsal arch. Each of the listed signaling or transcription factors is color coded to depict the species from which genetic evidence was obtained to support its function. B. The putative intracellular signaling pathway induced by Ednra. PLCB4 is activated downstream of Gnaq/Gna11 to induce gene expression. Also shown is GNAI3, though its actual function downstream of EDNRA is not clear.
Differential Dependence on EDNRA Signaling in Establishing Distal and Intermediate Derivatives
Complete loss of Ednra signaling in mice leads to disrupted development of the lower jaw and middle ear ossicles, derivatives of the distal and intermediate domains. However, partial loss of Ednra signaling appears to more severely affect intermediate domain derivatives and the corresponding earlier gene expression patterns [Ruest and Clouthier, 2009; Tavares et al., 2012]. Similarly, loss of plcb3 and furinA in zebrafish leads to variable changes in arch structures, though fusion of arch joints (intermediate domain derivatives) are always observed [Walker et al., 2007; Walker et al., 2006]. Until recently, the basis of this graded dependence on Ednra signaling was unclear. While several hypotheses have been put forth to account for this sensitivity [Kimmel et al., 2003], it now appears that Bmp signaling in the distal domain may provide a compensatory mechanism to downregulation of Ednra signaling after the signaling has been initiated [Alexander et al., 2011]. In zebrafish, Bmp signaling participates in patterning the distal arch by inducing edn1 expression and later maintaining Edn1-induced expression of hand2 [Alexander et al., 2011]. This correlates with the finding that overexpression of Bmp4 in mice leads to upregulation of Hand2 expression independent of Dlx5/Dlx6 [Bonilla-Claudio et al., 2012]. Gremlin2 confines Bmp activity to the distal arch [Zuniga et al., 2011], partially explaining why this mechanism does not exist in the intermediate domain. This would result in the intermediate domain having a longer temporal requirement for Ednra signaling and thus making this region more susceptible to Ednra perturbation. Supporting this idea of a compensatory distal mechanism is the finding that, unlike in edn1-/- mutants, expression of the distal domain gene hand2 recovers in plcb3 and furinA mutants by 55 hpf [Miller et al., 2000b; Miller et al., 2003]. Thus, these findings may explain why first arch skeletal defects in ACS patients are often most severe around the jaw joint, an intermediate domain-derived structure.
Thus far, the genetic defects observed in ACS patients are predicted to result from reduction of EDNRA signaling. However, it should be expected that other craniofacial syndromes may result from overactivation of EDNRA signaling. All cranial NCCs express Ednra [Clouthier et al., 1998a] (or both ednra genes in zebrafish [Nair et al., 2007]) and they appear competent to respond to Ednra signaling. Overexpression of Bmp4 in zebrafish results in upregulation of ventral gene expression in dorsal arch domains [Alexander et al., 2011]. Likewise, injection of high levels of human EDN1 into the arches of zebrafish results in homeotic transformations of dorsal arch structures into ventral arch-like structures [Kimmel et al., 2007]. Further, introducing one copy of an Edn1 cDNA into the Ednra locus results in homeotic transformation of the maxilla into a mandible [Sato et al., 2008]. As Edn1 is not expressed in the maxillary portion of the first arch [Clouthier et al., 1998b; Yanagisawa et al., 1998], this further points to the competency of more maxillary (dorsal) NCCs to respond to Ednra signaling. Thus Edn1 appears to be both necessary and sufficient for dictating the DV identities of skeletogenic NCCs, consistent with its role as a key mediator of NCC development and skeletal patterning in the mandibular domain of the first arch. Future studies aimed at examining the spatio-temporal regulation of Ednra signaling are thus critical to help understand the role of this signaling pathway in human craniofacial development and disease.
Acknowledgments
The authors would like to thank Michael Cunningham and Paul Trainor for unpublished data. This work was supported in part from grants from the National Institutes of Health (DEC; DE014181, DE018899 and DE020076 (FaceBase Consortium)), CEPID/FAPESP and CNPq (MRP) and E-Rare CRANIRARE (SL).
Biographies
David E. Clouthier is an Associate Professor in the School of Dental Medicine at the University of Colorado Anschutz Medical Campus. His lab focuses on the regulation of neural crest cell patterning and facial morphogenesis, utilizing both mouse and zebrafish models.
Maria Rita Passos-Bueno is a full professor in the Institute of Biosciences at University of São Paulo. Her lab is dedicated to the study of the genetic mechanisms of craniofacial syndromes and autism spectrum disorders.
Andre L.P. Tavares is a post-doctoral researcher in the School of Dental Medicine at the University of Colorado Anschutz Medical Campus. His research interests include embryonic patterning of the craniofacial complex and the basis of human facial dysmorphologies.
Stanislas Lyonnet and Jeanne Amiel are clinical geneticists at the Hôpital Necker-Enfants Malades. Their research interests are focused on the genetic causes of rare malformative syndromes, particularly those involving defects in neural crest cells
Christopher Gordon is a post-doctoral researcher at the Hôpital Necker-Enfants Malades. His research interests include craniofacial developmental biology and the genetic causes of human craniofacial disorders.
Footnotes
The authors have no conflict of interest to declare.
References
- Al-Qattan MM. Cosman (question mark) ear: congenital auricular cleft between the fifth and sixth hillocks. Plast Reconstr Surg. 1998;102:439–441. doi: 10.1097/00006534-199808000-00023. [DOI] [PubMed] [Google Scholar]
- Alexander C, Zuniga E, Blitz IL, Wada N, LePabic P, Javidan Y, Zhang T, Cho KW, Crump JG, Schilling TF. Combinatorial roles for Bmps and Endothelin 1 in patterning the dorsal-ventral axis of the craniofacial skeleton. Development. 2011;138:5135–5146. doi: 10.1242/dev.067801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker PA, Aftimos S, Anderson BJ. Airway management during an EXIT procedure for a fetus with dysgnathia complex. Paediatr Anaesth. 2004;14:781–786. doi: 10.1111/j.1460-9592.2004.01284.x. [DOI] [PubMed] [Google Scholar]
- Barron F, Woods C, Kuhn K, Bishop J, Howard MJ, Clouthier DE. Downregulation of Dlx5 and Dlx6 expression by Hand2 is essential for initiation of tongue morphogenesis. Development. 2011;138:2249–2259. doi: 10.1242/dev.056929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baynash AG, Hosoda K, Giaid A, Richardson JA, Emoto N, Hammer RE, Yanagisawa M. Interaction of endothelin-3 with endothelin-B receptor is essential for development of epidermal melanocytes and enteric neurons. Cell. 1994;79:1277–1285. doi: 10.1016/0092-8674(94)90018-3. [DOI] [PubMed] [Google Scholar]
- Bonilla-Claudio M, Wang J, Bai Y, Klysik E, Selever J, Martin JF. Bmp signaling regulates a dose-dependent transcriptional program to control facial skeletal development. Development. 2012;139:709–719. doi: 10.1242/dev.073197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodovsky S, Westreich M. Question mark ear: a method for repair. Plast Reconstr Surg. 1997;100:1254–1257. doi: 10.1097/00006534-199710000-00028. [DOI] [PubMed] [Google Scholar]
- Charité J, McFadden DG, Merlo GR, Levi G, Clouthier DE, Yanagisawa M, Richardson JA, Olson EN. Role of Dlx6 in regulation of an endothelin-1-dependent, dHAND branchial arch enhancer. Genes Dev. 2001;15:3039–3049. doi: 10.1101/gad.931701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charité J, McFadden DG, Olson EN. The bHLH transcription factor dHAND controls Sonic hedgehog expression and establishment of the zone of polarizing activity during limb development. Development. 2000;127:2461–2470. doi: 10.1242/dev.127.11.2461. [DOI] [PubMed] [Google Scholar]
- Cheong E, Zheng Y, Lee K, Lee J, Kim S, Sanati M, Lee S, Kim YS, Shin HS. Deletion of phospholipase C beta4 in thalamocortical relay nucleus leads to absence seizures. Proc Natl Acad Sci U S A. 2009;106:21912–21917. doi: 10.1073/pnas.0912204106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clouthier DE, Garcia E, Schilling TF. Regulation of facial morphogenesis by endothelin signaling: insights from mouse and fish. Am J Med Genet A. 2010;152A:2962–2973. doi: 10.1002/ajmg.a.33568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clouthier DE, Hosoda K, Richardson JA, Williams SC, Yanagisawa H, Kuwaki T, Kumada M, Hammer RE, Yanagisawa M. Cranial and cardiac neural crest defects in endothelin-A receptor-deficient mice. Development. 1998a;125:813–824. doi: 10.1242/dev.125.5.813. [DOI] [PubMed] [Google Scholar]
- Clouthier DE, Hosoda K, Richardson JA, Williams SC, Yanagisawa H, Kuwaki T, Kumada M, Hammer RE, Yanagisawa M. Cranial and cardiac neural crest defects in endothelin-A receptor-deficient mice. Development. 1998b;125:813–824. doi: 10.1242/dev.125.5.813. [DOI] [PubMed] [Google Scholar]
- Clouthier DE, Williams SC, Yanagisawa H, Wieduwilt M, Richardson JA, Yanagisawa M. Signaling pathways crucial for craniofacial development revealed by endothelin-A receptor-deficient mice. Dev Biol. 2000;217:10–24. doi: 10.1006/dbio.1999.9527. [DOI] [PubMed] [Google Scholar]
- Cosman B, Bellin H, Crikelair GF. The Question Mark ear. Plast Reconstr Surg. 1970;46:454–457. doi: 10.1097/00006534-197011000-00006. [DOI] [PubMed] [Google Scholar]
- Cote JF, Vuori K. GEF what? Dock180 and related proteins help Rac to polarize cells in new ways. Trends Cell Biol. 2007;17:383–393. doi: 10.1016/j.tcb.2007.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couly GF, Coltey PM, Le Douarin NM. The triple origin of skull in higher vertebrates: a study in chick-quail chimeras. Development. 1993;117:409–429. doi: 10.1242/dev.117.2.409. [DOI] [PubMed] [Google Scholar]
- Depew MJ, Simpson CA, Morasso M, Rubenstein JLR. Reassessing the Dlx code: the genetic regulation of branchial arch skeletal pattern and development. J Anat. 2005;207:501–561. doi: 10.1111/j.1469-7580.2005.00487.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Divizia MT, Cordone A, Bado M, Rosaia L, Cirillo Silengo M, Ravazzolo R, Lerone M. Auriculo-condylar syndrome or new syndrome? Clin Dysmorphol. 2002;11:143–144. doi: 10.1097/00019605-200204000-00015. [DOI] [PubMed] [Google Scholar]
- Emoto N, Yanagisawa M. Endothelin converting enzyme-2: a membrane-bound, phosphoramidon-sensitive metalloprotease with acidic pH optimum. J Biol Chem. 1995;70:15262–15268. doi: 10.1074/jbc.270.25.15262. [DOI] [PubMed] [Google Scholar]
- Erlich MS, Cunningham ML, Hudgins L. Transmission of the dysgnathia complex from mother to daughter. Am J Med Genet. 2000;95:269–274. [PubMed] [Google Scholar]
- Fraser SE, Keynes RJ, Lumsden AGS. Segmentation in the chick embryo hindbrain is defined by cell lineage restriction. Nature. 1990;344:431–435. doi: 10.1038/344431a0. [DOI] [PubMed] [Google Scholar]
- Fumiiri M, Hyakusoku H. Congenital auricular cleft. Plast Reconstr Surg. 1983;71:249–250. doi: 10.1097/00006534-198302000-00019. [DOI] [PubMed] [Google Scholar]
- Gaultier C, Amiel J, Dauger S, Trang H, Lyonnet S, Gallego J, Simonneau M. Genetics and early disturbances of breathing control. Pediatr Res. 2004;55:729–733. doi: 10.1203/01.PDR.0000115677.78759.C5. [DOI] [PubMed] [Google Scholar]
- Gerkes EH, van Ravenswaaij CM, van Essen AJ. Question mark ears and post-auricular tags. Eur J Med Genet. 2008;51:264–267. doi: 10.1016/j.ejmg.2008.01.002. [DOI] [PubMed] [Google Scholar]
- Gordon CT, Vuillot A, Marlin S, Gerkes E, Henderson A, Alkindy A, Holder-Espinasse M, Park SS, Omarjee A, Sanchis-Borja M, Bdira EB, Oufadem M, Sikkema-Raddatz B, Stewart A, Palmer R, McGowan R, Petit F, Delobel B, Speicher MR, Aurora P, Kilner D, Pellerin P, Simon M, Bonnefont JP, Tobias ES, Garcia-Minaur S, Bitner-Glindzicz M, Lindholm P, Meijer BA, Abadie V, Denoyelle F, Vazquez MP, Rotky-Fast C, Couloigner V, Pierrot S, Manach Y, Breton S, Hendriks YM, Munnich A, Jakobsen L, Kroisel P, Lin A, Kaban LB, Basel-Vanagaite L, Wilson L, Cunningham ML, Lyonnet S, Amiel J. Heterogeneity of mutational mechanisms and modes of inheritance in auriculocondylar syndrome. Journal of medical genetics. 2013;50:174–186. doi: 10.1136/jmedgenet-2012-101331. [DOI] [PubMed] [Google Scholar]
- Greig AV, Podda S, Thorne CH, McCarthy JG. The question mark ear in patients with mandibular hypoplasia. Plast Reconstr Surg. 2012;129:368e–369e. doi: 10.1097/PRS.0b013e31823af031. [DOI] [PubMed] [Google Scholar]
- Guion-Almeida ML, Kokitsu-Nakata NM, Zechi-Ceide RM, Vendramini S. Auriculo-condylar syndrome: further evidence for a new disorder. Am J Med Genet. 1999;86:130–133. doi: 10.1002/(sici)1096-8628(19990910)86:2<130::aid-ajmg8>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- Guion-Almeida ML, Zechi-Ceide RM, Vendramini S, Kokitsu-Nakata NM. Auriculo-condylar syndrome: additional patients. Am J Med Genet. 2002;112:209–214. doi: 10.1002/ajmg.10631. [DOI] [PubMed] [Google Scholar]
- Heude E, Bouhali K, Kurihara Y, Kurihara H, Couly G, Janvier P, Levi G. Jaw muscularization requires Dlx expression by cranial neural crest cells. Proc Natl Acad Sci U S A. 2010;107:11441–11446. doi: 10.1073/pnas.1001582107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho PC, Tsui YC, Lin YW, Persaud SD, Wei LN. Endothelin-1 promotes cytoplasmic accumulation of RIP140 through a ET(A)-PLCbeta-PKCepsilon pathway. Molecular and cellular endocrinology. 2012;351:176–183. doi: 10.1016/j.mce.2011.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosoda K, Hammer RE, Richardson JA, Baynash AG, Cheung JC, Giaid A, Yanagisawa M. Targeted and natural (piebald-lethal) mutations of endothelin-B receptor gene produce megacolon associated with spotted coat color in mice. Cell. 1994;79:1267–1276. doi: 10.1016/0092-8674(94)90017-5. [DOI] [PubMed] [Google Scholar]
- Ikle JM, Artinger KB, Clouthier DE. Identification and characterization of the zebrafish pharyngeal arch-specific enhancer for the basic helix-loop-helix transciption factor Hand2. Dev Biol. 2012;368:118–126. doi: 10.1016/j.ydbio.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jampol M, Repetto G, Keith DA, Curtin H, Remensynder J, Holmes LB. New syndrome? Prominent, constricted ears with malformed condyle of the mandible. Am J Med Genet. 1998;75:449–452. doi: 10.1002/(sici)1096-8628(19980217)75:5<449::aid-ajmg1>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- Jeong J, Li X, McEvilly RJ, Rosenfeld MG, Lufkin T, Rubenstein JLR. Dlx genes pattern mammalian jaw primordium by regulating both lower jaw-specific and upper jaw-specific genetic programs. Development. 2008;135:2905–2916. doi: 10.1242/dev.019778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Lyubarsky A, Dodd R, Vardi N, Pugh E, Baylor D, Simon MI, Wu D. Phospholipase C beta 4 is involved in modulating the visual response in mice. Proc Natl Acad Sci U S A. 1996;93:14598–14601. doi: 10.1073/pnas.93.25.14598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JM, Moonis G, Green GE, Carmody R, Burbank HN. Syndromes of the first and second branchial arches, part 2: syndromes. AJNR Am J Neuroradiol. 2011;32:230–237. doi: 10.3174/ajnr.A2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley GG, Kaproth-Joslin KA, Reks SE, Smrcka AV, Wojcikiewicz RJ. G-protein-coupled receptor agonists activate endogenous phospholipase Cepsilon and phospholipase Cbeta3 in a temporally distinct manner. J Biol Chem. 2006;281:2639–2648. doi: 10.1074/jbc.M507681200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Jun KS, Lee SB, Kang NG, Min DS, Kim YH, Ryu SH, Suh PG, Shin HS. Phospholipase C isozymes selectively couple to specific neurotransmitter receptors. Nature. 1997;389:290–293. doi: 10.1038/38508. [DOI] [PubMed] [Google Scholar]
- Kimmel CB, Ullmann B, Walker M, Miller CT, Crump JG. Endothelin 1-mediated regulation of pharyngeal bone development in zebrafish. Development. 2003;130:1339–1351. doi: 10.1242/dev.00338. [DOI] [PubMed] [Google Scholar]
- Kimmel CB, Walker MB, Miller CT. Morphing the hyomandibular skeleton in development and evolution. J Exp Zool (Mol Dev Evol) 2007;308B:609–624. doi: 10.1002/jez.b.21155. [DOI] [PubMed] [Google Scholar]
- Kokitsu-Nakata NM, Zechi-Ceide RM, Vendramini-Pittoli S, Romanelli Tavares VL, Passos-Bueno MR, Guion-Almeida ML. Auriculo-condylar syndrome. Confronting a diagnostic challenge. Am J Med Genet A. 2011;158A:59–65. doi: 10.1002/ajmg.a.34337. [DOI] [PubMed] [Google Scholar]
- Kurihara Y, Kurihara H, Suzuki H, Kodama T, Maemura K, Nagai R, Oda H, Kuwaki T, Cao WH, Kamada N, Jishage K, Ouchi Y, Azuma S, Toyoda Y, Ishikawa T, Kumada M, Yazaki Y. Elevated blood pressure and craniofacial abnormalities in mice deficient in endothelin-1. Nature. 1994;368:703–710. doi: 10.1038/368703a0. [DOI] [PubMed] [Google Scholar]
- Le Douarin NM, Ziller C, Couly GF. Patterning of neural crest derivatives in the avian embryo: in vivo and in vitro studies. Dev Biol. 1993;159:24–49. doi: 10.1006/dbio.1993.1219. [DOI] [PubMed] [Google Scholar]
- Lumsden A, Sprawson N, Graham A. Segmental origin and migration of neural crest cells in the hindbrain region of the chick embryo. Development. 1991;113:1281–1291. doi: 10.1242/dev.113.4.1281. [DOI] [PubMed] [Google Scholar]
- Masotti C, Oliveira KG, Poerner F, Splendore A, Souza J, Freitas Rda S, Zechi-Ceide R, Guion-Almeida ML, Passos-Bueno MR. Auriculo-condylar syndrome: mapping of a first locus and evidence for genetic heterogeneity. Eur J Hum Genet. 2008;16:145–152. doi: 10.1038/sj.ejhg.5201955. [DOI] [PubMed] [Google Scholar]
- McGowan R, Murday V, Kinning E, Garcia S, Koppel D, Whiteford M. Novel features in auriculo-condylar syndrome. Clin Dysmorphol. 2011;20:1–10. doi: 10.1097/MCD.0b013e32833e56f5. [DOI] [PubMed] [Google Scholar]
- Medeiros DM, Crump JG. New perspectives on pharyngeal dorsoventral patterning in development and evolution of the vertebrate jaw. Dev Biol. 2012;371:121–135. doi: 10.1016/j.ydbio.2012.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller CT, Kimmel CB. Morpholino phenocopies of endothelin 1 (sucker) and other anterior arch class mutations. Genesis. 2001;30:186–187. doi: 10.1002/gene.1061. [DOI] [PubMed] [Google Scholar]
- Miller CT, Schilling TF, Lee K, Parker J, Kimmel CB. sucker encodes a zebrafish Endothelin-1 required for ventral pharyngeal arch development. Development. 2000a;127:3815–3828. doi: 10.1242/dev.127.17.3815. [DOI] [PubMed] [Google Scholar]
- Miller CT, Schilling TF, Lee K-H, Parker J, Kimmel CB. sucker encodes a zebrafish Endothelin-1 required for ventral pharyngeal arch development. Development. 2000b;127:3815–3838. doi: 10.1242/dev.127.17.3815. [DOI] [PubMed] [Google Scholar]
- Miller CT, Swartz ME, Khuu PA, Walker MB, Eberhart JK, Kimmel CB. mef2ca is required in cranial neural crest to effect Endothelin1 signaling in zebrafish. Dev Biol. 2007;308:144–157. doi: 10.1016/j.ydbio.2007.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller CT, Yelon D, Stainier DY, Kimmel CB. Two endothelin 1 effectors, hand2 and bapx1, pattern ventral pharyngeal cartilage and the jaw joint. Development. 2003;130:1353–1365. doi: 10.1242/dev.00339. [DOI] [PubMed] [Google Scholar]
- Nagano K, Fukami K, Minagawa T, Watanabe Y, Ozaki C, Takenawa T. A novel phospholipase C delta4 (PLCdelta4) splice variant as a negative regulator of PLC. J Biol Chem. 1999;274:2872–2879. doi: 10.1074/jbc.274.5.2872. [DOI] [PubMed] [Google Scholar]
- Nair S, Li W, Cornell R, Schilling TF. Requirements for endothelin type-A receptors and endothelin-1 signaling in the facial ectoderm for the patterning of skeletogenic neural crest cells in zebrafish. Development. 2007;134:335–345. doi: 10.1242/dev.02704. [DOI] [PubMed] [Google Scholar]
- Ozeki H, Kurihara Y, Tonami K, Watatani S, Kurihara H. Endothelin-1 regulates the dorsoventral branchial arch patterning in mice. Mech Dev. 2004;121:387–395. doi: 10.1016/j.mod.2004.02.002. [DOI] [PubMed] [Google Scholar]
- Ozturk S, Sengezer M, Isik S, Gul D, Zor F. The correction of auricular and mandibular deformities in auriculo-condylar syndrome. J Craniofac Surg. 2005;16:489–492. doi: 10.1097/01.scs.0000147655.94656.0d. [DOI] [PubMed] [Google Scholar]
- Pan B, Jiang H, Zhao Y, Lin L, Guo D, Zhuang H. Clinical analysis, repair and aetiology of question mark ear. J Plast Reconstr Aesthet Surg. 2010;63:28–35. doi: 10.1016/j.bjps.2008.08.039. [DOI] [PubMed] [Google Scholar]
- Park C. Correction of the unilateral question mark ear. Plast Reconstr Surg. 1998;101:1620–1623. doi: 10.1097/00006534-199805000-00030. [DOI] [PubMed] [Google Scholar]
- Plummer NW, Spicher K, Malphurs J, Akiyama H, Abramowitz J, Nurnberg B, Birnbaumer L. Development of the mammalian axial skeleton requires signaling through the Galpha(i) subfamily of heterotrimeric G proteins. Proc Natl Acad Sci U S A. 2012;109:21366–21371. doi: 10.1073/pnas.1219810110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posso CM, Wolff GA, Suarez LD. Question mark ear deformity: a combined method for correction. Aesthetic Plast Surg. 2011;35:646–649. doi: 10.1007/s00266-010-9619-2. [DOI] [PubMed] [Google Scholar]
- Priolo M, Lerone M, Rosaia L, Calcagno EP, Sadeghi AK, Ghezzi F, Ravazzolo R, Silengo M. Question mark ears, temporo-mandibular joint malformation and hypotonia: auriculo-condylar syndrome or a distinct entity? Clin Dysmorphol. 2000;9:277–280. doi: 10.1097/00019605-200009040-00009. [DOI] [PubMed] [Google Scholar]
- Propst EJ, Ngan BY, Mount RJ, Martin-Munoz D, Blaser S, Harrison RV, Cushing SL, Papsin BC. Ossicular fusion and cholesteatoma in auriculo-condylar syndrome: in vivo evidence of arrest of embryogenesis. Laryngoscope. 2013;123:528–532. doi: 10.1002/lary.23492. [DOI] [PubMed] [Google Scholar]
- Rieder MJ, Green GE, Park SS, Stamper BD, Gordon CT, Johnson JM, Cunniff CM, Smith JD, Emery SB, Lyonnet S, Amiel J, Holder M, Heggie AA, Bamshad MJ, Nickerson DA, Cox TC, Hing AV, Horst JA, Cunningham ML. A human homeotic transformation resulting from mutations in PLCB4 and GNAI3 causes auriculocondylar syndrome. American journal of human genetics. 2012;90:907–914. doi: 10.1016/j.ajhg.2012.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruest LB, Dager M, Yanagisawa H, Charité J, Hammer RE, Olson EN, Yanagisawa M, Clouthier DE. dHAND-Cre transgenic mice reveal specific potential functions of dHAND during craniofacial development. Dev Biol. 2003;257:263–277. doi: 10.1016/s0012-1606(03)00068-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruest LB, Clouthier DE. Elucidating timing and function of endothelin-A receptor signaling during craniofacial development using neural crest cell-specific gene deletion and receptor antagonism. Dev Biol. 2009;328:94–108. doi: 10.1016/j.ydbio.2009.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruest LB, Xiang X, Lim KC, Levi G, Clouthier DE. Endothelin-A receptor-dependent and -independent signaling pathways in establishing mandibular identity. Development. 2004;131:4413–4423. doi: 10.1242/dev.01291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto A, Yanagisawa M, Sawamura T, Enoki T, Ohtani T, Sakurai T, Nakao K, Toyo-oka T, Masaki T. Distinct subdomains of human endothelin receptors determine their selectivity to EndothelinA-selective antagonist and EndothelinB-selective agonists. J Biol Chem. 1993;268:8547–8553. [PubMed] [Google Scholar]
- Sato T, Kurihara Y, Asai R, Kawamura Y, Tonami K, Uchijima Y, Heude E, Ekker M, Levi G, Kurihara H. An endothelin-1 switch specifies maxillomandibular identity. Proc Natl Acad Sci U S A. 2008;105:18806–18811. doi: 10.1073/pnas.0807345105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiekel J, Lindner M, Hetzel A, Wemhoner K, Renigunta V, Schlichthorl G, Decher N, Oliver D, Daut J. The inhibition of the potassium channel TASK-1 in rat cardiac muscle by endothelin-1 is mediated by phospholipase C. Cardiovascular research. 2013;97:97–105. doi: 10.1093/cvr/cvs285. [DOI] [PubMed] [Google Scholar]
- Shamseldin HE, Faden MA, Alashram W, Alkuraya FS. Identification of a novel DLX5 mutation in a family with autosomal recessive split hand and foot malformation. Journal of medical genetics. 2012;49:16–20. doi: 10.1136/jmedgenet-2011-100556. [DOI] [PubMed] [Google Scholar]
- Shkalim V, Eliaz N, Linder N, Merlob P, Basel-Vanagaite L. Autosomal dominant isolated question mark ear. Am J Med Genet A. 2008;146A:2280–2283. doi: 10.1002/ajmg.a.32452. [DOI] [PubMed] [Google Scholar]
- Storm AL, Johnson JM, Lammer E, Green GE, Cunniff C. Auriculo-condylar syndrome is associated with highly variable ear and mandibular defects in multiple kindreds. Am J Med Genet A. 2005;138A:141–145. doi: 10.1002/ajmg.a.30883. [DOI] [PubMed] [Google Scholar]
- Stuffken MJ, Tuinzing DB. Dysgnathia complex, a rare deviation. Ned Tijdschr Tandheelkd. 2008;115:394–396. [PubMed] [Google Scholar]
- Takato T, Takeda H, Kamei M, Uchiyama K. The question mark ear (congenital auricular cleft): a familial case. Ann Plast Surg. 1989;22:69–73. doi: 10.1097/00000637-198901000-00013. [DOI] [PubMed] [Google Scholar]
- Talbot JC, Johnson SL, Kimmel CB. hand2 and Dlx genes specify dorsal, intermediate and ventral domains within zebrafish pharyngeal arches. Development. 2010;137:2507–2517. doi: 10.1242/dev.049700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavares ALP, Garcia EL, Kuhn K, Woods CM, Williams T, Clouthier DE. Ectodermal-derived Endothelin1 is required for patterning the distal and intermediate domains of the mouse mandibular arch. Dev Biol. 2012 doi: 10.1016/j.ydbio.2012.08.003. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travieso R, Chang CC, Terner JS, Beckett J, Wong K, Teng E, Steinbacher DM. A range of condylar hypoplasia exists in Treacher Collins syndrome. J Oral Maxillofac Surg. 2013;71:393–397. doi: 10.1016/j.joms.2012.04.031. [DOI] [PubMed] [Google Scholar]
- Tucker AS, Watson RP, Lettice LA, Yamada G, Hill RE. Bapx1 regulates patterning in the middle ear: altered regulatory role in the transition from the proximal jaw during vertebrate evolution. Development. 2004;131:1235–1245. doi: 10.1242/dev.01017. [DOI] [PubMed] [Google Scholar]
- Uuspaa V. Combined bilateral external ear deformity and hypoplastic mandible. Case report. Scand J Plast Reconstr Surg. 1978;12:165–167. doi: 10.3109/02844317809012989. [DOI] [PubMed] [Google Scholar]
- Vayvada H, Karaca C, Menderes A, Yilmaz M. Question mark ear deformity and a modified surgical correction method: a case report. Aesthetic Plast Surg. 2005;29:251–254. doi: 10.1007/s00266-004-0136-z. discussion 255. [DOI] [PubMed] [Google Scholar]
- Verzi MP, Agarwar P, Brown C, McCulley DJ, Schwarz JJ, Black BL. The transcription factor MEF2C is required for craniofacial development. Dev Cell. 2007;12:645–652. doi: 10.1016/j.devcel.2007.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent RW, Ryan RF, Longenecker CG. Malformation of ear associated with urogenital anomalies. Plast Reconstr Surg Transplant Bull. 1961;28:214–220. doi: 10.1097/00006534-196108000-00006. [DOI] [PubMed] [Google Scholar]
- Walker MB, Miller CT, Swartz ME, Eberhart JK, Kimmel CB. phospholipase C, beta 3 is required for Endothelin1 regulation of pharyngeal arch patterning in zebrafish. Dev Biol. 2007;304:194–207. doi: 10.1016/j.ydbio.2006.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker MB, Miller CT, Talbot JC, Stock DW, Kimmel CB. Zebrafish furin mutants reveal intricacies in regulating Endothelin1 signaling in craniofacial patterning. Dev Biol. 2006;295:194–205. doi: 10.1016/j.ydbio.2006.03.028. [DOI] [PubMed] [Google Scholar]
- Wennerberg K, Rossman KL, Der CJ. The Ras superfamily at a glance. J Cell Sci. 2005;118:843–846. doi: 10.1242/jcs.01660. [DOI] [PubMed] [Google Scholar]
- Xu D, Emoto N, Giaid A, Slaughter C, Kaw S, deWit D, Yanagisawa M. ECE-1: a membrane-bound metalloprotease that catalyzes the proteolytic activation of big endothelin-1. Cell. 1994;78:473–485. doi: 10.1016/0092-8674(94)90425-1. [DOI] [PubMed] [Google Scholar]
- Yanagisawa H, Yanagisawa M, Kapur RP, Richardson JA, Williams SC, Clouthier DE, de Wit D, Emoto N, Hammer RE. Dual genetic pathways of endothelin-mediated intercellular signaling revealed by targeted disruption of endothelin converting enzyme-1 gene. Development. 1998;125:825–836. doi: 10.1242/dev.125.5.825. [DOI] [PubMed] [Google Scholar]
- Yanagisawa M. The endothelin system: a new target for therapeutic intervention. Circulation. 1994;89:1320–1322. doi: 10.1161/01.cir.89.3.1320. [DOI] [PubMed] [Google Scholar]
- Zuniga E, Rippen M, Alexander C, Schilling TF, Crump JG. Gremlin2 regulates distinct roles of Bmp and Endothelin 1 signaling in dorsoventral patterning of the facial skeleton. Development. 2011;138:5147–5156. doi: 10.1242/dev.067785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuniga E, Stellabotte F, Crump JG. Jagged-Notch signaling ensures dorsal skeletal identity in the vertebrate face. Development. 2010;137:1843–1852. doi: 10.1242/dev.049056. [DOI] [PMC free article] [PubMed] [Google Scholar]