

Abstract
The etiology of skeletal disease is driven by genetic and environmental factors. Genome-wide association studies (GWAS) of osteoporotic phenotypes have identified novel candidate genes, but have only uncovered a small proportion of the trait variance explained. This “missing heritability” is caused by several factors, including the failure to consider gene-by-environmental (G*E) interactions. Some G*E interactions have been investigated, but new approaches to integrate environmental data into genomic studies are needed. Advances in genotyping and meta-analysis techniques now allow combining genotype data from multiple studies, but the measurement of key environmental factors in large human cohorts still lags behind, as do the statistical tools needed to incorporate these measures in genome-wide association meta-studies. This review focuses on discussing ways to enhance G*E interaction studies in humans and how the use of rodent models can inform genetic studies. Understanding G*E interactions will provide opportunities to effectively target intervention strategies for individualized therapy.
Keywords: Bone mineral density, Candidate gene, Collaborative cross, Congenic strains, Diversity outcross, Fracture, Gene-by-environment interactions, Genome-wide association studies, Osteoporosis, Rodentmodels, Transgenic mice
Introduction
Osteoporosis is an aging-related disease characterized by micro-architectural deterioration of bone tissue, which increases skeletal fragility, leading to the occurrence of fractures. It is among the most common and debilitating diseases of the skeleton, resulting in over 9 million fractures annually worldwide [1]. Fragility fracture is the main and most detrimental outcome of osteoporosis, as fracture is associated with substantial increases in both mortality and morbidity. Bone mineral density (BMD) predictably declines with advanced age in both men and women, inversely correlates with fracture and, thus, remains the best clinical predictor for determining future fracture risk. Not surprisingly, the burden on healthcare systems and the associated costs are expected to increase with the aging of the population in western countries [2], and therefore early identification and treatment of subjects at high risk should help alleviate the burden of this disease.
A perpetual search for environmental factors (exposures) predisposing a patient to osteoporosis is underway [3]. In many instances, the environmental factors influencing bone physiology in a given individual can dominate genetic ones. Rarely is a patient subjected to a single environmental factor; as more commonly, multiple environmental exposures work in concert to impact bone health. Environmental factors include behaviors that the patient may have control over and factors that are outside of the patient's control. Thus, risk factors for fracture are both widespread and diverse and can include social disadvantage, high levels of social stress, nutritional inadequacies, and adverse behaviors such as smoking, physical inactivity, comorbidities, and in women, higher educational attainment and oral contraception use [4].
A multitude of data has been collected and data analysis conducted to facilitate the genetic exploration of the etiology of osteoporosis throughout the years. A traditional definition of “environment” embraces the circumstances where genes are operating, but recently, the chromatin environment has become recognized as an important part of the definition. As such the study of the etiology of osteoporosis has included both conventional genomics work, as well as incorporation of other types of data such as proteomics and explorations of the role of DNA methylation [5, 6]. The recent rapid accumulation of functional-genomics and proteomics data provides additional insight into the evolutionary relationship between the genotypes and human diseases [7]. From the genetics point of view, osteoporosis is considered a complex heritable disease, as by definition, both environmental (E) and genetic (G) components contribute to its development. However, neither (G) nor (E) alone can explain the risk of osteoporotic fracture, thus, the third part of the puzzle, namely gene-by-environmental (G*E) contributions, are suspected to play a role in the disease etiology. Herein we will review the progress that has been made in the study of G*E interactions in the field of bone biology. The goal of this review article is to highlight the challenges that recent developments have brought to light and to describe how they extend or revise previous concepts in the field.
Genetic and Genomic Exploration of Osteoporosis-Related Phenotypes
The idea of “genetic determinism” is all but discredited; there are multiple indications that virtually all human diseases result from the interaction of genetic susceptibility and environmental factors [8]. Even penetrance of some rare Mendelian diseases long thought to be the result of a deficiency in a single gene product, may depend on the modifying effects of environment. In terms of the quantitative genetic theory, G*E interaction means that genotypes vary in their environmental sensitivity, leading to variable expression of genotypes under different environment. Genotype-phenotype relationship can be dependent on the presence of environmental factor, and vice versa. Figure 1 illustrates the statistical aspects of the theory of interaction.
Fig. 1.
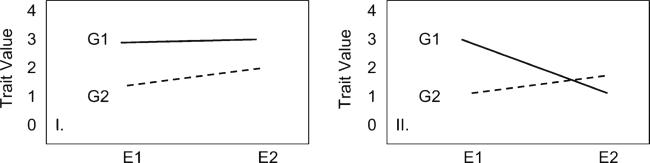
Graphical representation of some genotype-environment interactions. The y-axis represents a trait value; the x-axis represents 2 environmental conditions (E1 and E2). Under no interaction model I, genotype G1 is superior to G2 in each environment. Under interaction model II, the relative position of the 2 genotypes changes depending on the environment (crossover in the trait value of genotypes between environments). G1 is superior to G2 in 1 environment and inferior in the other, thus, the change in environment affects the 2 genotypes in opposite directions
In the last decade, the genetic exploration of human complex disease has become more robust. This was facilitated by the advent of technical advances allowing for large-scale genotyping coupled with the computational ability to analyze vast amounts of the DNA polymorphisms for association with disease and or phenotype. These so-called genome-wide association studies (GWAS) offer an unbiased approach to identify new candidate genes for human diseases. To date, the GWAS approach has proved productive in uncovering multiple genes responsible for complex diseases. However, the predictive ability of most of the GWAS findings is compromised by small percentage of the disease risk attributable to genetic factors, as detected by these studies. Moreover, the addition of polymorphisms (genetic risk score) on top of any model already saturated with environmental risk factors does not usually improve discrimination, but rather increases in the area-under-curve (AUC) are often modest [9, 10]. The AUC has a value for risk prediction, especially for studies into clinical outcomes. For example, in cardiovascular disease, traditional risk factors (such as blood pressure and serum cholesterol concentration) are more prominent than genetic factors (alterations in genes associated with the condition) for predicting disease susceptibility [11]. One can expect that the environment reflected by long-lasting risk factors would have consequences on penetrance of existing alleles and new mutations.
Which genetic variants should be expected to interact with the environment is still an unresolved question. Most GWAS report only significantly-associated genetic loci, which overwhelmingly are common single nucleotide polymorphisms (SNPs). These significantly-associated SNPs might have strong marginal effects, which propelled them to the top of the associated SNP set because their effects are not environment-dependent. Several studies that took into account main-effects of all common genome-wide polymorphisms still explain no more than half of the genetic predisposition [12••]. This phenomenon of “missing heritability” is partly due to the failure to take G*E interaction into account in the GWAS design and interpretation. Despite this recognized deficiency, few GWAS to date have incorporated G*E interactions into the analysis structure [13, 14] and this is primarily due to the challenges associated with doing so. There are at least 3 major issues currently hampering the incorporation of the G*E interaction term in GWAS: (1) we are often unable to discern what is a correct “environmental” exposure to interact with genome-wide variants, (2) the statistical power is usually low plus analytical methodology is under-developed or cumbersome, and (3) there are not yet developed criteria on how to replicate (statistically) and prove/validate the suggested interactions biologically. Here we provide more detailed view of these challenges and propose ways to overcome them, as pertains to osteoporosis and related skeletal pathology.
What Environment is Interacting with Individual Genetic Background to Predispose to Osteoporosis?
Reciprocity Between Changing Environment and Evolutionary Outcome: Specific Case of Skeleton
There is an ongoing interaction between a species’ genome and its environment over the course of many generations, which eventually shapes the species’ phenome [15]. Changes to genes produce phenotypes or traits that either do or do not suit an organism in the environment in which that mutated organism finds itself and some mutant alleles produce traits that enable an organism to better fit (survive) in a particular environment. Those same alleles, in a different environment, might not be advantageous [16, 17]. A change in environment can lead to maladaptations, which are reflected in prevalent chronic diseases [15, 18]. However, outcomes of skeletal aging, such as osteoporosis, should not serve as an evolutionary target as osteoporosis occurs past reproductive years. Osteoporosis can occur as a byproduct of selection on other skeletal traits, including those that are beneficial in early-life. For example, changes in vertebral bone size and shape were linked to evolutionary adaptations associated with bipedalism and in turn, they are associated with reduced bone strength in modern humans [19]. As such, the lighter and less robust bones of a modern Homo Sapiens have a distinct biomechanical advantage early on, yet become a risk factor for fractures later in life.
Evolutionary medicine can help discern which aspects of the human-specific environment one should be focused on. As it applies to skeletal traits, the modern Homo Sapiens environment can probably be traced to an effect of the exposure to farming and increased sedentarization. Gene-by-culture interaction in recent human evolution seems to reflect a long-lasting effect of dietary and physical activity changes during the last 15 millennia [20], including the invention of cooking [21] and increased fat intake. A variety of human-specific environmental factors that might affect osteoporosis risk have been proposed; some of which have been widely studied over the years in the context of bone disease. These factors include: diet (including microelements, maternal nutrition, and intra-uterine environment, with a foray into epigenetics); age and aging (ontogenetic stage); exercise/load-related; gender (sex hormones); modern medicines (such as hormones, beta-blockers, NSAIDS, and obviously, bisphosphonates and rosiglitazone); behavior (beyond diet and exercise), including smoking and alcohol consumption and social factors. The impact of some environmental factors is multifactorial. For example dietary fat intake is also a risk factor for obesity, which in turn is associated with local toxicity and general inflammation (negatively impacts bone), and biomechanically contributes to loading (positively impacts bone).
Interpreting these environmental influences from a disease risk prediction point of view both for an individual and for a population is complicated. A good review for calcium and vitamin D interactions with genetic variants on bone pheno-types has been recently published [22]. The conclusion was that adequate calcium and vitamin D intake can reduce the genetic risk of certain SNPs and improve bone health. The authors cite recommended consensus levels [23], which may or may not be applicable to all genotypes. Dietary recommendations, although trying to account for presence of subpopulations and differences between the reference cohorts, are still oriented to a majority of targeted populations, whereas genetics deals with rare variants and extreme phenotypes.
Studies of Genes-By-Environment Interactions in Human Samples
Single gene (SNP)-by-environment association studies are well established. Among the earliest studies, polymorphic differences in the following candidate genes were specifically interrogated for interactions with environmental conditions: VDR with age and dietary calcium [24], IL6 with diet and lifestyle [25], LRP5 with physical activity [26], and PPARG with dietary fat [27]. In Table 1 we present several more recent studies of G*E interactions with regard to osteoporotic relevant phenotypes, including BMD. Papers reporting candidate genes in community-dwelling adult participants where G*E was studied were included in this Table, but it is not an exhaustive list of all literature reported studies. Rather we applied the STREGA criteria [28] to select only the most robust studies for reporting in this summary.
Table 1.
Recent studies of G*E interactions with osteoporosis-related phenotypes in humans
| Gene | Tested marker(s) | Sample: age, sex, ethnic composition | Phenotype (skeletal site for BMD; how OP was defined) | Main effect: OR or RR aP value b | Interaction | Ref. |
|---|---|---|---|---|---|---|
| APOE | Alleles ε2/ε3/ε4 | Scottish women, early postmenopausal (n=2721) | LS BMD, FN BMD; BMD change | P≤0.05 for LS BMD and change in LS BMD | Tested for an interaction between the APOE polymorphism and dietary vitamin Kl intake | [78] |
| COLIA1 | –1997G/T (rs1107946),- 1663in/delT (rs2412298), and +1245G/T | 1717 Caucasian perimenopausal women (age 50.0±2.9 y) | LS BMD, Hip BMD; All Frx & vertebral Frx; turnover markers (25-hydroxyvitamin D, osteocalcin, bone specific alkaline phosphatase, hydroxyproline) | P<.05-0.005 | No association with pMP change in BMD (10-y follow-up), fracture risk; no interaction with the hormone therapy | [79] |
| ESR1 | 4 SNPs, incl. rs2077647 and rs2234693 | Japanese women (cases: 114 postmenopausal with a confirmed osteoporosis) and controls: 171 healthy (mean age of 39.0 y) | Osteoporosis risk | OR = 3.15, 95 % CI = 1.83-5.41 (Haplotype of 2 SNPs) | Statistically significant interaction (P=0.03) with alcohol drinking | [80] |
| IL6 | –634C/G | 176 Chinese girls aged 9–11 y (Pre-menarche) | BMD and BMC at total body, total hip and FN | <0.05 | Interactions observed with physical activity (P<0.05) and calcium intake | [81] |
| LRP5 | 4 SNPs (incl. rs4988321) | Greek postmenopausal women (n = 578); stratified by calcium intake revealed that in the low calcium intake group (at 680 mg/d) | LS and hip BMD osteoporotic Frx | rs4988321 with LS BMD (P=0.002) | Interaction of the rs4988321 with calcium intake (P=0.016); In low calcium intake group, rs4988321 Associated with LS BMD (P=0.001) | [82] |
| MTHFR | rs1801133 (C677T) | 5035 Dutch men and women, age 55+ y-old | Fracture | n.s. | Found interaction with dietary riboflavin intake on Frx risk | [83] |
| MTHFR | rs1801133 (C677T) | 5816 U.K. Caucasian children, 9.9 y-old | BMD: total body less head region (N=5816), lumbar spine (N=3196) | P<0.001 (spine BMD) | SNP*sex interaction: P=0.04 (spine BMD) | [84] |
| PPARG | 13 SNPs (incl. rs2028760, rs1801282, rs1805192) | US Caucasian men (n=867, 62.2±9.1 y) and women (n=925, 60.5±9.1 y) | FN, TR, and LS BMD | <0.05 (mostly in men with FN) | Interaction with dietary fat intake on BMD: rs1151999 & rs709150 in men and rs1175381 and rs1186464 in women. | [27] |
| PPARG | 10 SNPs (incl. rs12497191, rs4135263, rs1151999, and rs1152003) | 2 Danish cohorts: a case-control (n=809) and 1716 perimenopausal women allocated to hormone therapy or not at baseline and followed for 10 y | vertebral Frx; LS & FN BMD | VertFrx risk (OR= 1.48-1.76, P=0.005-0.04 for rs12497191, rs4135263, and rs1151999) increased BMD (P≤0.02 for rs1151999) | An interaction between rs1151999 and diet and rs1152003 and weight on BMD | [85] |
| VDR | BsmI, TaqI and Cdx-2 | Greek postmenopausal women (n=578); stratified by calcium intake | LS & hip BMD osteoporotic Frx | n.s. | In low calcium intake group (<680 mg/d), all variants were associated with LS BMD (P<0.05) | [86] |
BMC bone mineral content, BMD bone mineral density, CI confidence interval, FN femoral neck, Frx fracture, LS lumbar spine, m.s. marginally significant, n.s. nonsignificant, OP osteoporosis, OR odds ratio, PMP postmenopausal (women), RR risk ratio, TR trochanter
OR and 95 % CI
Statistical significance: when there are more than one phenotype or SNP, lowest P value is provided
One of the requirements of all recent association studies is replication of the major findings. Unfortunately, replication of G*E interacting loci has met with limited success. Inconsistent association for any given allele might be due partly to differences inherent to the study populations, the measurement techniques used for data collection, and the statistical modeling algorithms used. The differences in the study populations may be due to the study-specific environment, which is difficult to identify and control. For example, there might be gross differences in levels of typical dietary intakes or the study-specific threshold could be very different among population samples (for example, median fat intake in a sample from Scandinavian population will be way too high for the Mediterranean region counterparts). Obviously, age differences between the discovery and replication samples must be considered, especially for aging-related traits such as BMD. Long-term exposure to stress or other adverse effects may contribute to the disparity between results in younger and older cohorts, in addition to age associated physiologic decline. Thus, these inconsistencies could mask a true replication of an association, or a failure of replication simply could be because a true association does not exist (ie, the original finding was a spurious/false-positive association). The difficulty arises in determining the root cause of any failure to replicate.
Both association studies with predefined candidate genes and GWAS require inclusion of an interaction term with a covariate (exposure) to produce G*E interaction statistics. The analytical challenge for the association studies is that the sample size for the interactions assessment should be approximately 4 times larger than the sample used to detect genetic main effects only [29•]. Another limitation of G*E statistical exploration is that “environment” is usually heuristic and selected from a list of measurable data available to the researcher and the measurement of environment in large human samples still lags behind primary genotype and phenotype data collection. Many exposure measurements are perceived as “soft” in metrics, because they often rely on subjective reports or non-adjudicated physician assessment. Furthermore, there is no single tool like GWAS for environmental screening, although approaches like ‘Environment-wide Association Study’ (EWAS) have been proposed [30] but these studies are limited by the measurements already included in a dataset.
Issues to be Considered for Meta-Analysis Approaches
The combining of cohorts by meta-analyses approaches must often be employed to increase statistical power, but the lack of availability of common environmental data for all subjects or their assessment with different tools make the pooling of data very difficult, as is outlined above [22]. Furthermore, as is also outlined above, the exposure to some factors can be significantly different between different populations [22], which makes combining them for meta-analyses prone to error due to heterogeneity. The diversity of methods available to assess dietary intake and health habits pose problems for comparisons between different studies and populations. To generate a comparable G*E statistics in multiple studies, a threshold might be assigned based on the level of exposure to the environmental factor. For example, an exposure can be ranked, categorized, or dichotomization (high vs low level exposure) can be applied. Although intuitive for interpretation, such simplification of the environment is a limitation, since it weakens the statistical power by further stratification of the sample.
Long Way from GWAS to GEWIS
As follows from the above appraisal of the status in the field, a well-powered “GEWIS” – which stands for gene-environment-wide interaction study - is years away. A mid-way between the GWAS and GEWIS is “GWIS” - genome-wide interaction study, which models interaction of genome-wide polymorphisms with 1 covariate at a time. An example is that of Liu et al. [31], in which genome-wide SNP interactions with sex were studied for lumbar spine (LS) and femoral neck (FN) BMD. The question this study attempted to solve was whether different genes are at work in the male vs female skeleton, or whether expression and penetrance of the same genes are modulated by gender-specific environments, such as exposure to gonadal steroids, differences in physical activity, and muscle strength. Despite the large collaborative effort (25,353 individuals from 8 Caucasian cohorts were included), no genome-wide significant (P<5×10-8) evidence for gene-bysex interaction were found to influence BMD variation in that screen. The authors suggest that, if they exist, gene-by-sex interactions probably have weak effects on BMD, accounting for less than 0.08 % of its variation per implicated SNP.
Another study, by Koller et al. [32], can also be partly considered an example of GWIS, as it attempted to estimate an effect of 1 environmental stratum (by sex and menopause) on association between the genome-wide SNPs and BMD. In GWAS of premenopausal women from 4 Caucasian cohorts (n=4061), SNPs in the WNT16 and ESR1/C6ORF97 achieved genome-wide significant association. This study confirmed that several of the genes contributing to BMD variation across a broad age range in men and women had effects of similar magnitude on BMD in premenopausal women, which implies that no interaction takes place. This study is a response to a statistical challenge of analyzing 2-way interaction (G * sex * E), for which the power of discovery is almost zero. Indeed, some authors warned against arbitrarily hypothesizing and stratifying the samples, in absence of formally-tested (and significant) interaction term [33].
A Need for Biological Validation of G*E Findings
Replication of a G*E study in an independent human sample (reproducibility) is required, but statistically-derived associations are universally considered hypothetical unless functionally validated. Often times, functional validation cannot be accomplished and or molecular modes of action cannot be ascertained in humans or from human derived tissue samples. The use of animal models allows experiments to be conducted specifically to overcome many of the limitations associated with the study of disease in humans [34], since they provide both great precision for environmental measurement/manipulation and functional validation of interactions.
The use of Mouse Models in G*E Research: Advantages and Cautions
Use of animal models fill 2 specific needs particularly well in the context of the study of G*E interactions: they can be used to collect phenotype data that cannot be collected from human subjects and they can be used to study single environmental variables in isolation. Among model organisms, the mouse is considered important for the study of human diseases. The mouse genome is remarkably comparable to the human genome at the nucleotide level. At the gene level approximately 17,770 mouse genes have a known direct human ortholog (http://www. informatics.jax.org), and organizationally, the mouse and human genomes remain highly syntenic despite a quite long evolutionary distance between them [35]. Thus, genetic findings in mice are often concordant with genetic findings in humans [36].
Physiologically and anatomically, mice and humans are remarkably similar and while there are some marked differences between them, the abject number of differences is small compared with the number of similar if not identical systems. The question remains, among others: can human diet and physical exercise be correctly modeled in rodents? Evolutionarily mice and humans diverged 70–75 million years ago [35] and mice display distinct evolutionary patterns from humans. A blind following of discoveries made in mice without considering the differences between these 2 species could produce a bias but despite these differences, there is evidence of translatability between gene x diet studies conducted in mice and those using data from humans. We have shown that allelic differences in Pparg modulate the impact of total dietary fat on bone mass in both mice and humans [37], demonstrating that despite physiologic differences between these 2 species, G*E interactions found in mice are meaningful and informative for human health.
Genetic Loci by Environment Mapping Studies in Mice
Genetic loci can be identified using mice via a number of different approaches [38]. In the bone field, the 3 methods that have been most utilized are the 2-strain intercross [36, 39–44], recombinant inbred lines [45–50], and recombinant congenic strains [51–54]. The 2-strain intercross method for genetic mapping in mice involves creating a population of genetically unique mice by either intercrossing 2 strains of mice for at least 2 generations or intercrossing 2 strains followed by backcrossing to 1 of the founder strains. Once this population of mice is generated, each mouse is phenotyped and genotyped and genetic loci are identified by interval mapping [38].
Particularly interesting for gene by environment studies are the 2 crosses that have been made by intercrossing the C57BL/6J (B6) and C3H/HeJ (C3H) strains [36, 55, 56]. For both of these crosses, femoral BMD was measured in young adult mice (16 and 20 weeks respectively), however, the mice used in the “Beamer” cross were fed a low fat rodent chow diet whereas the “Farber” mice were fed a high fat Western diet. Although a number of genetic loci in common were identified between the 2 studies, such as on Chromosomes (Chr) 4, 10, and 18, their results were not perfectly concordant. For example, Beamer et al. identified a locus at 30 cM on mouse Chr 13 (corrected mouse genetic map position [57, 58]), whereas no locus was identified on the 13th Chr in the Farber cross [36, 56]. There are slight technical differences between the Farber and Beamer studies that could account for these mapping discrepancies, but the most profound dissimilarity remains the diets used, suggesting that the Chr 13 QTL may represent a diet interacting locus. This hypothesis is further substantiated by the observation that the Chr 6 locus which we previously demonstrated to interacted with dietary fat [27], was not observed in the Farber cross [36, 56].
A lingering issue involving the use of the 2 strain-F2 cross strategy is one of mapping resolution. The confidence intervals supporting the peak location for QTL mapped using this strategy tend to be large and incorporate several hundred genes. One method that can be used to overcome this problem is to continue to interbreed the mice past the second generation to create so called Advanced Intercross Lines (AIL) or advanced intercross populations [59, 60]. This effectively increases the density of the observable genetic recombinations between the 2 strains, thereby increasing mapping resolution [59]. An AIL created by inter-breeding the B6 and the High-runner strains was recently used to identify 2 dietary fat interacting loci for bone: 1 on Chr 19 for femoral length and 1 on Chr 7 for femur shape, but none of the BMD loci mapped in this study interacted with dietary fat. This study was very small, including only 466 mice, and it is possible that there was insufficient power to identify all interacting QTL after incorporating other covariates such as gender and age into the mapping model [61•]. In a much larger study, using an AIL descended from the LG/J and SM/J strains, Norgard et al. identified 17 loci for long bone length that interacted with dietary fat [62], but BMD was not examined in this study.
A variation on the intercross strategy is the recombinant inbred (RI) strain strategy. In the RI strategy, the goal is create a population of repeatable genetic diversity. In short, 2 strains are intercrosses for at least 2 generations and then de novo lines or strains of mice are created by inbreeding these lines to the point where each mouse is homozygous or “fixed” for all genetic alleles and each mouse within a line is isogenic with all other mice of that same line. As a set, alleles from both parental strains are equally represented in the population at the majority of genetic locations, but each line is a unique combination of fixed founder alleles [63]. Using the LG/J by SM/J (LGXSM) RI population, Carson et al. showed that dietary fat impacted trabecular bone volume over total volume ratio (BV/TV) and trabecular shape in the proximal tibia and BV/TV, trabecular thickness, spacing, connectivity density, and shape in the lumbar vertebral body. Further, these investigators were able to map loci for various aspects of trabecular morphology to Chrs 2, 9, 10, and 14 [46]; (this decrease in trabecular bone mass is associated with increased osteoclast activity [64]). This work emphasizes that caution must be used when drawing overarching conclusions regarding how an environmental factor affects bone based on work conducted with a single inbred strain, as the impact an environment condition on bone is contingent on the alleles present.
New Mouse Genetic Reference Populations for G*E Studies in Mice
There remains a stigma attached to genetic mapping in mice based on the pervading belief that only loci can be identified but not actually genes [65]. This is simply no longer the case. The mouse genetics research community has made heavy investments into the creation of resources and the development of methodologies that are revolutionizing the landscape of genetic research using mouse models. For example, draft genome sequences for 17 strains of mice are now available, which serve to define the genetic differences present in these strains including simple polymorphisms such as SNP, small insertion/deletions (indels), as well as larger differences such as long deletions, insertions, copy number variations (CNVs), and rearrangements [66]. Based on the known ancestral relationships between different strains, this genome sequence data can be used to impute the polymorphic differences in a much wider number of inbred mouse strains than just the 17 that have been sequenced to date [67]. The time and resources have been devoted to the creation of new sets of isogenic stocks [68•, 69–72], genetically modified mice (Table 2) and new outbred mouse stocks [73] that facilitate designing a number of different studies germane to addressing questions regarding G*E interactions. These resources only just begun to be used in skeletal biology, but they represent significant advance for the field [74•].
Table 2.
Recent finding demonstrating diet by gene interactions impacting bone in mice
| Dietary factor | Gene | Findings | Reference |
|---|---|---|---|
| Calcium | Trpv6 | Increases gut calcium absorption in situations of low dietary calcium | [87] |
| Casr | Is required in neonates to respond to low calcium situations | [88•] | |
| Pth1r | Phosphorylation of Pthr1 is required for normal calcium homeostasis in low calcium situations | [89] | |
| Pth1r | Pth1r signaling through phospholipase C is protective for bone mass in low calcium situations. | [90] | |
| Phosphate | Galnt3 | A low phosphate diet may be a viable treatment option for familial tumoral calcinosis | [91•] |
| High fat intake | Spry1 | Adipocyte specific over expression of this gene protects against high fat diet induced bone loss | [92] |
| Alox5 | Loss of Alox5 exacerbates high fat diet induced bone loss, presumably by increasing osteoclastic action | [93] | |
| Sparc | Increased cortical bone after high fat feeding was observed in mice lack Sparc (Osteonectin). | [94] | |
| Protein | Igf1 | Over expression of Igf1 in osteoblasts mitigates bone loss induced by feeding a low protein diet. | [95] |
Use of Transgenic Mice for G*E Studies
One advantage to the use of mice to determine gene function is that their genome can be easily modified and transgenic mice are being used with increasing frequency to examine G*E effects. The mouse proves to be ideal for these studies as environmental factors can be controlled fairly tightly and 1 environmental perturbation at a time can be examined. Recent gene by diet studies in rodents have provided a wealth of information not only about basic gene function, but have also proven useful for testing putative treatment options (Table 2). The one caveat to broad scale comparisons of diet studies in mice is the lack of diet standardization in the field and often the details matter in these studies. For example a variety of high fat rodent diets are all commonly used. Not only is there no consensus regarding with what percent Kcal from fat constitutes “high fat”, the type of fat in these diets varies widely. C57BL/6J (B6) mice, are acutely sensitive to dietary fat with regards to skeletal response when fed a lard based diet [64], but show no skeletal response to high fat butter/corn oil based diets [27]. Further, it goes without saying that strain background matters in these studies. Sadly, little is known regarding how different strains will respond to different types of dietary challenge with regards to bone physiology. It must be remembered that a lack of change in the phenotype of a transgenic model after challenge with a dietary modification may well be a function of unappreciated epistatic factors.
Increasingly, mouse models are being used to understand the mechanisms underlying number of other environmental factor by gene interactors outside of just nutrition, including alcohol consumption, smoking, and to a lesser extent, exercise. While exercise can be modeled in mice, there has been little work in the G*E bone realm thus far. Examples of recent studies towards this end include the demonstration by Shimizu et al. that loss of Aldh2 increases the sensitivity of bone to ethanol-induced bone loss [75] and the work by Wahl et al., who showed that loss of Tnfr1 is actually protective against the suppressive effects of ethanol on fracture defect repair [76••].
Conclusions and Recommendations for Design of Future Genetic Epidemiologic Studies of Interaction
The knowledge of which environment can alter the expression of genes that cause or ameliorate disease is important, therefore it should be target of our investigations. To identify biomarkers that will allow early disease detection by stratification on exposure, there is a need to develop a new approach to augment existing methods, including enhancements from a statistical point of view [18] and integration of the animal models into this research topic. Also of note, according to an evolutionary view [7], disease genes affecting morphological traits such as anatomical structures evolve more slowly than those affecting physiological traits such as immune responses. If this observation is true also for phenome responses during shorter periods, there's a need to analyze “endophenotypes” (intermediate phenotypes), such as bone turnover measures, to catch up with short-term exposure changes. New imaging techniques and molecular markers might be more sensitive to capture environmental exposure that affects them and interacts with their underlying genetics. By identifying and characterizing gene-by-modifiable environment interactions, we have more opportunities to effectively target intervention strategies [77].
There is a genuine need for investment into the study of G*E interactions, as a widespread screening for osteoporosis is already being performed in clinical practice and this level of knowledge will be crucial for making recommendations for treatments and or interventions at a personalized medicine level. For example, exercise and physical therapy may be beneficial for some genotypes more than for others. G*E interactions, if robustly identified and biologically significant, could prove useful in primary health care to persuade the genetically vulnerable patients to modify their behavior with far reaching benefits in health and the cost of health care. In summary, understanding gene-by-modifiable environment interactions will afford more opportunities to effectively target intervention strategies and deliver individual therapy, by providing the evidence needed to improve prevention of skeletal diseases through exposure-based interventions.
Footnotes
Compliance with Ethics Guidelines
Conflict of Interest CL Ackert-Bicknell's institution has received grant from NIAMS/NIH (AR060234) to support her work. D Karasik declares that he has no conflicts of interest.
Human and Animal Rights and Informed Consent This article does not contain any studies with human or animal subjects performed by any of the authors.
Contributor Information
Cheryl L. Ackert-Bicknell, The Jackson Laboratory, 600 Main St, Bar Harbor, ME 04609, USA cheryl.ackertb@jax.org
David Karasik, Faculty of Medicine in the Galilee, Bar-Ilan University, H. Szold St. 14, POB 1589, Safed 1311502, Israel; Hebrew SeniorLife and Harvard Medical School, Boston MA 02131, USA.
References
Papers of particular interest, published recently, have been highlighted as:
• Of importance
•• Of major importance
- 1.Johnell O, Kanis JA. An estimate of the worldwide prevalence and disability associated with osteoporotic fractures. Osteoporos Int. 2006;17(12):1726–33. doi: 10.1007/s00198-006-0172-4. [DOI] [PubMed] [Google Scholar]
- 2.Burge R, Dawson-Hughes B, Solomon DH, et al. Incidence and economic burden of osteoporosis-related fractures in the United States, 2005–2025. J Bone Miner Res. 2007;22(3):465–75. doi: 10.1359/jbmr.061113. [DOI] [PubMed] [Google Scholar]
- 3.Samelson EJ, Cupples LA, Hannan MT, et al. Long-term effects of serum cholesterol on bone mineral density in women and men: the Framingham Osteoporosis Study. Bone. 2004;34(3):557–61. doi: 10.1016/j.bone.2003.11.024. [DOI] [PubMed] [Google Scholar]
- 4.Leslie WD. Clinical review: ethnic differences in bone mass– clinical implications. J Clin Endocrinol Metab. 2012;97(12):4329–40. doi: 10.1210/jc.2012-2863. [DOI] [PubMed] [Google Scholar]
- 5.Xu XH, Dong SS, Guo Y, et al. Molecular genetic studies of gene identification for osteoporosis: the 2009 update. Endocr Rev. 2010;31(4):447–505. doi: 10.1210/er.2009-0032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Easwaran H, Johnstone SE, Van Neste L, et al. A DNA hypermethylation module for the stem/progenitor cell signature of cancer. Genome Res. 2012;22(5):837–49. doi: 10.1101/gr.131169.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Park S, Yang JS, Kim J, et al. Evolutionary history of human disease genes reveals phenotypic connections and comorbidity among genetic diseases. Sci Rep. 2012;2:757. doi: 10.1038/srep00757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Khoury MJ, McCabe LL, McCabe ER. Population screening in the age of genomic medicine. N Engl J Med. 2003;348(1):50–8. doi: 10.1056/NEJMra013182. [DOI] [PubMed] [Google Scholar]
- 9.Pencina MJ, D'Agostino Sr RB, D'Agostino Jr RB, Vasan RS. Evaluating the added predictive ability of a new marker: from area under the ROC curve to reclassification and beyond. Stat Med. 2008;27(2):157–72. doi: 10.1002/sim.2929. discussion 207–12. [DOI] [PubMed] [Google Scholar]
- 10.Lee SH, Lee SW, Ahn SH, et al. Multiple gene polymorphisms can improve prediction of non-vertebral fracture in postmenopausal women. J Bone Miner Res. 2013 doi: 10.1002/jbmr.1955. doi:10.1002/jbmr.1955. [DOI] [PubMed] [Google Scholar]
- 11.Burke W, Psaty BM. Personalized medicine in the era of genomics. JAMA. 2007;298(14):1682–4. doi: 10.1001/jama.298.14.1682. [DOI] [PubMed] [Google Scholar]
- 12••.Yang J, Benyamin B, McEvoy BP, et al. Common SNPs explain a large proportion of the heritability for human height. Nat Genet. 2010;42(7):565–9. doi: 10.1038/ng.608. [Using data from a large-size GWAS of human height, this paper demonstrates that much of the so-called ‘missing heritability’ can be explained by large numbers of polymorphisms of small effect. By including allelic data at lesser minor allelic frequency and a restricted maximum likelihood model, they were able to explain approximately one-half heritability of human height.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Beaty TH, Ruczinski I, Murray JC, et al. Evidence for gene-environment interaction in a genome wide study of nonsyndromic cleft palate. Genet Epidemiol. 2011;35(6):469–78. doi: 10.1002/gepi.20595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu Y, Xu H, Chen S, et al. Genome-wide interaction-based association analysis identified multiple new susceptibility Loci for common diseases. PLoS Genet. 2011;7(3):e1001338. doi: 10.1371/journal.pgen.1001338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cordain L, Eaton SB, Sebastian A, et al. Origins and evolution of the Western diet: health implications for the 21st century. Am J Clin Nutr. 2005;81(2):341–54. doi: 10.1093/ajcn.81.2.341. [DOI] [PubMed] [Google Scholar]
- 16.Scott EC. This I, believe: we need to understand evolution, adaptation, and phenotype. Front Genet. 2012;3:303. doi: 10.3389/fgene.2012.00303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Varki A, Geschwind DH, Eichler EE. Explaining human uniqueness: genome interactions with environment, behavior and culture. Nat Rev Genet. 2008;9(10):749–63. doi: 10.1038/nrg2428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Booth FW, Lees SJ. Fundamental questions about genes, inactivity, and chronic diseases. Physiol Genomics. 2007;28(2):146–57. doi: 10.1152/physiolgenomics.00174.2006. [DOI] [PubMed] [Google Scholar]
- 19.Cotter MM, Loomis DA, Simpson SW, et al. Human evolution and osteoporosis-related spinal fractures. PLoS One. 2011;6(10):e26658. doi: 10.1371/journal.pone.0026658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Karasik D. Osteoporosis: an evolutionary perspective. Hum Genet. 2008;124(4):349–56. doi: 10.1007/s00439-008-0559-8. [DOI] [PubMed] [Google Scholar]
- 21.Stedman HH, Kozyak BW, Nelson A, et al. Myosin gene mutation correlates with anatomical changes in the human lineage. Nature. 2004;428(6981):415–8. doi: 10.1038/nature02358. [DOI] [PubMed] [Google Scholar]
- 22.Stathopoulou MG, Grigoriou E, Dedoussis G. Calcium and vitamin D intake interactions with genetic variants on bone phenotype. Curr Nutr Rep. 2012;1:169–74. [Google Scholar]
- 23.Dawson-Hughes B, Mithal A, Bonjour JP, et al. IOF position statement: vitamin D recommendations for older adults. Osteoporos Int. 2010;21(7):1151–4. doi: 10.1007/s00198-010-1285-3. [DOI] [PubMed] [Google Scholar]
- 24.Ferrari S, Rizzoli R, Manen D, et al. Vitamin D receptor gene start codon polymorphisms (FokI) and bone mineral density: interaction with age, dietary calcium, and 3′-end region polymorphisms. J Bone Miner Res. 1998;3(6):925–30. doi: 10.1359/jbmr.1998.13.6.925. [DOI] [PubMed] [Google Scholar]
- 25.Ferrari SL, Karasik D, Liu J, et al. Interactions of interleukin-6 promoter polymorphisms with dietary and lifestyle factors and their association with bone mass in men and women from the framing-ham osteoporosis study. J Bone Miner Res. 2004;19(4):552–9. doi: 10.1359/JBMR.040103. [DOI] [PubMed] [Google Scholar]
- 26.Kiel DP, Ferrari SL, Cupples LA, et al. Genetic variation at the low-density lipoprotein receptor-related protein 5 (LRP5) locus modulates Wnt signaling and the relationship of physical activity with bone mineral density in men. Bone. 2007;40(3):587–96. doi: 10.1016/j.bone.2006.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ackert-Bicknell CL, Demissie S, Marin de Evsikova C, et al. PPARG by dietary fat interaction influences bone mass in mice and humans. J Bone Miner Res. 2008;23(9):1398–408. doi: 10.1359/JBMR.080419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Little J, Higgins JP, Ioannidis JP, et al. STrengthening the REporting of Genetic Association Studies (STREGA)–an extension of the STROBE statement. Genet Epidemiol. 2009;33(7):581–98. doi: 10.1002/gepi.20410. [DOI] [PubMed] [Google Scholar]
- 29•.Thomas D. Gene—environment-wide association studies: emerging approaches. Nat Rev Genet. 2010;11(4):259–72. doi: 10.1038/nrg2764. [A useful review with forays into the randomized clinical trials and pharmacogenomics.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Patel CJ, Cullen MR, Ioannidis JP, Butte AJ. Systematic evaluation of environmental factors: persistent pollutants and nutrients correlated with serum lipid levels. Int J Epidemiol. 2012;41(3):828–43. doi: 10.1093/ije/dys003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu CT, Estrada K, Yerges-Armstrong LM, et al. Assessment of gene-by-sex interaction effect on bone mineral density. J Bone Miner Res. 2012;27(10):2051–64. doi: 10.1002/jbmr.1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Koller DL, Zheng HF, Karasik D, et al. Meta-analysis of genome-wide studies identifies WNT16 and ESR1 SNPs associated with bone mineral density in premenopausal women. J Bone Miner Res. 2013;28(3):547–58. doi: 10.1002/jbmr.1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Patsopoulos NA, Tatsioni A, Ioannidis JP. Claims of sex differences: an empirical assessment in genetic associations. JAMA. 2007;298(8):880–93. doi: 10.1001/jama.298.8.880. [DOI] [PubMed] [Google Scholar]
- 34.Aitman TJ, Boone C, Churchill GA, et al. The future of model organisms in human disease research. Nat Rev Genet. 2011;12(8):575–82. doi: 10.1038/nrg3047. [DOI] [PubMed] [Google Scholar]
- 35.Waterston RH, Lindblad-Toh K, Birney E, et al. Initial sequencing and comparative analysis of the mouse genome. Nature. 2002;420(6915):520–62. doi: 10.1038/nature01262. [DOI] [PubMed] [Google Scholar]
- 36.Ackert-Bicknell CL, Karasik D, Li Q, et al. Mouse BMD quantitative trait loci show improved concordance with human genome-wide association loci when recalculated on a new, common mouse genetic map. J Bone Miner Res. 2010;25(8):1808–20. doi: 10.1002/jbmr.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Paigen K. A miracle enough: the power of mice. Nat Med. 1995;1(3):215–20. doi: 10.1038/nm0395-215. [DOI] [PubMed] [Google Scholar]
- 38.Broman KW. Review of statistical methods for QTL mapping in experimental crosses. Lab Anim (NY) 2001;30(7):44–52. [PubMed] [Google Scholar]
- 39.Li X, Masinde G, Gu W, et al. Genetic dissection of femur breaking strength in a large population (MRL/MpJ × SJL/J) of F2 Mice: single QTL effects, epistasis, and pleiotropy. Genomics. 2002;79(5):734–40. doi: 10.1006/geno.2002.6760. [DOI] [PubMed] [Google Scholar]
- 40.Masinde GL, Wergedal J, Davidson H, et al. Quantitative trait loci for periosteal circumference (PC): identification of single loci and epistatic effects in F2 MRL/SJL mice. Bone. 2003;32(5):554–60. doi: 10.1016/s8756-3282(03)00063-2. [DOI] [PubMed] [Google Scholar]
- 41.Kesavan C, Mohan S, Srivastava AK, et al. Identification of genetic loci that regulate bone adaptive response to mechanical loading in C57BL/6J and C3H/HeJ mice intercross. Bone. 2006;39(3):634–43. doi: 10.1016/j.bone.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 42.Wergedal J, Ackert-Bicknell C, Tsaih S, et al. Femur mechanical properties in the F2 progeny of an NZB/B1NJ × RF/J cross are regulated predominantly by genetic loci that regulate bone geometry. J Bone Miner Res. 2006;21(8):1256–66. doi: 10.1359/jbmr.060510. [DOI] [PubMed] [Google Scholar]
- 43.Shimizu M, Higuchi K, Bennett B, et al. Identification of peak bone mass QTL in a spontaneously osteoporotic mouse strain. Mamm Genome. 1999;10(2):81–7. doi: 10.1007/s003359900949. [DOI] [PubMed] [Google Scholar]
- 44.Nakanishi R, Shimizu M, Mori M, et al. Secreted frizzled-related Protein 4 is a negative regulator of peak BMD in SAMP6 Mice. J Bone Miner Res. 2006;21(11):1713–21. doi: 10.1359/jbmr.060719. [DOI] [PubMed] [Google Scholar]
- 45.Bower AL, Lang DH, Vogler GP, et al. QTL analysis of trabecular bone in BXD F2 and RI mice. J Bone Miner Res. 2006;21(8):1267–75. doi: 10.1359/jbmr.060501. [DOI] [PubMed] [Google Scholar]
- 46.Carson EA, Kenney-Hunt JP, Pavlicev M, et al. Weak genetic relationship between trabecular bone morphology and obesity in mice. Bone. 2012;51(1):46–53. doi: 10.1016/j.bone.2012.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lang DH, Sharkey NA, Mack HA, et al. Quantitative trait loci analysis of structural and material skeletal phenotypes in C57BL/6J and DBA/2 second-generation and recombinant inbred mice. J Bone Miner Res. 2005;20(1):88–99. doi: 10.1359/JBMR.041001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Reich MS, Jarvis JP, Silva MJ, Cheverud JM. Genetic relationships between obesity and osteoporosis in LGXSM recombinant inbred mice. Gen Res. 2008;90(5):433–44. doi: 10.1017/S0016672308009798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Klein R, Mitchell S, Phillips T, et al. Quantitative trait loci affecting peak bone mineral density in mice. J Bone Miner Res. 1998;13:1648–56. doi: 10.1359/jbmr.1998.13.11.1648. [DOI] [PubMed] [Google Scholar]
- 50.Lang DH, Sharkey NA, Lionikas A, et al. Adjusting data to body size: a comparison of methods as applied to quantitative trait loci analysis of musculoskeletal phenotypes. J Bone Miner Res. 2005;20(5):748–57. doi: 10.1359/JBMR.041224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Saless N, Litscher SJ, Lopez Franco GE, et al. Quantitative trait loci for biomechanical performance and femoral geometry in an inter-cross of recombinant congenic mice: restriction of the Bmd7 candidate interval. FASEB J. 2009;23(7):2142–54. doi: 10.1096/fj.08-118679. [DOI] [PubMed] [Google Scholar]
- 52.Saless N, Litscher SJ, Houlihan MJ, et al. Comprehensive skeletal phenotyping and linkage mapping in an intercross of recombinant congenic mouse strains HcB-8 and HcB-23. Cells Tissues Organs. 2011;194(2–4):244–8. doi: 10.1159/000324774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Saless N, Litscher SJ, Vanderby R, et al. Linkage mapping of principal components for femoral biomechanical performance in a reciprocal HCB-8 × HCB-23 intercross. Bone. 2011;48(3):647–53. doi: 10.1016/j.bone.2010.10.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Saless N, Lopez Franco GE, Litscher S, et al. Linkage mapping of femoral material properties in a reciprocal intercross of HcB-8 and HcB-23 recombinant mouse strains. Bone. 2010;46(5):1251–9. doi: 10.1016/j.bone.2010.01.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Beamer WG, Shultz KL, Donahue LR, et al. Quantitative trait loci for femoral and lumbar vertebral bone mineral density in C57BL/6J and C3H/HeJ inbred strains of mice. J Bone Miner Res. 2001;16:1195–206. doi: 10.1359/jbmr.2001.16.7.1195. [DOI] [PubMed] [Google Scholar]
- 56.Farber CR, van Nas A, Ghazalpour A, et al. An integrative genetics approach to identify candidate genes regulating BMD: combining linkage, gene expression, and association. J Bone Miner Res. 2009;24(1):105–16. doi: 10.1359/JBMR.080908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cox A, Ackert-Bicknell CL, Dumont B, et al. A new standard genetic map for the laboratory mouse. Genetics. 2009;182(4):1335–44. doi: 10.1534/genetics.109.105486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ackert-Bicknell C, Paigen B, Korstanje R. Recalculation of 23 mouse HDL QTL datasets improves accuracy and allows for better candidate gene analysis. J Lipid Res. 2013;54(4):984–94. doi: 10.1194/jlr.M033035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Darvasi A, Soller M. Advanced intercross lines, an experimental population for fine genetic mapping. Genetics. 1995;141(3):1199–207. doi: 10.1093/genetics/141.3.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Darvasi A. Experimental strategies for the genetic dissection of complex traits in animal models. Nat Genet. 1998;18(1):19–24. doi: 10.1038/ng0198-19. [DOI] [PubMed] [Google Scholar]
- 61•.Leamy LJ, Kelly SA, Hua K, et al. Quantitative trait loci for bone mineral density and femoral morphology in an advanced intercross population of mice. Bone. 2013;55(1):222–9. doi: 10.1016/j.bone.2013.02.014. [This paper clearly highlights that dietary fat interacts with genetic loci to impact bone morphology, but also serves to highlight the complexity and power issue trade-offs that arise from incorporating covariates into mapping models.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Norgard EA, Lawson HA, Pletscher LS, et al. Genetic factors and diet affect long-bone length in the F34 LG, SM advanced intercross. Mamm Genome. 2011;22(3–4):178–96. doi: 10.1007/s00335-010-9311-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Silver LM. Mouse genetics. Oxford University Press; New York: 1995. [Google Scholar]
- 64.Cao JJ, Sun L, Gao H. Diet-induced obesity alters bone remodeling leading to decreased femoral trabecular bone mass in mice. Ann N Y Acad Sci. 2010;1192:292–7. doi: 10.1111/j.1749-6632.2009.05252.x. [DOI] [PubMed] [Google Scholar]
- 65.Flint J, Valdar W, Shifman S, Mott R. Strategies for mapping and cloning quantitative trait genes in rodents. Nat Rev Genet. 2005;6(4):271–86. doi: 10.1038/nrg1576. [DOI] [PubMed] [Google Scholar]
- 66.Keane TM, Goodstadt L, Danecek P, et al. Mouse genomic variation and its effect on phenotypes and gene regulation. Nature. 2011;477(7364):289–94. doi: 10.1038/nature10413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wang JR, de Villena FP, Lawson HA, et al. Imputation of single-nucleotide polymorphisms in inbred mice using local phylogeny. Genetics. 2012;190(2):449–58. doi: 10.1534/genetics.111.132381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68•.Welsh CE, Miller DR, Manly KF, et al. Status and access to the Collaborative Cross population. Mamm Genome. 2012;23(9–10):706–12. doi: 10.1007/s00335-012-9410-6. [The Collaborative Cross represents a powerful new mouse genetic reference population. Because of the inclusion of wild derived strains as founders, over 38 million polymorphisms are represented in this population. Further as these lines are near isogenic stock, genetically balanced environmental interaction studies can be conducted with these mice, thus, these mice represent a powerful future resource for G*E studies.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Churchill GA, Airey DC, Allayee H, et al. The Collaborative Cross, a community resource for the genetic analysis of complex traits. Nat Genet. 2004;36(11):1133–7. doi: 10.1038/ng1104-1133. [DOI] [PubMed] [Google Scholar]
- 70.Iraqi FA, Churchill G, Mott R. The Collaborative Cross, developing a resource for mammalian systems genetics: a status report of the Wellcome Trust cohort. Mamm Genome. 2008;19(6):379–81. doi: 10.1007/s00335-008-9113-1. [DOI] [PubMed] [Google Scholar]
- 71.Morahan G, Balmer L, Monley D. Establishment of “The Gene Mine”: a resource for rapid identification of complex trait genes. Mamm Genome. 2008;19(6):390–3. doi: 10.1007/s00335-008-9134-9. [DOI] [PubMed] [Google Scholar]
- 72.Chesler E, Miller D, Branstetter L, et al. The Collaborative Cross at Oak Ridge National Laboratory: developing a powerful resource for systems genetics. Mamm Genome. 2008;19(6):382–9. doi: 10.1007/s00335-008-9135-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Churchill GA, Gatti DM, Munger SC, Svenson KL. The Diversity Outbred mouse population. Mamm Genome. 2012;23(9–10):713–8. doi: 10.1007/s00335-012-9414-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74•.Philip VM, Sokoloff G, Ackert-Bicknell CL, et al. Genetic analysis in the Collaborative Cross breeding population. Genome Res. 2011;21(8):1223–38. doi: 10.1101/gr.113886.110. [This paper represents the first use of the Collaborative Cross in skeletal biology and emphasizes the superior genetic loci mapping resolution possible when using this population. This paper also emphasizes the increased phenotypic variation represented by these mice, suggesting that these mice will be more sensitive to environmental stressors.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Shimizu Y, Sakai A, Menuki K, et al. Reduced bone formation in alcohol-induced osteopenia is associated with elevated p21 expression in bone marrow cells in aldehyde dehydrogenase 2-disrupted mice. Bone. 2011;48(5):1075–86. doi: 10.1016/j.bone.2011.01.008. [DOI] [PubMed] [Google Scholar]
- 76••.Wahl EC, Aronson J, Liu L, et al. Distraction osteogenesis in TNF receptor 1 deficient mice is protected from chronic ethanol exposure. Alcohol. 2012;46(2):133–8. doi: 10.1016/j.alcohol.2011.08.007. [Fracture healing studies from a genetics point of view are difficult to conduct using human subjects and social behaviour environmental factors such as alcohol-use are equally complicated to study due to difficulties associated with accurate data collection. This study powerfully demonstrates that animal models will allow for the meaningful study of G*E interactions impacting extremely complex phenotypes.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Karasik D, Ferrari SL. Contribution of gender-specific genetic factors to osteoporosis risk. Ann Hum Genet. 2008;72(Pt 5):696–714. doi: 10.1111/j.1469-1809.2008.00447.x. [DOI] [PubMed] [Google Scholar]
- 78.Macdonald HM, McGuigan FE, Lanham-New SA, et al. Vitamin K1 intake is associated with higher bone mineral density and reduced bone resorption in early postmenopausal Scottish women: no evidence of gene-nutrient interaction with apolipoprotein E polymorphisms. Am J Clin Nutr. 2008;87(5):1513–20. doi: 10.1093/ajcn/87.5.1513. [DOI] [PubMed] [Google Scholar]
- 79.Gonzalez-Bofill N, Husted LB, Harslof T, et al. Effects of COLIA1 polymorphisms and haplotypes on perimenopausal bone mass, postmenopausal bone loss and fracture risk. Osteoporos Int. 2011;22(4):1145–56. doi: 10.1007/s00198-010-1292-4. [DOI] [PubMed] [Google Scholar]
- 80.Sonoda T, Takada J, Iba K, et al. Interaction between ESRalpha polymorphisms and environmental factors in osteoporosis. J Orthop Res. 2012;30(10):1529–34. doi: 10.1002/jor.22083. [DOI] [PubMed] [Google Scholar]
- 81.Li X, He GP, Zhang B, et al. Interactions of interleukin-6 gene polymorphisms with calcium intake and physical activity on bone mass in pre-menarche Chinese girls. Osteoporos Int. 2008;19(11):1629–37. doi: 10.1007/s00198-008-0613-3. [DOI] [PubMed] [Google Scholar]
- 82.Stathopoulou MG, Dedoussis GV, Trovas G, et al. Low-density lipo-protein receptor-related protein 5 polymorphisms are associated with bone mineral density in Greek postmenopausal women: an interaction with calcium intake. J Am Diet Assoc. 2010;110(7):1078–83. doi: 10.1016/j.jada.2010.04.007. [DOI] [PubMed] [Google Scholar]
- 83.Yazdanpanah N, Uitterlinden AG, Zillikens MC, et al. Low dietary riboflavin but not folate predicts increased fracture risk in postmenopausal women homozygous for the MTHFR 677 T allele. J Bone Miner Res. 2008;23(1):86–94. doi: 10.1359/jbmr.070812. [DOI] [PubMed] [Google Scholar]
- 84.Steer C, Emmett P, Lewis S, et al. THe Methylenetetrahydrofolate Reductase (MTHFR) C677T polymorphism is associated with spinal BMD in nine-year-old children. J Bone Miner Res. 2009;24(1):117–24. doi: 10.1359/jbmr.080814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Harslof T, Tofteng CL, Husted LB, et al. Polymorphisms of the peroxisome proliferator-activated receptor gamma (PPARgamma) gene are associated with osteoporosis. Osteoporos Int. 2011;22(10):2655–66. doi: 10.1007/s00198-010-1491-z. [DOI] [PubMed] [Google Scholar]
- 86.Stathopoulou MG, Dedoussis GV, Trovas G, et al. The role of vitamin D receptor gene polymorphisms in the bone mineral density of Greek postmenopausal women with low calcium intake. J Nutr Biochem. 2011;22(8):752–7. doi: 10.1016/j.jnutbio.2010.06.007. [DOI] [PubMed] [Google Scholar]
- 87.Lieben L, Benn BS, Ajibade D, et al. Trpv6 mediates intestinal calcium absorption during calcium restriction and contributes to bone homeostasis. Bone. 2010;47(2):301–8. doi: 10.1016/j.bone.2010.04.595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88•.Shu L, Ji J, Zhu Q, et al. The calcium-sensing receptor mediates bone turnover induced by dietary calcium and parathyroid hormone in neonates. J Bone Miner Res. 2011;26(5):1057–71. doi: 10.1002/jbmr.300. [This study demonstrates the complexity of environmental interactions on pathways and systems.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Datta NS, Samra TA, Mahalingam CD, et al. Role of PTH1R internalization in osteoblasts and bone mass using a phosphorylation-deficient knock-in mouse model. J Endocrinol. 2010;207(3):355–65. doi: 10.1677/JOE-10-0227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Guo J, Liu M, Yang D, et al. Phospholipase C signaling via the parathyroid hormone (PTH)/PTH-related peptide receptor is essential for normal bone responses to PTH. Endocrinology. 2010;151(8):3502–13. doi: 10.1210/en.2009-1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91•.Ichikawa S, Austin AM, Gray AK, et al. Dietary phosphate restriction normalizes biochemical and skeletal abnormalities in a murine model of tumoral calcinosis. Endocrinology. 2011;152(12):4504–13. doi: 10.1210/en.2011-1137. [This is a preclinical trial demonstrating how G*E interaction studies may direct and shape future treatment options.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Urs S, Henderson T, Le P, et al. Tissue-specific expression of Sprouty1 in mice protects against high-fat diet-induced fat accumulation, bone loss and metabolic dysfunction. Br J Nutr. 2012;108(6):1025–33. doi: 10.1017/S0007114511006209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Le P, Kawai M, Bornstein S, et al. A high-fat diet induces bone loss in mice lacking the Alox5 gene. Endocrinology. 2012;153(1):6–16. doi: 10.1210/en.2011-0082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Nie J, Bradshaw AD, Delany AM, Sage EH. Inactivation of SPARC enhances high-fat diet-induced obesity in mice. Connect Tissue Res. 2011;52(2):99–108. doi: 10.3109/03008207.2010.483747. [DOI] [PubMed] [Google Scholar]
- 95.Brennan-Speranza TC, Rizzoli R, Kream BE, et al. Selective osteo-blast overexpression of IGF-I in mice prevents low protein-induced deterioration of bone strength and material level properties. Bone. 2011;49(5):1073–9. doi: 10.1016/j.bone.2011.07.039. [DOI] [PubMed] [Google Scholar]