

Abstract
Combining computer-assisted drug design and synthetic efforts, we generated compounds with potent and balanced activities toward both D3 dopamine receptor and fatty acid amide hydrolase (FAAH) enzyme. Concurrently modulating these targets, our compounds hold a great potential toward exerting a disease-modifying effect on nicotine addiction and other forms of compulsive behavior.
Keywords: Drug Design, Multitarget, MTDL, Molecular Modeling, Polypharmacology
Tobacco smoking is a chronic syndrome that represents one of the most severe global health threats.1 While it is the prolonged exposure to harmful substances contained in cigarette smoke that eventually leads to cardiovascular and respiratory conditions, cancer, and other disorders, tobacco addiction is caused by nicotine. Nicotine is a psychoactive alkaloid that elevates the levels of dopamine in areas of the brain connected to reward,2 thus leading to addiction. Available treatments for nicotine addiction are partially effective in attenuating the symptoms of withdrawal but their success in preventing relapse has only been very limited.3 Dopamine receptor D3 (DRD3) is a member of the GPCR superfamily that is mainly expressed in the mesolimbocortical system, a neural pathway implicated in reward and motivated behavior.4 DRD3 has been extensively investigated to develop new medications for nicotine addiction.5
In animal models, DRD3 partial agonists decrease the compulsion for nicotine self-administration under reinforcement schedules and prevent the establishment of drug-seeking behavior.6, 7 However, DRD3 modulators do not display any significant effect on the rewarding properties of nicotine, and have only mild effects on withdrawal. It has been suggested, therefore, that an effective medication could be obtained coupling the modulation of DRD3 with additional effects on other relevant targets.6 Recent studies have shown that inhibition of the fatty acid amide hydrolase (FAAH) enzyme is effective in counteracting the abuse-related effects of nicotine.8 In animal models, URB597, a selective FAAH inhibitor,9 reduces the nicotine-induced elevation of dopamine in the brain, preventing self-administration and preferential behaviours.
Herein, we report on the rational design, synthesis, and biological evaluation of the first set of dual DRD3 partial agonists and FAAH inhibitors.
In Figure 1, examples of known D3 selective modulators (1-3) 5 and FAAH inhibitors (4-6) 9 are reported. We realized that it was possible to devise a dual-target pharmacophore model exploiting the overlap between the pharmacophoric features of DRD3 partial agonists and those of the O-aryl carbamate derivatives (Figure S1 in Supporting Information).5, 10, 11
Figure 1.

Known DRD3 antagonists/partial agonists NGB2904 (1), CJB090 (2) and BP-897 (3) and known FAAH inhibitor derivatives URB524 (4), URB597 (5) and PF-622 (6).
Ideally, molecules matching this description should be able to concurrently modulate both targets. Querying 263 annotated structures of O-aryl carbamate derivatives and 4298 DRD3 modulators retrieved from ChEMBL,12 we could not find any match to the combined pharmacophore. Hence, we pursued the generation of novel, purposely conceived compounds. We assembled an in silico library of 280 compounds, in which each molecule had a univocal arrangement of chemical features rationally selected to display activity and selectivity on both targets. These compounds were docked into the crystal structures of rat FAAH (r-FAAH) 13 and human DRD3 14 (see Supporting Information for details).
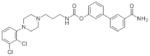
Two compounds, 7 and 8 (Table 1), displayed good predicted binding affinities at both targets. In r-FAAH, compound 7 adopted the orientation suggested for O-aryl carbamates by the crystal structure of URB597-carbamoylated humanized rat FAAH,15 as well as by quantum mechanical studies carried out on 416 (Figure 2a). The docked pose of 7 at DRD3 (Figure 2b) was in good agreement with the binding mode previously proposed for DRD3 selective modulators.14 The aryl-piperazine is lodged in the same region occupied by eticlopride in the crystal and the O-biphenyl moiety projected toward the less conserved region of the pocket. Compound 8 established similar interactions with both targets (see Supporting Information). Encouraged by these results, we synthesized 7 and 8. The syntheses are reported in Supporting Information. The biological activities of the new compounds were evaluated on r-FAAH, human FAAH (h-FAAH) and in a human DRD3 functional assay (see Supporting Information). Results are reported in Table 1. Known DRD3 modulators 2 and 3 did not show any significant inhibitory activity on r-FAAH and h-FAAH. FAAH inhibitor 5 had no activity on DRD3 (see Supporting Information). Interestingly, compounds 7 and 8 turned out to be very potent FAAH inhibitors with 0.3 nM and 0.1 nM on r-FAAH and 1.6 nM and 1.3 nM activities on h-FAAH, respectively. In agreement with the SAR reported by Mor et al.,19 an elongated substituent at the nitrogen side of the carbamate was beneficial for potency. The presence of a basic nitrogen atom in the lipophilic acyl chain binding pocket was already reported for PF-622 (6, Figure 1) and was not detrimental for activity.20 At the same time, compounds 7 and 8 showed potent modulatory activity on DRD3, with a partial agonist profile and median effective concentration (EC50) of 6.5 nM and 3.9 nM (see Table 1). The length of the linker did not influence potency. This first set of data confirmed our initial hypothesis that a seamless combination of the pharmacophoric features of FAAH inhibitors and DRD3 partial agonists in a single molecular entity can lead to dual-target modulators. Next, the compounds were tested against human dopamine D2 receptor (DRD2) short isoform to evaluate the DRD2/DRD3 selectivity ratio, as the simultaneous activation of DRD2 and DRD3 could lead to severe side effects.21 Both derivatives resulted selective for DRD3, with DRD2/DRD3 affinity ratios greater than 80. The second off-target that we tested was the cannabinoid receptor CB1. CB1 receptors are highly expressed in regions of the brain implicated in dopamine-mediated reward,22, 23 and the CB1 antagonist rimonabant (SR141716A) was evaluated in clinical trials as a potential new medication for nicotine addiction.24 Despite its effectiveness, rimonabant was not further developed due to neuropsychiatric side effects attributed to the blockade of intrinsic endocannabinoid signalling.24 CB1 direct agonism can be also detrimental, because of the pleiotropic functions served by this receptor in the brain and peripheral tissues.22 Surprisingly, compounds 7 and 8 showed CB1 activation in the picomolar range (see Table 1). While it is known that 5 has no effect on CB1,25 we tested known DRD3 partial agonists 2 and 3 for CB1 activity. 3 did not show any activity but 2 turned out to be a rather potent CB1 agonist with an EC50 of 840 nM (Table S1 in the Supporting Information).
Table 1.
Biological data of activities of known compounds and synthesized molecules.
| Compound | Structure | rat FAAH IC50 (nM) |
human FAAH IC50 (nM) |
DRD3 EC50 (nM) |
DRD3% Efficacy* |
DRD2 EC50 (nM) |
DRD2% Efficacy** |
Ratio D2/D3 |
CB1 EC50 (nM) |
Ratio CB1/D3 |
|---|---|---|---|---|---|---|---|---|---|---|
| 7 |
|
0.3 | 1.6 | 6.5 | 51.7 | >1000 | 41.2 | >154 | 0.9 | 0.1 |
| 8 |
|
0.1 | 1.3 | 3.9 | 64.8 | 320.0 | 61.1 | 82 | 0.3 | 0.1 |
| 15 |
|
0.7 | 0.6 | 1.0 | 55.6 | 23.0 | 25.2 | 23 | 14.0 | 14 |
| 16 |
|
13.0 | 2.7 | 7.7 | 81.2 | 240.0 | 32.3 | 31 | 64.0 | 8 |
| 17 |
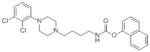
|
22.0 | 6.1 | 1.3 | 50.4 | 209.0 | A | 161 | 420.0 | 323 |
vs. 300 nM dopamine;
vs. 3 μM dopamine;
N.C., value not calculable, concentration – response curve show less than 25% effect at the highest concentration; A, antagonist
Figure 2.

a) Structures of the selected molecules 7 and 8 matching the combined pharmacophore; b) Docked pose of 7 in the crystal structure of rat FAAH; c) Docked pose of 7 in the crystal structure of human DRD3.
To design-out CB1 activity, three additional derivatives were prepared modifying the O-aryl group according to Scheme S1. Since 7 showed a classic partial agonist profile on DRD3 and a greater selectivity toward DRD2 with respect to 8, the length of the linker was kept at 4 methylene units. To modulate the orientation of the aryl-substituent, we introduced a p-biphenyl moiety (15). Although the latter moiety was reported as detrimental for FAAH activity,9 compound 15 maintained a good potency, showed a partial agonist profile on DRD3, and acquired a small but significant selectivity ratio over CB1. In this case, the main issue was the selectivity over DRD2, which, dropping from over 150- to 23-fold, was negatively affected by this substitution. Next, considering that: i) 3 is completely devoid of CB1 activity and that ii) the naphthyl group was already reported on both DRD3 modulators 5 and FAAH inhibitors,26 the two naphthyl-substituted regioisomers 16 and 17 were synthesized. These compounds were endowed with good and balanced activities in the low nanomolar range. However, 16 did not show any improvement in CB1 selectivity relative to 15, had only a moderate 31-fold selectivity over DRD2, and the functional assay on DRD3 highlighted an almost full agonist activity profile. Conversely, 17 succeeded in the CB1 designing-out effort, showing a good selectivity with a CB1/DRD3 ratio over 300-fold and 420 nM EC50 on CB1, over 450-fold lower than the prototype 7. Together with potent and balanced activities (6.1 nM on h-FAAH and 1.3 nM on DRD3), compound 17 also had 161-fold selectivity over DRD2, a clear partial agonist profile, and interesting physicochemical calculated features (see Table S1 in the Supporting Information). The docked poses of compounds 8 and 15-17 at FAAH and DRD3 are reported in the Supporting Information.
Conclusions
Here, confirming the feasibility of our recently reported strategy,27 we have combined computational methods and synthetic efforts to successfully discover novel, potent and balanced dual-target molecules. The described compounds are an example of dual modulators rationally designed to display activity toward a GPCR and an enzyme, which are structurally unrelated but involved in a common biological function.28 While the vast majority of known drugs have been developed as selective modulators of a single target, this approach has shown several limitations in treating complex and multifaceted pathologies.29 The Multi-Target Directed Ligand (MTDL) strategy is based on the idea that a single molecular entity can be devised to hit multiple targets that cooperate in the framework of the same disease.30 MTDLs may have a superior therapeutic effect with respect to single target compounds and might prevent unwanted compensations.31-34 Being able to modulate DRD3 and inhibit FAAH, this class of compounds might hold great potential as disease-modifying agents for the treatment of nicotine addiction.
Supplementary Material
Acknowledgment
This work was supported in part by the National Institute for Drug Abuse Grant No. DP1 DA031387 (to Piomelli). The authors thank Dr. Angelo Reggiani for his comments on the experimental setup and Dr. Giuseppe Giardina for the useful discussions. Bottegoni, De Simone, Ruda, Bandiera, Piomelli, and Cavalli are inventors in a patent application in which the novel structures described in this study are claimed.
Footnotes
Experimental procedures; characterizations; spectral data for all compounds. See DOI: 10.1039/c000000x
Notes and references
- 1.Syed BA, Chaudhari K. Nat. Rev. Drug Discov. 2013;12:97–98. doi: 10.1038/nrd3914. [DOI] [PubMed] [Google Scholar]
- 2.Escobar-Chávez JJ, Domínguez-Delgado CL, Rodríguez-Cruz IM. Drug Des., Dev. Ther. 2011;5:211–224. doi: 10.2147/DDDT.S10033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Polosa R, Benowitz NL. Trends Pharmacol. Sci. 2011;32:281–289. doi: 10.1016/j.tips.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Everitt BJ, Robbins TW. Nat. Neurosci. 2005;8:1481–1489. doi: 10.1038/nn1579. [DOI] [PubMed] [Google Scholar]
- 5.Micheli F. ChemMedChem. 2011;6:1152–1162. doi: 10.1002/cmdc.201000538. [DOI] [PubMed] [Google Scholar]
- 6.Mugnaini M, Iavarone L, Cavallini P, Griffante C, Oliosi B, Savoia C, Beaver J, Rabiner EA, Micheli F, Heidbreder C, Andorn A, Pich E. Merlo, Bani M. Neuropsychopharmacology. 2013;38:302–312. doi: 10.1038/npp.2012.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hackling A, Ghosh R, Perachon S, Mann A, Holtje HD, Wermuth CG, Schwartz JC, Sippl W, Sokoloff P, Stark H. J. Med. Chem. 2003;46:3883–3899. doi: 10.1021/jm030836n. [DOI] [PubMed] [Google Scholar]
- 8.Scherma M, Panlilio LV, Fadda P, Fattore L, Gamaleddin I, Le Foll B, Justinova Z, Mikics E, Haller J, Medalie J, Stroik J, Barnes C, Yasar S, Tanda G, Piomelli D, Fratta W, Goldberg SR. J. Pharmacol. Exp. Ther. 2008;327:482–490. doi: 10.1124/jpet.108.142224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mor M, Rivara S, Lodola A, Plazzi PV, Tarzia G, Duranti A, Tontini A, Piersanti G, Kathuria S, Piomelli D. J. Med. Chem. 2004;47:4998–5008. doi: 10.1021/jm031140x. [DOI] [PubMed] [Google Scholar]
- 10.Deng H. Expert Opin. Drug Discovery. 2010;5:961–993. doi: 10.1517/17460441.2010.513378. [DOI] [PubMed] [Google Scholar]
- 11.Newman AH, Beuming T, Banala AK, Donthamsetti P, Pongetti K, LaBounty A, Levy B, Cao J, Michino M, Luedtke RR, Javitch JA, Shi L. J. Med. Chem. 2012;55:6689–6699. doi: 10.1021/jm300482h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gaulton A, Bellis LJ, Bento a. P., Chambers J, Davies M, Hersey A, Light Y, McGlinchey S, Michalovich D, Al-Lazikani B, Overington JP. Nucleic Acids Res. 2012;40:D1100–1107. doi: 10.1093/nar/gkr777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Min X, Thibault ST, Porter AC, Gustin DJ, Carlson TJ, Xu H, Lindstrom M, Xu G, Uyeda C, Ma Z, Li Y, Kayser F, Walker NP, Wang Z. Proc. Natl. Acad. Sci. U. S. A. 2011;108:7379–7384. doi: 10.1073/pnas.1016167108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chien EY, Liu W, Zhao Q, Katritch V, Han GW, Hanson MA, Shi L, Newman AH, Javitch JA, Cherezov V, Stevens RC. Science. 2010;330:1091–1095. doi: 10.1126/science.1197410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mileni M, Kamtekar S, Wood DC, Benson TE, Cravatt BF, Stevens RC. J. Mol. Biol. 2010;400:743–754. doi: 10.1016/j.jmb.2010.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lodola A, Mor M, Rivara S, Christov C, Tarzia G, Piomelli D, Mulholland AJ. Chem. Commun. 2008:214–216. doi: 10.1039/b714136j. [DOI] [PubMed] [Google Scholar]
- 17.Del Zotto A, Amoroso F, Baratta W, Rigo P. Eur. J. Org. Chem. 2008;2009:110–116. [Google Scholar]
- 18.Knolker HJ, Braxmeier T. Tetrahedron Lett. 1996;4039:5861–5864. [Google Scholar]
- 19.Mor M, Lodola A, Rivara S, Vacondio F, Duranti A, Tontini A, Sanchini S, Piersanti G, Clapper JR, King AR, Tarzia G, Piomelli D. J. Med. Chem. 2008;51:3487–3498. doi: 10.1021/jm701631z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ahn K, Johnson DS, Fitzgerald LR, Liimatta M, Arendse A, Stevenson T, Lund ET, Nugent R. a., Nomanbhoy TK, Alexander JP, Cravatt BF. Biochemistry. 2007;46:13019–13030. doi: 10.1021/bi701378g. [DOI] [PubMed] [Google Scholar]
- 21.Joyce JN, Millan MJ. Drug Discov. Today. 2005;10:917–925. doi: 10.1016/S1359-6446(05)03491-4. [DOI] [PubMed] [Google Scholar]
- 22.Di Marzo V. Nat. Rev. Drug Discov. 2008;7:438–455. doi: 10.1038/nrd2553. [DOI] [PubMed] [Google Scholar]
- 23.Maldonado R, Berrendero F. Curr. Drug Targets. 2010;11:440–449. doi: 10.2174/138945010790980358. [DOI] [PubMed] [Google Scholar]
- 24.Moreira FA, Crippa JA. Rev. Bras. Psiquiatr. 2009;31:145–153. doi: 10.1590/s1516-44462009000200012. [DOI] [PubMed] [Google Scholar]
- 25.Piomelli D, Tarzia G, Duranti A, Tontini A, Mor M, Compton TR, Dasse O, Monaghan EP, Parrott JA, Putman D. CNS Drug Rev. 2006;12:21–38. doi: 10.1111/j.1527-3458.2006.00021.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tarzia G, Duranti A, Tontini A, Piersanti G, Mor M, Rivara S, Plazzi PV, Park C, Kathuria S, Piomelli D. J. Med. Chem. 2003;46:2352–2360. doi: 10.1021/jm021119g. [DOI] [PubMed] [Google Scholar]
- 27.Bottegoni G, Favia AD, Recanatini M, Cavalli A. Drug Discov. Today. 2012;17:23–34. doi: 10.1016/j.drudis.2011.08.002. [DOI] [PubMed] [Google Scholar]
- 28.Peters JU. J. Med. Chem. 2013 Article ASAP. [Google Scholar]
- 29.Csermely P, Agoston V, Pongor S. Trends Pharmacol. Sci. 2005;26:178–182. doi: 10.1016/j.tips.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 30.Cavalli A, Bolognesi ML, Minarini A, Rosini M, Tumiatti V, Recanatini M, Melchiorre C. J. Med. Chem. 2008;51:347–372. doi: 10.1021/jm7009364. [DOI] [PubMed] [Google Scholar]
- 31.Mohr K, Schmitz J, Schrage R, Trankle C, Holzgrabe U. Angew. Chem. 2013;52:508–516. doi: 10.1002/anie.201205315. [DOI] [PubMed] [Google Scholar]
- 32.Butini S, Gemma S, Campiani G, Franceschini S, Trotta F, Borriello M, Ceres N, Ros S, Coccone SS, Bernetti M, De Angelis M, Brindisi M, Nacci V, Fiorini I, Novellino E, Cagnotto A, Mennini T, Sandager-Nielsen K, Andreasen JT, Scheel-Kruger J, Mikkelsen JD, Fattorusso C. J. Med. Chem. 2009;52:151–169. doi: 10.1021/jm800689g. [DOI] [PubMed] [Google Scholar]
- 33.Morphy R, Kay C, Rankovic Z. Drug Discov. Today. 2004;9:641–651. doi: 10.1016/S1359-6446(04)03163-0. [DOI] [PubMed] [Google Scholar]
- 34.Hopkins AL. Nat. Chem. Biol. 2008;4:682–690. doi: 10.1038/nchembio.118. [DOI] [PubMed] [Google Scholar]
- 35.Newman AH, Cao J, Bennett CJ, Robarge MJ, Freeman RA, Luedtke RR. Bioorg. Med. Chem. Lett. 2003;13:2179–2183. doi: 10.1016/s0960-894x(03)00389-5. [DOI] [PubMed] [Google Scholar]
- 36.Garcia-Ladona FJ, Cox BF. CNS Drug Rev. 2003;9:141–158. doi: 10.1111/j.1527-3458.2003.tb00246.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.