

Abstract
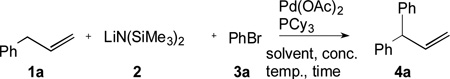
The combination of aryl bromides, allylbenzene, base and a palladium catalyst usually results in a Heck reaction. Herein we combine these same reagents, but override the Heck pathway by employing a strong base. In the presence of LiN(SiMe3)2, allylbenzene derivatives undergo reversible deprotonation. Transmetallation of the resulting allyllithium intermediate to LPdAr(Br) and reductive elimination provide the 1,1-diarylprop-2-enes, which are not accessible via the Heck reaction. The regioselectivity in this deprotonative cross-coupling process is catalyst-controlled and very high.
Keywords: chemoselectivity, regioselectivity, deprotonative-cross-coupling reaction, C-H functionalization, palladium-catalyzed cross-coupling
Catalytic functionalization of unactivated sp3-hybridized C–H bonds in the absence of directing groups is highly desirable, but remains challenging.[1, 2] The level difficulty of these functionalizations increases dramatically when regio- and chemoselectivity issues are present. In our efforts to address such challenges, we recently initiated a program for the functionalization of weakly acidic sp3-hybridized C–H bonds by palladium catalyzed deprotonative cross-coupling processes (DCCP). Substrates that have been successfully functionalized using this approach include diarylmethanes,[3, 4] sulfoxides,[5] sulfones,[6] amides,[7] and chromium-activated benzylic amines (to produce enantioenriched diarylmethylamines).[8] Based on these results, we hypothesized that it might be possible to functionalize allylbenzene derivatives and control chemo- and regioselectivity. Successful development of such a process would require: 1) conditions for the deprotonation of allylbenzene that are amenable to catalysis, 2) catalysts that can promote the regioselective arylation, and 3) control of base reactivity such that the more acidic product is not deprotonated and isomerized or further functionalized.
Typically, reactions of allylbenzenes with aryl bromides in the presence of palladium catalysts and base afford Heck–type γ-selective products, often as mixtures of regio- and geometric isomers (Scheme 1, right).[9, 10] We envisioned that a strong base could divert the chemoselectivity from olefin coordination and insertion of allylbenzene in the Heck coupling to transmetallation of the metallated allyl (Scheme 1, left).[11, 12] The catalyst/ligand combination would control the regioselectivity of the arylation in the DCCP, thus enabling the formation of α-arylated products that are inaccessible via the Heck pathway. It is noteworthy that this approach is distinct from known C–H activation/arylations of allylbenzenes and related substrates.[13]
Scheme 1.

Overriding Heck Coupling: Heck Reaction (right) vs. DCCP of Allylbenzene with Strong Base (left).
Herein, we disclose the first metal-catalyzed C(sp3)–H arylation of allylbenzenes (pKa ~ 34 in DMSO)[14] with aryl bromides to afford 1,1-diarylprop-2-enes. A base/catalyst combination [LiN(SiMe3)2/Pd-PCy3] is advanced that efficiently controls the chemoselectivity and promotes regioselective DCCP of allylbenzenes in good to excellent yields (51–97%).
Our first challenge was to identify conditions for the deprotonation of allylbenzene’s C(sp3)–H. The benzylic C−H’s in allylbenzene have traditionally been deprotonated with n- and sec-BuLi at −78 °C or with n-BuMgCl.[15] These strong bases, however, are impractical for cross-coupling reactions because of their limited compatibility with catalysts and coupling partners. We, therefore, focused on reversible in situ deprotonation of allylbenzene.
As a surrogate for the transmetallation step in the arylation reaction in Scheme 1, we substituted reaction of metallated allylbenzene with benzyl chloride (Scheme 2). To perform the benzylation, we screened 6 bases [LiN(SiMe3)2, NaN(SiMe3)2, KN(SiMe3)2, LiOt-Bu, NaOt-Bu and KOt-Bu] at room temperature in CPME (cyclopentylmethyl ether). As illustrated in Scheme 2, the bases leading to benzylation products were: KN(SiMe3)2 affording a 5:1 ratio of α:γ (80% yield), NaN(SiMe3)2 generating a 4:1 ratio (71% yield), and LiN(SiMe3)2 leading to a 1:1 ratio (13% yield). The α:γ ratios observed suggest that the nature of the metal plays a significant role in the regioselectivity.[16] None of the MO-t-Bu (M = Li, Na, K) bases generated detectable amounts of benzylation products. Unlike bases previously used to deprotonate allylbenzene (n- and sec-BuLi at −78 °C or n-Bu-MgCl), MN(SiMe3)2 has a high likelihood of compatibility with catalyst, reagents, and products in the DCCP (Scheme 1, left).
Scheme 2.

Benzylation Used as Surrogate for the Transmetallation Step in DCCP.
We next turned our attention to catalyst identification for the DCCP of allylbenzene. We tested 29 sterically and electronically diverse mono- and bidentate phosphine ligands, 3 bases [LiN(SiMe3)2, NaN(SiMe3)2, KN(SiMe3)2] and different Pd(0) and Pd(II) precursors at 110 °C using the microscale high-throughput experimentation (HTE) techniques (see Supporting Information for details). Interestingly, the results of the HTE indicated the only base leading to the α-arylated product (4a) was LiN(SiMe3)2 (Table 1). Note that the main group metal is involved in both the deprotonation and the transmetallation steps in the arylation reaction. As such, the best base for the benzylation may not be the best for the palladium-catalyzed reaction. Of the 29 ligands examined, PCy3 and Brettphos afforded very high regioselectivity in the coupling, giving exclusively the α-arylated products (see Supporting Information). NiXantphos, the only ligand that we found to perform well in the DCCP of diphenylmethane (pKa = 32 in DMSO)[17] with aryl bromides,[3] gave a 2.6:1 ratio of α- and γ-arylated products. Because PCy3 is less expensive than Brettphos,[18] we chose PCy3 as the ligand for optimization of the arylation reaction.
Table 1.
Optimization of Palladium-catalyzed DCCP of Allylbenzene.
 | |||||||
|---|---|---|---|---|---|---|---|
| entry | 1a:2:3a | solvent | temp. (°C) |
conc. (M) |
time (h) |
Pd(OAc)2/ PCy3 mol (%) |
4a[a] (%) |
| 1 | 1:3:3 | THF | 110 | 0.1 | 24 | 5/10 | 0 |
| 2 | 1:3:3 | DME | 110 | 0.1 | 24 | 5/10 | 0 |
| 3 | 1:3:3 | Dioxane | 110 | 0.1 | 24 | 5/10 | 10 |
| 4 | 1:3:3 | CPME | 110 | 0.1 | 24 | 5/10 | 15 |
| 5 | 1:3:3 | CPME | 80 | 0.1 | 24 | 5/10 | 30 |
| 6 | 1:3:3 | CPME | 60 | 0.1 | 24 | 5/10 | 20 |
| 7 | 1:3:3 | CPME | 80 | 0.2 | 24 | 5/10 | 40 |
| 8 | 3:3:1 | CPME | 80 | 0.2 | 24 | 5/10 | 65 |
| 9 | 3:3:1 | CPME | 80 | 0.3 | 24 | 5/10 | 68 |
| 10 | 3:3:1 | CPME | 80 | 0.4 | 24 | 5/10 | 69 |
| 11 | 4:4:1 | CPME | 80 | 0.2 | 24 | 5/10 | 74 |
| 12 | 4:4:1 | CPME | 80 | 0.2 | 24 | 5/15 | 80 |
| 13 | 4:4:1 | CPME | 80 | 0.2 | 24 | 5/20 | >99 |
Yield determined by 1H NMR analysis of crude mixture with internal standard CH2Br2; less than 4% of gamma-arylated product was detected by NMR.
Translation of the microscale lead outlined above to laboratory scale (0.1 mmol) using 1 equiv. of allylbenzene (1a), 3 equiv. of aryl bromide (2a), 3 equiv. of LiN(SiMe3)2, 5 mol % of Pd(OAc)2 and 10 mol % of PCy3 in CPME at 110 °C rendered the α-arylated product (4a) in 15% yield (entry 4, Table 1). Examination of four etheral solvents [THF, DME, dioxane and CPME] indicated that CPME was the best choice (entries 1–4, Table 1). During the optimization we observed the conversion of allylbenzene to trans-β- methylstyrene (major) and cis–β-methylstyrene (minor) (Scheme 3).[19] Unfortunately, these isomers are less acidic than allyl benzene and do not undergo deprotonation under our conditions at 110 °C. Decreasing the temperature of the reaction to 80 °C increased the product yield to 30% (entry 5). Lowering of the temperature to 60 °C decreased the product yield to 20% (entry 6). Given the weak acidity of allylbenzene, and the resulting low concentration of the allyl anion, we increased the reaction concentration from 0.1 M to 0.2 M. At the higher concentration, the yield of α-arylated product (4a) increased to 40% (entry 7). We, therefore, increased the amount of allylbenzene to 3 equiv. while using 3 equiv. of LiN(SiMe3)2 and 1 equiv. of bromobenzene at 0.2 M. The excess allylbenzene compensates for what appears to be an irreversible isomerization of some allylbenzene to unreactive β-methylstyrenes. Under these conditions, the α-arylated product (4a) was obtained in 65% yield (entry 8). Further increasing the concentration of the reaction mixture (0.3 M and 0.4 M) did not have an appreciable effect on the product yield (entries 9 and 10). We, therefore, chose 0.2 M as the reaction concentration. When 4 equiv. of allylbenzene, 4 equiv. of LiN(SiMe3)2 and 1 equiv. of bromobenzene were used, the product was obtained in 74% yield (entry 11). Further optimization was performed by changing the ratio of PCy3 with respect to the amount of palladium (entries 12 and 13). Finally, the arylation product (4a) was obtained in quantitative yield when employing Pd(OAc)2 (5 mol %), PCy3 (20 mol %) and a ratio of 4:4:1 of allylbenzene : LiN(SiMe3)2 : bromobenzene at 80 °C for 24 h (entry 13).
Scheme 3.

Isomerization of Allylbenzene to Unreactive β–Methylstyrenes.
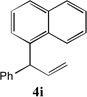
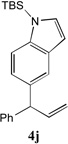
With our optimized conditions (entry 13, Table 1), we examined the substrate scope of the arylation of allylbenzene with aryl bromides (Table 2). The DCCP showed excellent reactivity with aryl bromides possessing electron-donating groups (81–97% yields, entries 2–5). A range of other substrates exhibited good reactivity, including those with substituents in the meta (85% yield, entry 6) and ortho positions (83% yield, entry 7) as well as 1- and 2-bromo naphthalene (86 and 74% yields, entries 8 and 9). Nitrogen protected 5-bromoindole was also a good coupling partner and furnished the α-arylated product in 86% yield (entry 10).[20] The yields were typically lower, however, with electron-deficient aryl bromides (52–66%, entries 11 and 12). Ketones are well known to undergo 1,2-carbonyl addition reactions with reactive organometallics. Additionally, p-bromoacetophenone (pKa in DMSO is 24.7[21]) can participate in competitive aldol chemistry[22] and Pd-catalyzed α-arylation of the enolate under basic condition.[2, 23] Yet the α-arylated product 4m derived from DCCP of allylbenzene was produced in reasonable yield (65%, entry 13). Acetals are known to undergo C–O cleavage with reactive organometallics,[24] however, the α-arylated product 4n was produced in 87% yield (entry 14).
Table 2.
Cross-coupling of Allylbenzene with Aryl bromides.
| entry | product | yield (%)[a] |
entry | product | yield (%)[a] |
|---|---|---|---|---|---|
| 1 | 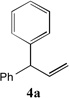 |
91[b] | 8 | 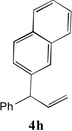 |
86[c] |
| 2 | 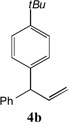 |
88[b] | 9 |
|
74[c] |
| 3 | 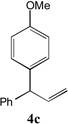 |
97[b] | 10 |
|
86[c,d] |
| 4 | 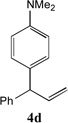 |
81[b] | 11 | 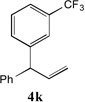 |
66[c,d] |
| 5 | 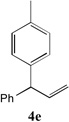 |
86[c] | 12 | 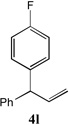 |
52[b] |
| 6 | 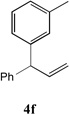 |
85[c,d] | 13 | 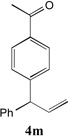 |
65[c,d] |
| 7 | 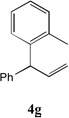 |
83[c,e] | 14 | 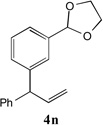 |
87[c,d] |
Less than 4 % of the gamma products were detected by NMR and no Heck product was observed under our conditions.
24 h.
36 h.
6 eq. of base used.
Concentration is 0.3M
We next turned our attention to the allylbenzene scope (Table 3). Electron donating 4-allylanisole exhibited good reactivity (66–88% yields, entries 1–4). Meta substituted 3-allyl toluene furnished the desired coupling products in 80–91% yield (entries 5 and 6). Protected 5-bromoindole underwent the α-arylation with 3-allyltoluene in 82% yield (entry 7). Ortho substituted 2-allyltoluene gave the desired product in 60% yield despite the additional steric hindrance at the α-center (entry 8). Electron-deficient 4-fluoro allylbenzene gave 64 and 66% yield with bromobenzene and protected 5-bromoindole (entries 9 and 10, respectively). Allyl derivatives 2- or 3-allylpyridine did not give the α-arylated products, but only underwent isomerization to the more stable vinyl pyridine derivatives. 2-Allylthiophene, on the other hand, underwent DCCP with 4-bromo tert-butylbenzene to afford the α-arylated product 4u in 51% yield (entry 11). These systems are significantly more acidic than allyl benzenes and will require different catalysts for to afford synthetically useful yields. It is noteworthy that excellent regioselectivity is observed in the substrates in Table 3, even when the steric and electronic parameters of the allylbenzene starting materials are varied.[10, 12]
Table 3.
Cross-coupling of Allylbenzenes with Aryl bromides.
| entry | product | yield (%)[a] | entry | product | yield (%)[a] |
|---|---|---|---|---|---|
| 1 | 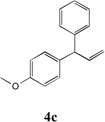 |
88 | 7 | 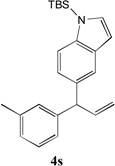 |
82 |
| 2 | 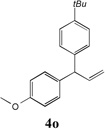 |
81 | 8 | 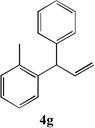 |
60[b] |
| 3 | 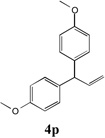 |
72 | 9 | 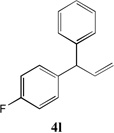 |
64 |
| 4 | 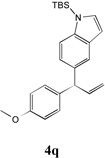 |
66[b] | 10 | 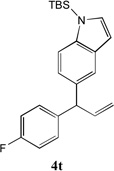 |
66 |
| 5 | 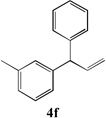 |
80[b] | 11 |
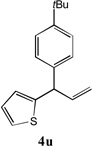
|
51[c] |
| 6 |
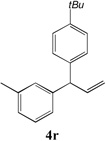
|
91 |
Reaction ran for 36 h.
6 eq. of base used.
5 eq. of thiophene allyl and 3 eq. of base used; obtained product along with 12 % of linear products.
When the DCCP with TBS protected bromoindole (3j) was scaled to 1 mmol with a ratio of 4:5:1 of 1a:2:3j in the presence of 5 mol % Pd(OAc)2 and 20 mol % PCy3, the product 4j was isolated in 77% yield (Scheme 4).
Scheme 4.

Scaled up DCCP of Allylbenzene with TBS-Protected Bromoindole.
In summary, we have developed the first direct α-arylation of unactivated allylbenzenes with aryl bromides via deprotonative cross-coupling processes. The significance of this work is it demonstrates that very weakly acidic hydrocarbon frameworks can be functionalized under DCCP conditions.
The palladium-catalyzed arylation proceeded efficiently in the presence of PCy3 and produces α-arylated 1,1-diarylprop-2-enes with very high regioselectivity (>95:5). It is noteworthy that our approach overrides the ubiquitous Heck reaction pathway by controlling the chemoselectivity. This is accomplished by use of a strong base, LiN(SiMe3)2, that reversibly deprotonates the allylbenzene. The lithiated allyl then undergoes transmetallation with the catalyst in a process that is faster than coordination and insertion of allylbenzene in the Heck pathway. The regiochemistry of the arylation is controlled by the ligand/palladium combination and is key to the success of this process. The fact that the α-arylated diarylallyl does not undergo base promoted isomerization to the more conjugated 1,1-diaryl-1-propene suggests that an enantioselective DCCP of allylbenzenes is possible and has inspired us to investigate this possibility.
Supplementary Material
Footnotes
P. J. W. acknowledges the NIH (National Institute of General Medical Sciences GM58101) and the NSF (CHE-1152488). G. F. acknowledges the Brazillian Science Without Borders program (237849/2012-7).
References
- 1.a) Niwa T, Yorimitsu H, Oshima K. Org. Lett. 2007;9:2373–2375. doi: 10.1021/ol0708119. [DOI] [PubMed] [Google Scholar]; b) Burton PM, Morris JA. Org. Lett. 2010;12:5359–5361. doi: 10.1021/ol102276e. [DOI] [PubMed] [Google Scholar]; c) Duez S, Steib AK, Manolikakes SM, Knochel P. Angew. Chem. 2011;123:7828–7832. doi: 10.1002/anie.201103074. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2011;50:7686–7690. doi: 10.1002/anie.201103074. [DOI] [PubMed] [Google Scholar]; d) Mousseau JJ, Larivee A, Charette AB. Org. Lett. 2008;10:1641–1643. doi: 10.1021/ol800396v. [DOI] [PubMed] [Google Scholar]
- 2.a) Baudoin O. Chem. Soc. Rev. 2011;40:4902–4911. doi: 10.1039/c1cs15058h. [DOI] [PubMed] [Google Scholar]; b) Jazzar R, Hitce J, Renaudat A, Sofack-Kreutzer J, Baudoin O. Chem. Eur. J. 2010;16:2654–2672. doi: 10.1002/chem.200902374. [DOI] [PubMed] [Google Scholar]; c) Bellina F, Rossi R. Chem. Rev. 2009;110:1082–1146. doi: 10.1021/cr9000836. [DOI] [PubMed] [Google Scholar]; d) Johansson CCC, Colacot TJ. Angew. Chem. 2010;122:686–718. [Google Scholar]; Angew. Chem. Int. Ed. 2010;49:676–707. doi: 10.1002/anie.200903424. [DOI] [PubMed] [Google Scholar]
- 3.Zhang J, Bellomo A, Creamer AD, Dreher SD, Walsh PJ. J. Am. Chem. Soc. 2012;134:13765–13772. doi: 10.1021/ja3047816. [DOI] [PubMed] [Google Scholar]
- 4.Bellomo A, Zhang J, Trongsiriwat N, Walsh PJ. Chem. Sci. 2013;4:849–857. [Google Scholar]
- 5.Jia T, Bellomo A, El Baina K, Dreher SD, Walsh PJ. J. Am. Chem. Soc. 2013;135:3740–3743. doi: 10.1021/ja4009776. [DOI] [PubMed] [Google Scholar]
- 6.Zheng B, Jia T, Walsh PJ. Org. Lett. 2013;15:1690–1693. doi: 10.1021/ol400472v. [DOI] [PubMed] [Google Scholar]
- 7.Zheng B, Jia T, Walsh PJ. Org. Lett. 2013;15:4190–4193. doi: 10.1021/ol4019002. [DOI] [PubMed] [Google Scholar]
- 8.McGrew GI, Stanciu C, Zhang J, Carroll PJ, Dreher SD, Walsh PJ. Angew. Chem. 2012;124:11678–11681. doi: 10.1002/anie.201201874. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2012;51:11510–11513. doi: 10.1002/anie.201201874. [DOI] [PubMed] [Google Scholar]
- 9.a) Berthiol F, Doucet H, Santelli M. Tetrahedron Lett. 2003;44:1221–1225. [Google Scholar]; b) Sawant D, Wagh Y, Bhatte K, Panda A, Bhanage B. Tetrahedron Lett. 2011;52:2390–2393. [Google Scholar]; c) Baig RBN, Varma RS. Green Chem. 2013;15:398–417. [Google Scholar]
- 10.Sigman MS, Werner EW. Acc. Chem. Res. 2012;45:874–884. doi: 10.1021/ar200236v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.a) Zhang SJ, Zhen JA, Reith MEA, Dutta AK. J. Med. Chem. 2005;48:4962–4971. doi: 10.1021/jm049021k. [DOI] [PubMed] [Google Scholar]; b) Muraoka K, Nojima M, Kusabayashi S, Nagase S. J. Chem. Soc., Perkin Trans. 2. 1986:761–767. [Google Scholar]; c) Kamigata N, Satoh A, Kondoh T, Kameyama M. Bull. Chem. Soc. Jpn. 1988;61:3575–3580. [Google Scholar]; d) Kobashi Y, Minowa T, Mukaiyama T. Chemistry Lett. 2005;34:756–757. [Google Scholar]
- 12.Hirashita T, Hayashi Y, Mitsui K, Araki S. Tetrahedron Lett. 2004;45:3225–3228. [Google Scholar]
- 13.a) Young AJ, White MC. J. Am. Chem. Soc. 2008;130:14090–14091. doi: 10.1021/ja806867p. [DOI] [PubMed] [Google Scholar]; b) Lin S, Song CX, Cai GX, Wang WH, Shi ZJ. J. Am. Chem. Soc. 2008;130:12901–12903. doi: 10.1021/ja803452p. [DOI] [PubMed] [Google Scholar]; c) Le C, Kunchithapatham K, Henderson WH, Check CT, Stambuli JP. Chem. Eur. J. 2013;19:11153–11157. doi: 10.1002/chem.201301787. [DOI] [PubMed] [Google Scholar]; d) Reed SA, Mazzotti AR, White MC. J. Am. Chem. Soc. 2009;131:11701–11706. doi: 10.1021/ja903939k. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Qin C, Jiao N. J. Am. Chem. Soc. 2010;132:15893–15895. doi: 10.1021/ja1070202. [DOI] [PubMed] [Google Scholar]; f) White MC. Synlett. 2012;23:2746–2748. [Google Scholar]; g) Bigi MA, White MC. J. Am. Chem. Soc. 2013;135:8460–8463. doi: 10.1021/ja402891m. [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Liu G, Yin G, Wu L. Angew. Chem. 2008;120:4811–4814. [Google Scholar]; Angew. Chem. Int. Ed. 2008;47:4733–4736. doi: 10.1002/anie.200801009. [DOI] [PubMed] [Google Scholar]; i) Nahra F, Liron F, Prestat G, Mealli C, Messaoudi A, Poli G. Chem. Eur. J. 2009;15:11078–11082. doi: 10.1002/chem.200901946. [DOI] [PubMed] [Google Scholar]; j) Delcamp JH, Gormisky PE, White MC. J. Am. Chem. Soc. 2013;135:8460–8463. doi: 10.1021/ja402891m. [DOI] [PMC free article] [PubMed] [Google Scholar]; k) Trost BM, Hansmann MM, Thaisrivongs DA. Angew. Chem. 2012;124:5034–5037. [Google Scholar]; Angew. Chem. Int. Ed. 2012;51:4950–4953. doi: 10.1002/anie.201200601. [DOI] [PubMed] [Google Scholar]
- 14.Bowden K, Cook RS. J. Chem. Soc. Perkin Trans. 2. 1972;2:1407. [Google Scholar]
- 15.a) Fraenkel G, Chen X, Gallucci J, Ren YL. J. Am. Chem. Soc. 2008;130:4140–4145. doi: 10.1021/ja0765215. [DOI] [PubMed] [Google Scholar]; b) Fiorelli C, Maini L, Martelli G, Savoia D, Zazzetta C. Tetrahedron. 2002;58:8679–8688. [Google Scholar]; c) Tanaka J, Nojima M, Kusabayashi S. J. Am. Chem. Soc. 1987;109:3391–3397. [Google Scholar]; d) Lamothe S, Cook K, Chan T. Can. J. Chem. 1992;70:1733–1742. [Google Scholar]; e) Terao J, Jin Y, Torii K, Kambe N. Tetrahedron. 2004;60:1301–1308. [Google Scholar]
- 16.17% of isomerization of the α product to the more conjugated product ((E)-but-2-ene-1,2-diyldibenzene) was detected with KN(SiMe3)2 base. No isomerization of the α product was detected with either NaN(SiMe3)2 or LiN(SiMe3)2 base.
- 17.Bordwell FG. Acc. Chem. Res. 1988;21:456–463. [Google Scholar]
- 18.S.A.C. LLC., PCy3 = $22/g; BrettPhos = 199/g.
- 19.a) Luu TXT, Lam TT, Le TN, Duus F. Molecules. 2009;14:3411–3424. doi: 10.3390/molecules14093411. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Al-Maskery I, Girling K, Jackson SD, Pugh L, Spence RR. Top. Catal. 2010;53:1163–1165. [Google Scholar]
- 20.This direct arylation, did not render any product when coupled with 2-3- or 4-pyridyl bromides.
- 21.pKa of acetophenone in DMSO is 24.7: Matthews WS, Bares JE, Bartmess JE, Bordwell FG, Cornforth FJ, Drucker GE, Margolin Z, McCallum RJ, McCollum GJ, Vanier NR. J. Am. Chem. Soc. 1975;97:7006.
- 22.Trost BM, Brindle CS. Chem. Soc. Rev. 2010;39:1600. doi: 10.1039/b923537j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.For reviews on arylation of activated C(sp3)−H bonds: Bellina F, Rossi R. Chem. Rev. 2010;110:1082. doi: 10.1021/cr9000836. Culkin DA, Hartwig JF. Acc. Chem. Res. 2003;36:234. doi: 10.1021/ar0201106.
- 24.Müller P, Nury P, Bernardinelli G. Eur. J. Org. Chem. 2001;21:4137. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.