

Abstract
Reversible modifications of cysteine thiols play a significant role in redox signaling and regulation. A number of reversible redox modifications, including disulfide formation, S-nitrosylation, and S-glutathionylation, have been recognized for their significance in various physiological and pathological processes. Here we describe a procedure for the enrichment of peptides containing reversible cysteine modifications. Starting with tissue or cell lysate samples, all of the unmodified free thiols are blocked using N-ethylmaleimide (NEM). This is followed by the selective reduction of those cysteines bearing the reversible modification(s) of interest. The reduction is achieved by using different reducing reagents that react specifically with each type of cysteine modification (e.g., ascorbate for S-nitrosylation). This protocol serves as a general approach for enrichment of thiol-containing proteins or peptides derived from reversibly modified proteins. The approach utilizes a commercially available thiol-affinity resin (Thiopropyl Sepharose 6B) to directly capture free thiol-containing proteins through a disulfide exchange reaction followed by on-resin protein digestion and multiplexed isobaric labeling to facilitate LC–MS/MS based quantitative site-specific analysis of cysteine-based reversible modifications. The overall approach requires a simpler workflow with increased specificity compared to the commonly used biotinylation-based assays. The procedure for selective enrichment and analyses of S-nitrosylation and the level of total reversible cysteine modifications (or total oxidation) is presented to demonstrate the utility of this general strategy. The entire protocol requires approximately 3 days for sample processing with an additional day for LC-MS/MS and data analysis.
Keywords: Mass spectrometry, post-translational modification, cysteine modification, redox modification, cysteine derivatisation, on-resin digestion, on-resin reaction, thiol enrichment, S-nitrosylation, reversibly oxidized cysteines, S-glutathionylation, S-acylation, iTRAQ, isobaric tags for relative and absolute quantification, tandem mass tags, TMT, MASIC
INTRODUCTION
Protein cysteine (Cys) residues are one of the most reactive amino acid residues where the cysteine free thiol serves as an electron donor in different reactions. Cysteine thiols in proteins not only frequently participate in enzymatic reactions, but are also subjected to a variety of covalent posttranslational modifications (PTMs), which function as important mediators of redox signaling and regulation1–4. The biological significance of several types of reversible cysteine PTMs has increasingly been recognized, including disulfide formation5, S-nitrosylation (SNO)6, 7, S-glutathionylation (SSG)8 and S-sulfenylation (SOH)9, as well as the non-redox S-acylation (the reversible attachment of fatty acids through a thiol-ester group mainly known as palmitoylation10, 11). These reversible modifications have been found to modulate protein function in different biological pathways including gene transcription, metabolism, signal transduction, apoptosis, and protein trafficking and subcellular localization3, 12, 13. However, our overall knowledge of cysteine-based reversible modifications is still quite limited due to the challenges associated with isolation, site-specific identification, and quantification of these labile and dynamic modifications.
Traditionally, investigation of reversible cysteine modifications was most commonly performed by utilization of the biotin-switch technique (BST), an indirect approach initially developed for the detection of S-nitrosylated proteins6, 14, 15, and other versions of modified BST. This technique has greatly advanced redox and Cys-PTM based biology by extending its applications to other types of reversible modifications including S-glutathionylation16, 17, and reversible thiol oxidation18, as well as S-acylation10, 11. Typically, the BST and modified BST consist of the following steps: 1) initial blocking of unmodified cysteine thiols by alkylation, 2) selective reduction of modified cysteines (e.g., SNO), 3) labeling of the reduced cysteines with reagents containing a biotin tag, and 4) enrichment of labeled proteins by avidin affinity purification for biochemical (e.g., Western blotting) and proteomic analyses. When coupled with liquid chromatography-tandem mass spectrometry (LC-MS/MS), those proteins that had been modified as well as the specific cysteine residues at which the modifications occurred can be identified6, 19. Although popular, the BST is labor intensive and often suffers from the issue of non-specific binding during enrichment.
As an alternative, our laboratory has developed a resin-assisted approach for high-efficiency enrichment of cysteine-containing peptides using thiol-affinity resins20, 21. More recently, we and others have demonstrated the effectiveness of enriching several types of reversibly modified cysteine thiols using this approach22–26. Herein, we describe the detailed procedure for resin-assisted enrichment of thiols as a general strategy for site-specific profiling multiple types of reversible cysteine modifications. Figure 1a illustrates the sample preprocessing options that are mainly comprised of blocking free thiols using N-ethylmaleimide (NEM) and selectively reducing the modified cysteines to free thiols using different reducing reagents specific for each type of reversible modification. Figure 1b displays the overall workflow of enrichment and analysis following sample pre-processing. The workflow starts from direct capturing of derived free thiol-containing proteins onto the resins, followed by on-resin trypsin digestion, on-resin multiplex isobaric labeling using either 4-plex iTRAQ (isobaric tags for relative and absolute quantification)27 or 6-plex TMT (tandem mass tags)28 reagents, and final elution of the captured peptides. Labeled peptides are then subjected to LC–MS/MS for identification of specific modified cysteine residues and quantification of reversible modifications.
Figure 1.


Schematic of the pre-processing and enrichment strategy for different reversible cysteine modifications. (a) Sample pre-processing strategies for different types of reversible cysteine modifications. The free thiols are initially blocked by alkylation and the different types of modified cysteines are selectively reduced to free thiols by using individual sets of reagents. Note that the term “total oxidation” is not exact as S-acylation is not a form of oxidation; however, the level of S-acylation is negligible compared to other forms of redox modifications. Also note that the breaking of S-acylation thioester bonds by DTT may not be effective since the reaction between DTT and thioesters is a much slower reaction compared to hydroxylamine. (b) Enrichment method for quantitative analysis of reversible cysteine modifications. After preprocessing, the free thiols derived from modified cysteines are captured by Thiopropyl Sepharose resin. Enriched proteins can be directly eluted for gel electrophoresis. On-resin digestion and on-resin isobaric labeling are performed for quantitative LC MS/MS analysis.
Compared to the BST, several advantages of the resin-assisted approach are notable. First, the resin-assisted procedure offers a simpler workflow than the biotin-avidin approach by facilitating the direct capture of thiol-containing proteins followed by on-resin tryptic digestion and on-resin isobaric labeling without the need of sample clean-up prior to the MS analysis (Figure 1b). In contrast, the BST requires the introduction of biotin-tags to protein thiols prior to enrichment on an avidin column. The eluted proteins are then digested and peptides are subjected to clean-up prior to MS analysis. Second, the covalent capture process provides a higher enrichment specificity (with more than 95% final identified peptides as Cys-containing peptides)23 and better sensitivity than avidin-based enrichment as demonstrated by a side-by-side comparison22. Lastly, this approach is more effective for enabling MS-based site-specific identification and quantification of modified cysteine residues by allowing on-resin protein digestion and multiplex isobaric labeling22, 23. Therefore, this resin-assisted approach should have broad applications in redox and Cys-PTM based biology by serving as a general enrichment strategy for multiple types of reversible cysteine redox modifications (e.g., SNO, SSG, and total reversible oxidation) as well as S-acylation when coupled to selective reduction (Figure 1).
Experimental design
In this protocol, we describe the detailed procedure for the enrichment of thiols using a thiol-affinity resin (Thiopropyl Sepharose 6B) as a general method for enrichment of reversible cysteine modifications. The procedure is presented mainly in the context of analyzing SNO-modified peptides enriched from mouse muscle. The following aspects should be considered when adapting the protocol to specific applications.
Different reversible cysteine modifications
For profiling different reversible cysteine modifications, the sample pre-processing procedure, including the blocking of free thiols and selective reduction of specific types of reversible modifications (Figure 1a), will need to be optimized. A number of previous reports have demonstrated selective reduction strategies for reversible cysteine modifications. For example, ascorbate is commonly used to reduce S-nitrosylation6, 23; DTT for total oxidation18, 26 (see Box 1 for a pre-processing procedure specific to total oxidation, i.e., total reversible cysteine modifications); glutaredoxin for S-glutathionylation29, 30; and hydroxylamine for S-acylation10, 25. Detailed conditions for selective reduction and possible negative or positive controls are listed in Supplementary Table 1. These strategies can be adapted and coupled with the resin-assisted enrichment described here for profiling specific reversible modifications. Besides those modifications shown on Figure 1a, protein S-sulfhydration has recently been reported as a physiological redox modification; a modified version of the BST method was applied to detect proteins modified by S-sulfhydration31. In principle, our protocol is also applicable to S-sulfhydration; however, the blocking reagent MMTS (methyl methanethiosulfonate) was shown to react with both free thiols and persulfides32. Further studies are necessary in order to develop a specific blocking reaction scheme for this modification.
BOX 1. PRE-PROCESSING FOR ENRICHING TOTAL REVERSIBLY OXIDIZED CYSTEINES FROM RAW 264.7 CELLS ● TIMING 4–5 h.
Here we present another example of the pre-processing procedure for enriching total reversibly oxidized cysteines and total cysteines in RAW 264.7 cells. The procedure can be used to analyze the total oxidation level of protein cysteines.
Cell culture and protein extraction
-
1
Culture adherent cells until 80–90% confluent in complete cell culture media in 100-mm cell culture dishes. Add desired treatment compound for the required length of time (e.g. 0.5 mM of diamide as an oxidant for 30 min).
-
2
Place cell culture dish on ice and aspirate growth media. Gently rinse cells twice with 6 ml of ice cold media containing no supplements to remove treatment compound, dead cells, and serum. Be sure all media is removed.
-
3
Add 1 ml of 20% (vol/vol) TCA in water, and rotate plate to make sure that TCA covers entire surface. Incubate cells on ice for 30 min.
! CAUTION TCA is very corrosive. Wear gloves.
-
4
Scrape cells and place into a 2-ml centrifuge tube and centrifuge at 13,000 g at 4 °C for 10 min. Wash cell pellet with 10% and 5% (vol/vol) TCA to effectively remove the majority of TCA. Aspirate the remainder of TCA carefully not to disturb the pellet.
Alkylation of free thiols
-
5
Dissolve the protein pellet, aided by brief sonication, in 400 μl of cell lysis buffer containing 8 M urea and 1% (vol/vol) SDS. Incubate the samples in the dark at 37 °C at 850 rpm in a Thermomixer for 1.5 h.
▲CRITICAL STEP By adding cell lysis buffer that contains NEM to block all free thiols, oxidized cysteine-containing peptides can be enriched. By adding cell lysis buffer without NEM, total cysteine-containing peptides can be enriched. Make sure that NEM alkylation is performed at ~pH 7 for effective blocking of free thiols. Be sure to degas buffers by sonication, and always keep samples at 4 °C before NEM blocking. Minimize the amount of bubbles generated during cell lysis/protein extraction in order to reduce artificial oxidation of samples.
-
6
Place samples on ice and add four times the volume (1.6 ml) of cold acetone (-20 °C) to remove excess NEM.
! CAUTION Acetone is flammable. Use in a well-ventilated space.
■ PAUSE POINT Vortex samples and place at −20 °C overnight.
DTT reduction of reversibly oxidized thiols
-
7
Centrifuge samples at 13,000 g at 4 °C for 10 min. Carefully remove acetone, and rinse the pellets with 500 μl of cold (-20 °C) acetone. Air dry the samples for about 2 min. Resuspend pellet in 400 μl resuspension buffer by brief sonication.
-
8
Add 4 μl of 1 M DTT to samples at a final concentration of 10 mM DTT. Incubate sample at 37 °C at 850 rpm for 1 h.
-
9
Remove excess DTT from samples with Amicon Ultra-4 ml filter units by washing one time with 3 ml of 8 M urea and one time with 3 ml of cold water with centrifugation at 4,000 g for ~30 min at 4 °C. Adjust the final sample volume to 50 μl in the Amicon filter.
-
10
Collect the sample, and rinse the Amicon filter with 50 μl enrichment coupling buffer. Combine with the collected sample. Perform BCA assay to determine protein concentration.
-
11
Take 100 μg proteins for each enrichment and readjust the final volume to be ~120 μl by adding coupling buffer.
-
12
Add 1.2 μl of 25 mM DTT and 1.2 μl of 10% (vol/vol) SDS to the sample to make a final concentration of 0.25 mM DTT and 0.1% (vol/vol) SDS. Transfer the sample to the preconditioned resin for enrichment, and continue with step 20 of the main procedure.
Protein versus peptide level enrichment
In principle, thiol-affinity enrichment can be performed at either the protein or peptide level. We have previously shown that both protein- and peptide-level enrichment provide comparable specificity and coverage of enriched cysteine-containing peptides23. In general, protein-level enrichment is simpler in its procedure; however, peptide-level enrichment has an advantage in terms of resin-binding capacity. If a large amount of starting materials is desired for a given experiment or the proteins are difficult to dissolve in the required reaction volume for capture, peptide-level enrichment may be preferred (see Box 2 for a procedure specific to peptide level enrichment).
BOX 2. PEPTIDE-LEVEL ENRICHMENT.
The protocol can also be adapted for peptide-level enrichment. Instead of directly protein-level capture, proteins with their free thiols blocked are subjected to in-solution trypsin digestion at a ratio of 1:50 (wt:wt, trypsin:protein). After the peptide samples are transferred to the spin column containing the preconditioned-resin (Step 20 in the PROCEDURE), peptides are selectively reduced using reducing reagents (e.g., ascorbate for SNO) and captured by the thiol-affinity resin. The enrichment protocol including pre-conditioning, selectively reduction, washing, on-resin isobaric labeling, and elution is essentially the same as the protein-level enrichment as described in the PROCEDURE.
Analysis by Western blot instead of LC-MS
This enrichment protocol can also be used for Western blotting applications to characterize specific cysteine-modified proteins by directly eluting the captured proteins from the resins.
Controls and replicates
Controls tailored for the modification in question are often necessary in the study of specific reversible modifications. In the case of SNO, controls may include negative control by SNO-specific UV photolysis6, 33, 34, and positive control treated with NO (nitric oxide) donors15. For other types of reversible modifications, similar control experiments should be employed (See Supplementary Table 1). UV-photolysis is considered as a superior method for generating a negative control without introducing transition metals such as CuCl. While the coupling of CuCl and ascorbate led to more efficient reduction of SNO23, the addition of transition metals to the BST remains controversial due to the potential of inducing artifacts33, 34. Biological replicates should also be included to ensure the confidence and consistency of the quantification. The multiplexed nature of isobaric labeling (iTRAQ or TMT) often facilitates the inclusion of several replicates in a single experiment.
Isotopic labeling strategy
For proteomic quantification of reversible modifications, different isotopic labeling strategies or label-free quantification may be applied depending on the applications. On-resin multiplex isobaric labeling (iTRAQ) is presented here because of the convenience of on-resin amine-reactive labeling and the multiplexing capability. Mass spectrometers capable of detecting low m/z range reporter ions of iTRAQ are needed for this approach. Alternatively, one can apply other isobaric labeling techniques (e.g., TMT), stable isotope labeling by amino acids in cell culture (SILAC)35, or label-free quantification26.
Limitations
Despite the advantages of this resin-assisted approach, its limitations are similar to those of the BST due to the nature of indirect detection for reversible PTMs. First, the efficiency and specificity of the free thiol blocking and selective reduction steps are critical for the confident identification of modified cysteine peptides. It is nearly unavoidable that a small portion of the identified cysteine-containing peptides may be false positives for any given type of modification; for example, disulfide formation can be falsely identified as SNO modification due to the imperfect specificity of ascorbate reduction23. However, well-designed controls (See Supplementary Table 1) and reliable quantification can help to effectively distinguish such potential artifacts34, 36, 37. Second, the sensitivity of this approach may still not be sufficient for effective detection of low-abundance endogenous redox modifications (e.g., SNO) in cells or tissues. If necessary, a positive control with elevated levels of modifications (e.g., treated with NO donors for SNO) can be included in a multiplex quantification experiment to increase the coverage of reversibly modified cysteine sites22, 23. Despite the potential limitations, the resin-assisted approach represents a unique method to identify novel cysteine sites sensitive to redox or other reversible modifications. Such discovery results could provide important guidance for functional studies of individual proteins by other orthogonal methods such as site-mutagenesis.
MATERIALS
REAGENTS
Phosphate buffered saline (PBS; Sigma-Aldrich, cat. no. P7059)
HEPES (Sigma-Aldrich, cat. no. H4034)
Neocuproine (Sigma-Aldrich, cat. no. N1501)
EDTA (Sigma-Aldrich, cat. no. E5134)
Triton X-100 (Sigma-Aldrich, cat. no. T8787)
Protease inhibitor cocktail (EMD Millipore, cat. no. 539134)
Dimethyl sulfoxide (DMSO; Sigma-Aldrich, cat. no. 276855)
S-Nitrosoglutathione (GSNO; Sigma-Aldrich, cat. no. N4148)
Sodium ascorbate (Sigma-Aldrich, cat. no. 11140)
CuCl (Sigma-Aldrich, cat. no. 212946) ! CAUTION Toxic, wear gloves.
N-ethylmaleimide (NEM; Sigma-Aldrich, cat. no. 04259)
Urea (Sigma-Aldrich, cat. no. U5378)
SDS (Sigma-Aldrich, cat. no. L6026)
Acetone (Fisher Scientific, cat. no. A949-1) ! CAUTION Flammable, use in a vented space.
Thiopropyl Sepharose 6B resin (GE Healthcare, cat. no. 17-0420-01)
BCA (bicinchoninic acid) protein assay reagent (Thermo Scientific, cat. no. 23227)
Sodium chloride (Sigma-Aldrich, cat. no. S3014)
Acetonitrile (ACN; Sigma-Aldrich, cat. no. 34998)
Trifluoroacetic acid (TFA; Sigma-Aldrich, cat. no. T6508) ! CAUTION Corrosive, prepare TFA solutions in a fume hood.
CaCl2 (Sigma-Aldrich, cat. no. C3306)
Trypsin (Promega, cat. no. V5280)
iTRAQ reagents kit (AB Sciex, cat. no. 4352135)
Triethylammonium bicarbonate buffer (TEAB; Sigma-Aldrich, cat. no. T7408)
Hydroxylamine (NH2OH, 50% (wt/wt) in water; Sigma-Aldrich, cat. no. 467804)
Dithiothreitol (DTT, Thermo Scientific, cat. no. 20291)
Ammonium bicarbonate (NH4HCO3; Sigma-Aldrich, cat. no. 09830)
Tris/Glycine/SDS buffer (Bio-Rad, cat. no. 161-0772)
Laemlli sample buffer (Bio-Rad, cat. no. 161-0737)
TCEP Solution (Thermo Scientific, cat. no. 77720)
Silver staining kit (Thermo Scientific, cat. no. 24612)
RPMI 1640 (Gibco, cat. no. 11835-030)
Fetal bovine serum (FBS; Atlanta Biologicals, cat. no. S12450)
Penicillin-Streptomycin (Gibco, cat. no. 15140)
L-Glutamine (Gibco, cat. no. 25030)
L-Cysteine (Sigma Aldrich, cat. no. 30089)
Sodium nitrite (NaNO2; Sigma Aldrich, cat. no. S-2252)
Hydrochloric acid solution (Fisher, cat. no. SA48-4)
Hank’s balanced salt solution (HBSS; Sigma Aldrich, cat. no. 55021C)
Diamide (Sigma Aldrich, cat. no. D3648)
Formic acid (Thermo Scientific, cat. no. 28905)
EQUIPMENT
Tissue-Tearor homogenizer (BioSpec Products, model #985370)
UV lamp Blak-Ray (Ultra-Violet Products)
Vortex mixer (Vortex-Genie 2)
Sonicator UTR200 (Hielscher)
Eppendorf centrifuge 5810R (Eppendorf)
Eppendorf centrifuge 5424 (Eppendorf) (only for washing the resin)
Thermomixer R (Thermo Scientific)
SpeedVac SC110 (Savant)
Round bottom tubes 5 ml polystyrene (BD Biosciences)
Microcentrifuge tubes (Fisherbrand)
Amicon Ultra-4 ml centrifugal filter units (10K molecular weight cut-off; Millipore)
Spin columns - screw cap (Fisher Scientific)
Precast 4–20% (vol/vol) Tris-HCl gradient gel (Bio-Rad)
Mini gel electrophoresis system (Bio-Rad)
Culture dish 100-mm (BD Falcon)
OMIX C18 Zip-Tip (Agilent Technologies)
LTQ-Orbitrap Velos mass spectrometer (Thermo Scientific) equipped with a Waters nanoACQUITY UPLC® system
In-house packed C18 column (65 cm × 75 μm id, 3-μm Jupiter C18 particles)
REAGENT SETUP
<CRITICAL> Most buffers can be stored at room temperature (20–25 °C) up to 1 month unless otherwise specified.
Mouse muscle tissue This procedure works well with quadriceps femoris muscle. In our lab, this was freshly isolated or stored at −80 °C (for a maximum of 12 months without problems). Other animal tissues may be similarly analyzed and ~1 mg starting protein amount is often required.
! CAUTION For animal studies, it is necessary to obtain permission and adhere to the appropriate institutional and governmental guidelines for all procedures.
RAW 264.7 cells In our experiments we have used Murine RAW 264.7 macrophages cultured with RPMI-1640 media. Other cultured cells may be similarly analyzed and 2–5 millions of cells are often required depending on the protein recovery for a given cell type.
Homogenization buffer 250 mM HEPES, 10 mM EDTA, 0.1 mM neocuproine, 1% (vol/vol) Triton X-100, pH 7.0, add protease inhibitor cocktail just before use.
SNO reaction buffer 250 mM HEPES, 1 mM EDTA, 0.1 mM neocuproine, pH 7.7
GSNO stock solution 10 mM GSNO in DMSO (20-μl aliquots, stored at −80 °C)
SNO reduction reagent stock solutions (1) 500 mM sodium ascorbate; (2) 500 μM CuCl (freshly prepared) ! CAUTION CuCl is toxic.
SNO reduction buffer 250 mM HEPES, pH 7.7
NEM blocking buffer 100 mM NEM, 2% (vol/vol) SDS, 250 mM HEPES, 10 mM EDTA, 0.1 mM neocuproine, pH 7.7
▲CRITICAL STEP NEM buffer should be freshly prepared immediately before the alkylation reaction.
Resuspension buffer 250 mM HEPES, 8 M urea, 0.1% (vol/vol) SDS; pH 7.7 (Add urea before use)
Enrichment coupling buffer 50 mM HEPES, 1 mM EDTA, pH 7.5
▲CRITICAL STEP It is recommended to degas the coupling buffer before the enrichment experiments.
Enrichment washing buffers (1) 8 M urea (freshly prepared), (2) 2 M NaCl, (3) 80% (vol/vol) ACN/0.1% (vol/vol) TFA, (4) 25 mM HEPES (pH 7.7)
Digestion buffer 25 mM HEPES buffer (pH 7.7), 0.1% (vol/vol) SDS, 1 mM CaCl2
Enrichment eluting buffer (1) 20 mM DTT in 25 mM ammonium bicarbonate (freshly made), (2) 80% (vol/vol) ACN/0.1% (vol/vol) TFA
C18 ZipTip clean-up buffers Starting buffer: 0.2% (vol/vol) TFA/H2O; Conditioning buffer: 100% (vol/vol) ACN; Equilibration buffer: 0.1% (vol/vol) TFA/H2O; Washing buffer: 5 % (vol/vol) ACN/0.1% (vol/vol) TFA; Elution buffer: 80% (vol/vol) ACN/0.1% (vol/vol) TFA
Cell culture media RPMI 1640, 10% (vol/vol) FBS, 100 U/ml penicillin, 100 μg/ml streptomycin, 2 mM L-glutamine ((Store at 4 °C, and warm to room temperature before use)
Cell lysis buffer 250 mM HEPES, 10 mM EDTA, 0.1 mM neocuproine, 1% (vol/vol) Triton X-100, 100 mM NEM, pH 7.0 (Add NEM just before use)
Nitrosocysteine (CysNO) stock solution Mix equal volumes of 200 mM cysteine in 1 M HCl with 200 mM NaNO2 in H2O at room temperature in the dark. Determine the concentration of CysNO immediately by spectrometry with absorbance at 334 nm and the extinction coefficient 900 M−1 cm−1. Neutralize by adding an equal amount of SNO reaction buffer to make ~50 mM CysNO stock solution. Dilute CysNO in HBSS with 10 mM HEPES to a 1 mM CysNO solution for immediate application to cells.
▲CRITICAL STEP Once diluted in HBSS, the CysNO solution is not stable and should be used immediately.
EQUIPMENT SETUP
LC MS/MS analysis
Different LC-MS/MS systems or settings can be used to analyze peptide samples. The coverage and sensitivity of peptide identifications will depend on the particular system and settings employed. When analyzing isobaric labeled peptide samples, HCD is necessary in order to obtain reporter ion intensity information38.
In our lab, we analyze peptide samples using an in-house developed nanoLC system (or a commercially available nanoLC system such as Waters nanoAquity UPLC®) coupled to an LTQ-Orbitrap Velos mass spectrometer via an electrospray ionization interface using chemically etched electrospray emitters (150-mm o.d. × 20-mm i.d.)39. The experiment requires a reversed phase C18 column (75 μm i.d. and 20–40 cm long) containing 3-μm C18 particles. In our lab, the HPLC system consists of a custom configuration of 100-ml Isco Model 100DM syringe pumps, two-position Valco valves, and a PAL autosampler40. For the experiment, operate the HPLC at a constant pressure (10,000 psi) with a gradient over the course of 100 min starting with 100% (vol/vol) of mobile phase A (0.1% (vol/vol) formic acid in nano-pure water) to 60% (vol/vol) of mobile phase B (0.1% (vol/vol) formic acid in ACN).
For MS analysis, record full MS spectra at a resolution of 30K (for ions at m/z 400) over the range of m/z 400–2,000 with an automated gain control (AGC) value of 1× 106. Data-dependent acquisition mode is used for MS/MS with an AGC target value of 3 × 104. Select the top six abundant parent ions for MS/MS using high-energy collisional dissociation (HCD) with 40% normalized collision energy and a resolution of 7,500. Perform precursor ion activation with an isolation width of 2 Da, a minimal intensity of 500 counts, and activation time of 10 ms. Use a dynamic exclusion time of 45 s.
PROCEDURE
Preparation of protein samples ● TIMING 4–5 h
-
1|
Cut frozen mouse quadriceps femoris muscle into 4 pieces, approximately 35 mg wet weight for each piece.
-
2|
Wash the tissue by rinsing twice with cold PBS. Mince muscle with a razor blade on a glass microscopy slide on ice.
-
3|
Transfer each piece of minced tissue to a 5 ml round bottom tube and homogenize in 500 μl of homogenization buffer at 13,000 rpm for approximately 1 minute, or until tissue is completely homogenized, using a tissue homogenizer. Transfer homogenate to a new 1.5 ml microcentrifuge tube.
-
4|
Centrifuge the homogenate at 10,000 g for 10 min at 4 °C and transfer supernatant to a new 1.5 ml microcentrifuge tube. Measure the protein concentration using the BCA assay.
-
5|
Dilute the muscle homogenates in SNO reaction buffer. Prepare four identical protein samples with 500 μg proteins in 1 ml volume for each sample.
▲CRITICAL STEP To minimize the photodecomposition of SNO during sample preparation, perform above steps at 4 °C under restrained light.
-
6|
(Optional) Induce S-nitrosylation (steps 6–8) To induce S-nitrosylation of proteins, add 10 μl DMSO (control), 1 μl of 10 mM GSNO, and 10 μl of 10 mM GSNO to three protein samples to obtain final concentrations of 0 μM, 10 μM, and 100 μM GSNO, respectively. Incubate at 37 °C with shaking at 800 rpm for 30 min. Cool down the samples to 4 °C after incubation to stop the reaction and minimize the alteration of thiol modifications.
▲ CRITICAL STEP When endogenous SNOs are of interest, steps 6–8 for inducing SNO can be omitted. The tissue can be directly homogenized in homogenization buffer followed by immediate blocking free thiols by adding NEM and SDS to reach a final concentration of 100 mM NEM and 2% (vol/vol) SDS. You would then continue to step 10.
-
7|
Prepare a negative control by adding SNO reduction buffer (5 mM sodium ascorbate and 5 μM CuCl) to one protein sample. Note that sodium ascorbate/CuCl reduces endogenous SNO before NEM alkylation.
▲CRITICAL STEP Negative control can also be generated by placing protein samples on ice in a 60-mm petri dish with 60 min exposure to a top-down UV transilluminator (365 nm) as previously described6, or by incubating the samples 2 cm from a 200-watt UV-visible mercury vapor lamp for 3 min (~4 watts of radiation from ~280–500 nm)33.
-
8|
Remove the excess GSNO by transferring the samples to 4 ml Amicon filters and by performing buffer exchange three times at 4 °C with 4 ml total volume of cold water. Centrifuge at 4,000 g for 15–30 min each time. The final sample volume is ~50 μl in the Amicon filter.
Blocking free thiols by alkylation ● TIMING 2.5 h
-
9|
Add 500 μl of NEM blocking buffer to samples. Incubate the sample in dark at 55 °C with shaking at 300 rpm in a Thermomixer for 30 min.
-
10|
Remove the excess NEM by buffer exchange using 4-ml Amicon filters with 3 ml of 8 M urea three times and with 3 ml of cold water once. Each time the filter is centrifuged at 4,000 g for 15–30 min at 4 °C. Concentrate the final sample to approximately 50 μl.
▲CRITICAL STEP A complete removal of NEM is critical for enriching modified thiols. For samples with low levels of reduced thiol-containing proteins (e.g., endogenous S-nitrosylated proteins), acetone precipitation is recommended as an alternative method to completely remove excess NEM.
? TROUBLESHOOTING
-
11|
Transfer the sample to a 0.6-ml microcentrifuge tube. Rinse the filter with 50 μl of 50 mM HEPES (pH 7.7) twice and combine the solution with the concentrated samples. Add 1 μl of 20% (vol/vol) SDS to the sample solution to reach a final SDS concentration of ~0.2%.
■ PAUSE POINT The protein samples can be stored at −80 °C overnight.
Pre-conditioning the Thiopropyl Sepharose resin ● TIMING 1 h
-
12|
Weigh 35 mg of Thiopropyl Sepharose 6B resin for each sample to be enriched and place in a 1.5-ml tube.
-
13|
Add 1 ml of water to each tube to rehydrate the resin for 15 min at room temperature.
-
14|
Cut 5 mm off the end of 1-ml pipette tip to make the hole larger for easier mixing. Use the modified pipette tip to resuspend the resin well and leave the tubes for another 10 min at room temperature. Note that the resin swells and forms chunks after rehydration. The final volume of rehydrated resin is ~100 μl.
-
15|
Remove and discard the upper 0.5 ml of water. Carefully resuspend the settled resin and transfer the suspension to a 1-ml spin column using the modified tip.
-
16|
Place the spin column in a 2-ml receiving tube and centrifuge at 1,000 g for 30 s to remove the water. Repeat this step by washing the resin 5 times with water and 5 times with coupling buffer.
▲CRITICAL STEP To minimize the thiol oxidation, degas the coupling buffer by sonication for 20 min before starting the enrichment experiment.
-
17|
Put the bottom plug and top cap on the spin column after the last wash. Keep the resin temporarily at 4 °C until the protein samples are prepared.
▲CRITICAL STEP Be sure to resuspend the resin when washing with coupling or washing buffer.
Selective reduction of S-nitrosylation ● TIMING 0.5 h
-
18|
Centrifuge the samples (from step 11) at 10,000 g for 2 min to remove any undissolved debris. Determine the protein concentration of the supernatant by the BCA assay. Adjust the volume of samples to ~100 μl with 50 mM HEPES (pH 7.7) so that protein concentrations are equal (400–500 μg) for each enrichment.
-
19|
Add 500 mM sodium ascorbate and 500 μM CuCl to each protein sample to reach a final concentration of 5 mM sodium ascorbate and 5 μM CuCl.
▲ CRITICAL STEP As an alternative strategy, peptide-level enrichment can be performed. Prior to reducing protein samples with sodium ascorbate/CuCl, proteins can be digested into peptides followed by SNO reduction for peptide-level enrichment (see BOX 2).
▲CRITICAL STEP Note that the procedure described here is for the enrichment and identification of SNO-modifications from mouse muscle treated with an NO donor as the primary example of profiling reversible cysteine modifications. BOX 1 presents another example of the pre-processing procedure for enriching reversibly oxidized cysteines in RAW 264.7 cells.
Enrichment of free thiol-containing proteins ● TIMING ~3 h
-
20|
Transfer each pre-processed (reduced) protein sample to the spin column that contains preconditioned resin with the bottom plug and top cap in place. Place each spin column into a 2-ml tube and incubate at room temperature with shaking at 800 rpm for 2 h.
? TROUBLESHOOTING
▲CRITICAL STEP Make sure the bottom plug is tightly fitted in the column to prevent sample leakage during incubation with shaking. We recommend checking for any potential leakage after 5 min of incubation.
-
21|
Place the spin column into a new 1.5-ml tube with both top cap and bottom plug removed. Spin at 1,500g for 1 min to collect the unbound portion (the proteins without free thiols or without formerly oxidized thiols).
▲CRITICAL STEP The unbound portion can be used for a second enrichment to ensure the resin capacity is sufficient for a complete enrichment.
-
22|
Thoroughly wash the resin five times with each of the enrichment washing buffers in the following order: 8 M urea; 2 M NaCl; 80% (vol/vol) ACN and 0.1% (vol/vol) TFA; and 25 mM HEPES. Use 0.5 ml buffer for each wash and replace both the top cap and bottom plug of the column after the last wash. The resin is washed 20 times in this step. ▲CRITICAL STEP Be sure to resuspend the resin during each wash step. Instead of on-resin digestion, the enriched proteins can be directly eluted (see step 32) for analyses using gel electrophoresis to evaluate protein-level enrichment and Western blot for measuring the levels of thiol modifications on a specific protein.
On-resin tryptic digestion ● TIMING 4 h
-
23|
Add 120 μl of digestion buffer containing 25 mM HEPES buffer (pH 7.7), 0.1% (vol/vol) SDS, 1 mM CaCl2, and 8 μg trypsin to each spin column. Close the top cap of each column. Incubate at 37 °C with shaking at 850 rpm for 3 h.
▲CRITICAL STEP This protocol is optimized using Promega Trypsin Gold. The digestion time may be different depending on the enzymes used. Digestion performance can be evaluated by eluting enriched peptides and running gel electrophoresis.
-
24|
Place the spin column into a new 1.5-ml tube with both top cap and bottom plug removed, and spin at 1,500 g for 1 min to collect the unbound portion (the non-cysteinyl peptides).
-
25|
Thoroughly wash the resin five times with each of the enrichment washing buffers: 1) 8 M urea, 2) 2 M NaCl, 3) 80% (vol/vol) ACN and 0.1% (vol/vol) TFA, and 4) 25 mM HEPES. Use 0.5 ml buffer for each wash and replace both the top cap and bottom plug of the column after the last wash.
▲CRITICAL STEP The last wash should be free of primary or secondary amines (e.g. Tris), while tertiary/quaternary amines such as HEPES or TEAB are suitable since they are not reactive towards iTRAQ or TMT reagents. TEAB is the dissolution buffer for iTRAQ or TMT labeling per manufacturer’s instruction.
■ PAUSE POINT Resin may be stored at 4 °C overnight.
On-resin isobaric labeling ● TIMING 3 h
-
26|
Warm one set of manufacturer-provided iTRAQ reagents, anhydrous ethanol, and dissolution buffer to room temperature.
-
27|
Add 70 μl of anhydrous ethanol to each iTRAQ reagent tube with gentle vortexing for 1 min. Spin the tubes at 1,000 g for 10 s to collect the reagents at the bottom of the tubes.
-
28|
Add 30 μl of manufacturer-provided dissolution buffer and 70 μl of iTRAQ reagents to each spin column. Close the top cap of the column. Incubate at room temperature with shaking at 850 rpm for 1 h to label all peptides on the resin.
? TROUBLESHOOTING
-
29|
Quench excess iTRAQ reagents by adding 8 μl of 5% (wt/wt) NH2OH in 200 mM TEAB to each column. Incubate at room temperature with shaking at 850 rpm for 15 min.
-
30|
Place the spin column in a new 2-ml tube with both top cap and bottom plug removed and spin at 1,500g for 1 min to remove the excess iTRAQ reagents.
-
31|
Wash the resin 5 times with 80% (vol/vol) ACN/0.1% (vol/vol) TFA, and 5 times with 25 mM NH4HCO3 (pH 7.8).
Elution of enriched cysteine-containing peptides ● TIMING 4–5 h
-
32|
Add 100 μl of 25 mM NH4HCO3 buffer containing 20 mM DTT to each column. Incubate at room temperature with shaking at 850 rpm for 30 min with both top cap and bottom plug in place.
-
33|
Use 2-ml receiving tubes to collect the eluted cysteine-containing peptides at 1,500g for 1 min with both top cap and bottom plug removed.
-
34|
Replace the bottom plug of the column. Add another 100 μl of 20 mM DTT elution buffer to resin, and mix with a pipet tip. Close the top cap of the column. Incubate at room temperature with shaking at 850 rpm for 10 min. Collect the eluted peptides in the same receiving tube at 1,500g for 1 min with both top cap and bottom plug removed. Repeat this step once.
▲CRITICAL STEP The resin tends to clump after DTT incubation because of its structural changes. We recommend using a pipette tip to stir the bead slurry after low-speed centrifugation.
-
35|
Wash the resin again with 100 μl of 80% (vol/vol) ACN/0.1% (vol/vol) TFA to collect the residue peptides to the same tube.
▲CRITICAL STEP Before combining the samples for LC-MS/MS analysis, an aliquot of each sample (50 μl each) can be dried and separated by gel electrophoresis to evaluate and verify the performance of overall enrichment.
-
36|
Combine all the samples from each iTRAQ-labeling tube. Dry down the sample in a SpeedVac to a reduced volume of ~15 μl.
-
37|
Clean up the combined iTRAQ-labeled samples with C18 Zip-Tips. Add 15 μl of 0.2% (vol/vol) TFA to the sample to make a final concentration of 0.1% (vol/vol) TFA. Condition the C18 ZipTip 3 times with 20 μl of ACN and 3 times with 20 μl of 0.1% (vol/vol) TFA. Load the sample to the ZipTip by 10 cycles of aspiration and dispensing. Wash samples 4 times with 5% ACN (vol/vol)/0.1% (vol/vol) TFA. Elute samples with 30 μl 80% (vol/vol) ACN/0.1% (vol/vol) TFA. Repeat the cleanup steps by using a new ZipTip to ensure maximum recovery of sample. Dry down the cleaned-up sample using a SpeedVac to a reduced volume of ~5 μl.
-
38|
Add 20 μl of 20 mM DTT to the final sample to prevent further oxidation of cysteine-containing peptides prior to LC-MS/MS analysis.
▲CRITICAL STEP The reducing reagent DTT is included in the final sample to prevent further thiol oxidation of the eluted cysteine-containing peptides prior to LC-MS/MS analysis.
■ PAUSE POINT Samples can be stored at −80 °C (stable for months) until LC MS/MS analysis.
SDS-PAGE gel electrophoresis and silver stain (optional) ● TIMING ~3 h
-
39|
Add 2x Laemlli sample buffer to the enriched samples (5 μl/sample) at 1:1 (vol/vol) dilution with 5 mM TCEP. Heat samples at 95 °C for 5 min. Run equal amounts of protein on a 4–20% (vol/vol) Tris-HCl gradient gel for 40 min at 170 V. Use silver staining to detect proteins/peptides.
? TROUBLESHOOTING
LC-MS/MS analysis ● TIMING ~3–5 h
-
40|
Inject 7 μl of the labeled peptide sample onto the LC column for LC MS/MS analysis. Run two technical replicates for each sample. See EQUIPMENT SETUP for details about the instrument configuration and settings.
Data analysis ● TIMING ~1 d
-
41|
Convert LC–MS/MS raw data into dta files using Extract_MSn in Bioworks 3.2. Extract iTRAQ reporter ions using MASIC, an in-house developed software (available at panomics.pnnl.gov).
-
42|
Search the tandem mass spectra (MS/MS) against a protein FASTA database (UniProt mouse database) using a database searching algorithm (e.g., Sequest)41. Set key searching parameters as follows: 50 ppm tolerance for precursor ion masses, 0.05 Da for fragment ion masses, a maximum of 2 missed tryptic cleavages, dynamic oxidation of methionine (+15.9949 Da), dynamic NEM modification of cysteine (+125.0477 Da), and static iTRAQ modification of lysine and N-termini (+144.1021 Da). Enable the decoy-database searching to limit the final false discovery rate at the unique peptide level to <1%42, 43. For filtering peptide identifications, set the mass measurement error within 5 ppm, the mass spectrometry generating function (MS-GF) score44 <1×10−8 for fully tryptic peptides, and the MS-GF score <1×10−10 for partially tryptic peptides.
-
43|
Link the final identified MS/MS spectra with the iTRAQ reporter ion intensity data and group the data at the unique peptide level by summing the reporter ion intensities for all spectra that identify the same peptide.
● TIMING
Step 1–8, Preparation of protein samples: 4–5 h
Step 9–11, Blocking free thiols by alkylation: 2.5 h
Step 12–17, Pre-conditioning the Thiopropyl Sepharose resin: 1 h
Step 18–19, Selective reduction of S-nitrosylation: 0.5 h
Step 20–22, Enrichment of free thiol-containing proteins: ~3 h
Step 23–25, On-resin tryptic digestion: 4 h
Step 26–31, On-resin isobaric labeling: 3 h
Step 32–38, Elution of enriched cysteine-containing peptides: 4–5 h
Step 39, SDS-PAGE gel electrophoresis and silver stain: ~3 h (optional)
Step 40, LC MS/MS analysis: ~3–5 h
Step 41–43, Data analysis: ~1 d
ANTICIPATED RESULTS
In a successful resin-assisted enrichment experiment, >95% of the final identified peptides should be cysteine-containing modified peptides based on the high specificity of this approach. The multiplexed quantification data that includes negative or positive controls will allow the discrimination of redox-sensitive cysteine sites from potential artifacts. In the example of quantitative reactivity profiling of S-nitrosylation in mouse muscle (Figure 2a), the levels of SNO on individual cysteine sites induced by different doses of NO donors (i.e., 10 μM and 100 μM of GSNO) were quantified based on the iTRAQ reporter ion intensities23. In this SNO reactivity profiling experiment, ~670 unique peptides were identified as potentially S-nitrosylated, covering 488 cysteine sites and 197 proteins. Figure 2b and 2c depicts an example of MS/MS fragmentation of the identified SNO-modified peptide from sarcoplasmic/endoplasmic reticulum calcium ATPase 1 (SERCA1) and the corresponding increases in SNO levels in response to GSNO treatments are displayed as normalized reporter ion intensities. Notably, Cys-12 of SERCA1 was reported to be susceptible to oxidation45, and our findings confirmed the presence of S-nitrosylation on Cys-12 and fifteen other cysteines in SERCA1.
Figure 2.


Enrichment of SNO-modified peptides from mouse muscle cells. (a) Experimental design and workflow with 4-plex iTRAQ labeling (114–117 m/z) for LC MS/MS analysis. (b) An MS/MS spectrum of the Cys-peptide #STEECLSYFGVSETTGLTPDQVK# from SERCA1 (UniProt:AT2A1_Mouse). The symbol # stands for the iTRAQ label on N-terminus and lysine. (c) The zoom-in spectrum of the reporter-ion region showing the increases of SNO levels on the identified peptide in response to GSNO treatment.
Figure 3a shows another example of S-nitrosylation in RAW 264.7 macrophage cells induced by different concentrations of CysNO. The displayed gel image of eluted proteins enriched by this protocol confirms the specificity of the enrichment as evident by the low background of the untreated sample, the induction of SNO by CysNO, as well as the photolysis of SNO by UV exposure following SNO induction. In another experiment, this protocol was also applied for profiling the level of total oxidation on individual cysteine residues where DTT was used to reduce all forms of reversibly oxidized thiols after the initial blocking of free thiols. The displayed gel image on Figure 3b confirms the increased levels of the final enriched oxidized cysteine-containing peptides (eluted from the resin after on-resin digestion and iTRAQ labeling) in response to diamide treatments. Following LC-MS/MS analysis of iTRAQ-labeled peptides, the average stoichiometry of thiol oxidation across ~1600 identified unique cysteine-containing peptides in this experiment (See Supplementary data 1) was presented in Figure 3c. Both Figure 3b and 3c support that the average level of thiol oxidation is relatively low in the untreated sample; however it increases significantly with low doses of diamide treatments. The results are consistent with a recent report of in vivo thiol oxidation46. These results confirm the effectiveness of this enrichment method for profiling the level of total cysteine oxidation. Similarly, the utility of this protocol for profiling S-acylation has been recently demonstrated25. Taken together, these results clearly illustrate the specificity and efficiency of the resin-assisted thiol-affinity approach and its general utility for quantitative profiling of reversible modifications on cysteine thiols.
Figure 3.
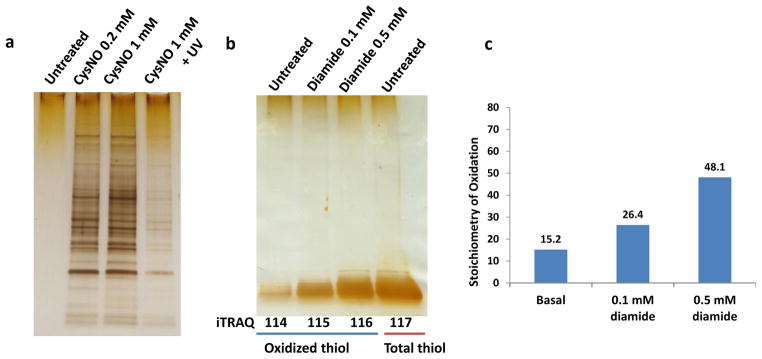
(a) SDS-gel image of enriched SNO-modified proteins from RAW 264.7 cells treated with CysNO for 15 min. UV exposure was performed in a 60 mm petri dish, 6 cm from a 15-watt UV lamp (λmax 365 nm) for 15 min. (b) SDS-gel image of enriched oxidized cysteine-containing peptides (the first 3 lanes) treated with diamide (0 mM, 0.1 mM and 0.5 mM) for 30 min and the total cysteine-containing peptides (lane 4) from RAW cells. (c) Stoichiometry of oxidation as measured by LC-MS/MS of the iTRAQ-labeled samples. Plotted here is the average stoichiometry across all identified peptides under different conditions. The stoichiometry of oxidation of each peptide was calculated as a percentage by dividing the reporter ion intensity for each channel (oxidized thiol) against the intensity of total thiol channel (enriched without NEM blocking).
Supplementary Material
Supplementary Table 1: Conditions for selective reduction, negative and positive controls for multiple reversible cysteine modifications
Supplemental Table 2: LC-MS/MS quantification data of total cysteine oxidation levels in RAW cells treated with 0.1 mM and 0.5 mM diamide
TABLE 1.
Troubleshooting table.
| Step | Problem | Possible reason | Solution |
|---|---|---|---|
| 20 | Low enrichment specificity (less than 95% of peptides are cysteine- containing peptides) | Insufficient resin wash before elution Very few modified Cys-peptides present in starting material |
Be sure the resin is well resuspended between washing steps Starting with more proteins before capture by the resin |
| 28 | Low iTRAQ labeling efficiency | Other amines present in the sample Inappropriate volume ratio of organic solvent to aqueous solution |
Washing the resin with amine-free buffers before labeling Keep the labeling solution volume at ~100 μl (70 μl ethanol and 30 μl dissolution buffer) because the iTRAQ is not stable in aqueous solution |
| 39 | Multiple bands on gel with molecular weights >6 kD for the eluted peptides after on-resin digestion Similar band intensities on peptide-gel between oxidized Cys and total Cys Low band intensities for all conditions |
Insufficient on-resin digestion Ineffective NEM blocking of free thiols before enrichment Residual NEM reacting with the reduced thiols |
Use more trypsin for digestion (A weak trypsin band may be visible at ~25 kD) Add NEM during cell lysis or tissue homogenization, or perform TCA precipitation to trap free thiols before alkylation Make sure buffer exchange is sufficient to remove excess NEM; recommend the use of acetone precipitation for samples with low-levels of reduced thiols |
Acknowledgments
Portions of this work were supported by the NIH Director’s New Innovator Award Program DP2OD006668 and a DOE Early Career Research Award (to W.J.Q.), NIH P41 GM103493 (to R.D.S.), and the DOE Office of Biological and Environmental Research Genome Sciences Program under the Pan-omics project. Experimental work was performed in the Environmental Molecular Science Laboratory, a DOE/BER national scientific user facility at PNNL in Richland, Washington. PNNL is operated by Battelle for the DOE under contract DE-AC05-76RLO-1830.
Footnotes
AUTHER CONTRIBUTIONS
J.G., M.J.G., and D.S. performed the experiments and optimized the protocol; T.L. developed the initial enrichment method; D.G.C. and R.D.S. contributed to development of the measurement capabilities used; W.J.Q. conceived and supervised the project. J.G., M.J.G., and W.J.Q. wrote the manuscript.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
Ontology:
Biological sciences / Cell biology / Post-translational modifications / Nitrosylation
Biological sciences / Biochemistry / Proteomics
Biological sciences / Biological techniques / Isolation, separation, purification / Protein enrichment
Biological sciences / Biological techniques / Spectroscopy / Mass spectrometry
Contributor Information
Jia Guo, Email: Jia.Guo@pnnl.gov.
Matthew J. Gaffrey, Email: Matthew.Gaffrey@pnnl.gov.
Dian Su, Email: su.dian@gene.com.
Tao Liu, Email: tao.liu@pnnl.gov.
David G. Camp, II, Email: dave.camp@pnnl.gov.
Richard D. Smith, Email: dick.smith@pnnl.gov.
Wei-Jun Qian, Email: Weijun.qian@pnnl.gov.
References
- 1.Giron P, Dayon L, Sanchez JC. Cysteine tagging for MS-based proteomics. Mass Spectrom Rev. 2011;30:366–395. doi: 10.1002/mas.20285. [DOI] [PubMed] [Google Scholar]
- 2.Held JM, Gibson BW. Regulatory control or oxidative damage? Proteomic approaches to interrogate the role of cysteine oxidation status in biological processes. Mol Cell Proteomics. 2012;11:R111 013037. doi: 10.1074/mcp.R111.013037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Antelmann H, Helmann JD. Thiol-based redox switches and gene regulation. Antioxid Redox Signal. 2011;14:1049–1063. doi: 10.1089/ars.2010.3400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bachi A, Dalle-Donne I, Scaloni A. Redox proteomics: chemical principles, methodological approaches and biological/biomedical promises. Chem Rev. 2013;113:596–698. doi: 10.1021/cr300073p. [DOI] [PubMed] [Google Scholar]
- 5.Sato Y, Inaba K. Disulfide bond formation network in the three biological kingdoms, bacteria, fungi and mammals. FEBS J. 2012;279:2262–2271. doi: 10.1111/j.1742-4658.2012.08593.x. [DOI] [PubMed] [Google Scholar]
- 6.Derakhshan B, Wille PC, Gross SS. Unbiased identification of cysteine S-nitrosylation sites on proteins. Nat Protoc. 2007;2:1685–1691. doi: 10.1038/nprot.2007.210. [DOI] [PubMed] [Google Scholar]
- 7.Greco TM, et al. Identification of S-nitrosylation motifs by site-specific mapping of the S-nitrosocysteine proteome in human vascular smooth muscle cells. Proc Natl Acad Sci USA. 2006;103:7420–7425. doi: 10.1073/pnas.0600729103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hess DT, Matsumoto A, Kim SO, Marshall HE, Stamler JS. Protein S-nitrosylation: purview and parameters. Nat Rev Mol Cell Biol. 2005;6:150–166. doi: 10.1038/nrm1569. [DOI] [PubMed] [Google Scholar]
- 9.Paulsen CE, et al. Peroxide-dependent sulfenylation of the EGFR catalytic site enhances kinase activity. Nat Chem Biol. 2012;8:57–64. doi: 10.1038/nchembio.736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wan J, Roth AF, Bailey AO, Davis NG. Palmitoylated proteins: purification and identification. Nat Protoc. 2007;2:1573–1584. doi: 10.1038/nprot.2007.225. [DOI] [PubMed] [Google Scholar]
- 11.Roth AF, et al. Global analysis of protein palmitoylation in yeast. Cell. 2006;125:1003–1013. doi: 10.1016/j.cell.2006.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brandes N, Schmitt S, Jakob U. Thiol-based redox switches in eukaryotic proteins. Antioxid Redox Signal. 2009;11:997–1014. doi: 10.1089/ars.2008.2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jortzik E, Wang L, Becker K. Thiol-based posttranslational modifications in parasites. Antioxid Redox Signal. 2012;17:657–673. doi: 10.1089/ars.2011.4266. [DOI] [PubMed] [Google Scholar]
- 14.Jaffrey SR, Erdjument-Bromage H, Ferris CD, Tempst P, Snyder SH. Protein S-nitrosylation: a physiological signal for neuronal nitric oxide. Nat Cell Biol. 2001;3:193–197. doi: 10.1038/35055104. [DOI] [PubMed] [Google Scholar]
- 15.Jaffrey SR, Snyder SH. The biotin switch method for the detection of S-nitrosylated proteins. Sci STKE. 2001;2001:pl1. doi: 10.1126/stke.2001.86.pl1. [DOI] [PubMed] [Google Scholar]
- 16.Lind C, et al. Identification of S-glutathionylated cellular proteins during oxidative stress and constitutive metabolism by affinity purification and proteomic analysis. Arch Biochem Biophys. 2002;406:229–240. doi: 10.1016/s0003-9861(02)00468-x. [DOI] [PubMed] [Google Scholar]
- 17.Reynaert NL, et al. In situ detection of S-glutathionylated proteins following glutaredoxin-1 catalyzed cysteine derivatization. Biochim Biophys Acta. 2006;1760:380–387. doi: 10.1016/j.bbagen.2006.01.006. [DOI] [PubMed] [Google Scholar]
- 18.Leichert LI, et al. Quantifying changes in the thiol redox proteome upon oxidative stress in vivo. Proc Natl Acad Sci USA. 2008;105:8197–8202. doi: 10.1073/pnas.0707723105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hao G, Derakhshan B, Shi L, Campagne F, Gross SS. SNOSID, a proteomic method for identification of cysteine S-nitrosylation sites in complex protein mixtures. Proc Natl Acad Sci USA. 2006;103:1012–1017. doi: 10.1073/pnas.0508412103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu T, et al. Improved proteome coverage using high-efficiency cysteinyl peptide enrichment: The mammary epithelial cell proteome. Proteomics. 2005;5:1263–1273. doi: 10.1002/pmic.200401055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu T, et al. High-throughput comparative proteome analysis using a quantitative cysteinyl-peptide enrichment technology. Anal Chem. 2004;76:5345–5353. doi: 10.1021/ac049485q. [DOI] [PubMed] [Google Scholar]
- 22.Forrester MT, et al. Proteomic analysis of S-nitrosylation and denitrosylation by resin-assisted capture. Nat Biotechnol. 2009;27:557–559. doi: 10.1038/nbt.1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Su D, et al. Quantitative site-specific reactivity profiling of S-nitrosylation in mouse skeletal muscle using cysteinyl peptide enrichment coupled with mass spectrometry. Free Radic Biol Med. 2013;57:68–78. doi: 10.1016/j.freeradbiomed.2012.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu M, et al. Site-specific proteomics approach for study protein s-nitrosylation. Anal Chem. 2010;82:7160–7168. doi: 10.1021/ac100569d. [DOI] [PubMed] [Google Scholar]
- 25.Forrester MT, et al. Site-specific analysis of protein S-acylation by resin-assisted capture. J Lipid Res. 2011;52:393–398. doi: 10.1194/jlr.D011106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Paulech J, et al. Large-Scale Capture of Peptides Containing Reversibly Oxidized Cysteines by Thiol-Disulfide Exchange Applied to the Myocardial Redox Proteome. Anal Chem. 2013;85:3774–3780. doi: 10.1021/ac400166e. [DOI] [PubMed] [Google Scholar]
- 27.Ross PL, et al. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol Cell Proteomics. 2004;3:1154–1169. doi: 10.1074/mcp.M400129-MCP200. [DOI] [PubMed] [Google Scholar]
- 28.Dayon L, et al. Relative quantification of proteins in human cerebrospinal fluids by MS/MS using 6-plex isobaric tags. Anal Chem. 2008;80:2921–2931. doi: 10.1021/ac702422x. [DOI] [PubMed] [Google Scholar]
- 29.Shelton MD, Chock PB, Mieyal JJ. Glutaredoxin: role in reversible protein s-glutathionylation and regulation of redox signal transduction and protein translocation. Antioxid Redox Signal. 2005;7:348–366. doi: 10.1089/ars.2005.7.348. [DOI] [PubMed] [Google Scholar]
- 30.Zhang C, Rodriguez C, Circu ML, Aw TY, Feng J. S-Glutathionyl quantification in the attomole range using glutaredoxin-3-catalyzed cysteine derivatization and capillary gel electrophoresis with laser-induced fluorescence detection. Anal Bioanal Chem. 2011;401:2165–2175. doi: 10.1007/s00216-011-5311-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mustafa AK, et al. H2S signals through protein S-sulfhydration. Sci Signal. 2009;2:ra72. doi: 10.1126/scisignal.2000464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pan J, Carroll KS. Persulfide Reactivity in the Detection of Protein S-Sulfhydration. ACS Chem Biol. 2013 doi: 10.1021/cb4001052. [DOI] [PMC free article] [PubMed]
- 33.Forrester MT, Foster MW, Stamler JS. Assessment and application of the biotin switch technique for examining protein S-nitrosylation under conditions of pharmacologically induced oxidative stress. J Biol Chem. 2007;282:13977–13983. doi: 10.1074/jbc.M609684200. [DOI] [PubMed] [Google Scholar]
- 34.Forrester MT, Foster MW, Benhar M, Stamler JS. Detection of protein S-nitrosylation with the biotin-switch technique. Free Radic Biol Med. 2009;46:119–126. doi: 10.1016/j.freeradbiomed.2008.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ong SE, et al. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics. 2002;1:376–386. doi: 10.1074/mcp.m200025-mcp200. [DOI] [PubMed] [Google Scholar]
- 36.Murray CI, Uhrigshardt H, O’Meally RN, Cole RN, Van Eyk JE. Identification and quantification of S-nitrosylation by cysteine reactive tandem mass tag switch assay. Mol Cell Proteomics. 2012;11:M111 013441. doi: 10.1074/mcp.M111.013441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Foster MW. Methodologies for the characterization, identification and quantification of S-nitrosylated proteins. Biochim Biophys Acta. 2012;1820:675–683. doi: 10.1016/j.bbagen.2011.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang Y, Ficarro SB, Li S, Marto JA. Optimized Orbitrap HCD for quantitative analysis of phosphopeptides. J Am Soc Mass Spectrom. 2009;20:1425–1434. doi: 10.1016/j.jasms.2009.03.019. [DOI] [PubMed] [Google Scholar]
- 39.Kelly RT, et al. Chemically etched open tubular and monolithic emitters for nanoelectrospray ionization mass spectrometry. Anal Chem. 2006;78:7796–7801. doi: 10.1021/ac061133r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Livesay EA, et al. Fully automated four-column capillary LC-MS system for maximizing throughput in proteomic analyses. Anal Chem. 2008;80:294–302. doi: 10.1021/ac701727r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Eng JK, Mccormack AL, Yates JR. An Approach to Correlate Tandem Mass-Spectral Data of Peptides with Amino-Acid-Sequences in a Protein Database. J Am Soc Mass Spectrom. 1994;5:976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- 42.Elias JE, Gygi SP. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat Methods. 2007;4:207–214. doi: 10.1038/nmeth1019. [DOI] [PubMed] [Google Scholar]
- 43.Qian WJ, et al. Probability-Based Evaluation of Peptide and Protein Identifications from Tandem Mass Spectrometry and SEQUEST Analysis: The Human Proteome. J Proteome Res. 2005;4:53–62. doi: 10.1021/pr0498638. [DOI] [PubMed] [Google Scholar]
- 44.Kim S, Gupta N, Pevzner PA. Spectral probabilities and generating functions of tandem mass spectra: a strike against decoy databases. J Proteome Res. 2008;7:3354–3363. doi: 10.1021/pr8001244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Viner RI, Williams TD, Schoneich C. Peroxynitrite modification of protein thiols: oxidation, nitrosylation, and S-glutathiolation of functionally important cysteine residue(s) in the sarcoplasmic reticulum Ca-ATPase. Biochemistry. 1999;38:12408–12415. doi: 10.1021/bi9909445. [DOI] [PubMed] [Google Scholar]
- 46.Knoefler D, et al. Quantitative in vivo redox sensors uncover oxidative stress as an early event in life. Mol Cell. 2012;47:767–776. doi: 10.1016/j.molcel.2012.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1: Conditions for selective reduction, negative and positive controls for multiple reversible cysteine modifications
Supplemental Table 2: LC-MS/MS quantification data of total cysteine oxidation levels in RAW cells treated with 0.1 mM and 0.5 mM diamide