

Abstract
Objective
Osteoarthritis (OA) is a degenerative disease resulting in severe joint cartilage destruction and disability. While the mechanisms underlying the development and progression of OA are poorly understood, gene mutations have been identified within cartilage-related signaling molecules implicating impaired cell signaling in OA and joint disease. The Notch pathway has recently been identified as a crucial regulator of growth plate cartilage development and components are expressed in joint tissues. Therefore, we set out to investigate a novel role for Notch signaling in joint cartilage development, maintenance, and the pathogenesis of joint disease.
Methods
We performed the first mouse genetic studies in which the core Notch signaling component, RBPjκ, was tissue-specifically deleted within joints. The Prx1Cre transgene removed Rbpjκ floxed alleles in mesenchymal joint precursor cells, while the Col2CreERT2 transgene specifically deleted Rbpjκ in postnatal chondrocytes. Articular chondrocyte cultures were also utilized to examine Notch regulation of gene expression.
Results
Loss of Notch signaling in mesenchymal joint precursor cells does not affect embryonic joint development, but rather results in an early, progressive OA-like pathology. Additionally, partial loss of Notch signaling in postnatal cartilage results in progressive joint cartilage degeneration and an age-related OA-like pathology. Inhibition of Notch signaling alters expression of the ECM-related factors: COL2A1, PRG4, COL10A1, MMP13, and ADAMTSs.
Conclusions
These data have identified the RBPjκ-dependent Notch pathway as: 1) a novel pathway involved in joint maintenance and articular cartilage homeostasis, 2) a critical regulator of articular cartilage ECM-related molecules, and 3) a potentially important therapeutic target for OA-like joint disease.
Keywords: RBPjκ, Notch, chondrocyte, articular cartilage, osteoarthritis
Introduction
Osteoarthritis (OA), a disease that primarily impacts diarthrodial joints, is characterized by fibrosis and degradation of the joint cartilages, osteophyte formation, subchondral bone sclerosis, and synovial dysplasia. Several risk factors contribute to the development and progression of OA including: age, sex, traumatic injury, obesity, metabolic dysfunction, and genetics (1). While the genetics of OA is complex and polygenic, a genetic contribution to OA has been well established. The hereditability of OA has been assessed in family and twin studies to be as high as 39–78% (2). Recently, genome-wide association screens (GWAS) have identified individual risk alleles associated with OA, including structural and extracellular matrix (ECM)-related factors expressed in joint cartilages (COL2A1, COL9A1, COL11A1, AGC1) and signaling molecules in the Wnt (FRZB), BMP (GDF5), and TGFβ (SMAD3, ASPN) pathways (2). Many of these targets have been mutated in mice to develop genetic models of OA, with signaling gain- or loss-of-function studies implicating dysregulated differentiation or maintenance of articular chondrocytes, downstream of an altered signaling modulator, as a central driver of OA-like disease (2).
Through the use of mouse genetic models, we have identified Notch signaling as a crucial regulator of chondrocyte proliferation and differentiation during embryonic cartilage development. In mammals, Notch signaling is initiated when one of the 11 Notch ligands (JAG1-2; DLL1,3,4; DLK1-2; MAGP1-2; DNER; and NB3) activates a single-pass transmembrane cell surface Notch receptor (NOTCH1-4) leading to a series of receptor cleavages mediated by ADAM proteases and the gamma-secretase complex. The final cleavage releases the Notch intra-cellular domain (NICD), which enters the nucleus and binds the transcriptional regulators, RBPjκ and MAML. An NICD-RBPjκ-MAML ternary complex then activates the expression of RBPjκ-dependent Notch target genes, including the Hes/Hey families (3). While several Notch receptors, ligands, and target genes are expressed in joint tissues in normal and diseased states of mice and humans (4–7), the functional role for this pathway in joint development, maintenance, and disease remains unknown. Here we have employed state-of-the-art mouse genetic loss-of-function approaches to establish, for the first time, that: 1) the RBPjκ–dependent Notch signaling pathway is required for postnatal joint cartilage maintenance, but not embryonic joint development, 2) Notch function in maintaining joint integrity is at least partially due to its signaling within postnatal articular/meniscal chondrocytes, and 3) the Notch signaling pathway may be an important genetic component in the pathogenesis of OA-like disease and joint failure.
Materials and Methods
Mice strains
Animal work was approved by the University of Rochester Committee on Animal Resources. All mouse strains including Rbpjκf/f, Prx1Cre and Col2CreERT2 have been previously described (8–10). Prx1Cre;Rbpjκf/f (RBPjκPrx1) and Col2CreERT2;Rbpjκf/f (RBPjκCol2TM) mice were viable and produced in Mendelian ratios. Tamoxifen (TM; 1mg/10g body weight) was administered daily via i.p. injection to all RBPjκCol2TM mice and littermate controls from P25–29 (sacrificed at P30, 2, and 8-months of age) in order to remove Rbpjκ floxed alleles.
Analyses of mice
Embryonic tissues at E15.5 or E18.5 and all postnatal tissues were harvested, fixed in 10% neutral-buffered formalin for 24 hours (embryonic) or 3 days (postnatal), decalcified in 14% EDTA overnight (embryonic) or 7–10 days in 14% EDTA or Immunocal (Decal Corp.) (postnatal), paraffin processed and embedded for sectioning. Tissues were sectioned at 5μm and standard alcian blue/hematoxylin/orange-g (ABH/OG) staining was performed in order to analyze tissue architecture. Polarized light microscopy was performed using an Axioskop40 microscope with polarizing filters (Zeiss). Beta-galactosidase staining was performed as previously described (11). ISH using radiolabeled riboprobes for Mmp13(12) and Prg4(13) was completed as previously described. IHC was performed on sections using traditional antigen retrieval and colorimetric development methodologies. The following primary antibodies were utilized: COL2A1 (Thermo Scientific, MS235-P), COL10A1 (Quartett, 2031501005), COL1A1 (Abcam, ab90395), PRG4 (Abcam, ab28484), TNC (Abcam, ab6346), MMP13 (Thermo Scientific, MS-825P), and NITEGE (MDBioProducts, 1042003). MicroCT analyses were performed on 2, 4, 6, and 8-month old mouse knees prior to decalcification using a VivaCT 40 scanner (Scanco USA, Inc.) as previously described (14).
Articular cartilage isolation, cell culture, and quantitative gene expression analyses
Articular cartilage was isolated from the knee joints of mutant and control mice as previously described with modifications (15). Briefly, articular cartilage was excised from femoral condyles and tibial plateaus with a scalpel and placed in 1X PBS. Cartilage fragments were digested using 3mg/ml Collagenase D in 10ml high glucose DMEM (Invitrogen) for 12 hours at 37°C. Murine articular chondrocyte (MAC) cell suspensions were filtered through 40μm filters and RNA was isolated using an RNeasy Mini kit (Qiagen). cDNA synthesis and real-time qPCR was performed on RNA extracted from the MACs as previously described (16). Primer sequences for Rbpjk, Hes1, Hey1, Col2a1, Agc1, Prg4, Col10a1, Mmp13, Adamts4, and Adamts5 are available upon request.
Primary MACs were also isolated from the knee joints of P19–P21 CD-1 mice as described above. Following digestion, MAC cell suspensions were filtered and plated at a density of 80,000 cells/well in 12-well tissue culture plates for 16 hours. Cells were then treated with 10μM DAPT or DMSO daily for two consecutive days. RNA was collected using the RNeasy Mini kit (Qiagen) and real-time qPCR was performed as described above.
Histomorphometry
Quantitative histomorphometry was performed on ABH/OG stained sections using the Osteomeasure Analysis System (Osteometrics). Cartilage thickness was measured from the middle of the femoral and tibial condyles. Cartilage area was traced from both articular cartilage surfaces using the bone area tool in the Osteomeasure software. The tide-mark was used to delineate between upper zone and deep zone articular cartilages. Three to five mice were analyzed in each group, and at least three slides were examined for each sample.
Statistical Analyses
Statistical analyses were performed using the Student’s t-test.
Results
Loss of Notch signaling in joint tissues does not impair articular cartilage during development
To determine the role for Notch signaling in joint development and maintenance we first generated a loss-of-function mouse model in which floxed alleles for the transcriptional Notch effector, Rbpjκ, were conditionally deleted. Since joint development requires communication from multiple tissues derived from a common mesenchymal progenitor and OA is a disease that affects the entire joint, we utilized the Prx1Cre transgene to delete floxed alleles in mesenchymal progenitors that give rise to all synovial joint tissues including the articular cartilage, meniscus, synovium, ligaments, and subchondral bone osteoblasts (16). Articular cartilage and joint formation occurs during embryonic development and proceeds through stages of joint site initiation, interzone formation, chondrogenesis, cavitation, and morphogenesis (17). Previously, we identified that Prx1Cre;Rbpjκf/f (RBPjκPrx1) mice exhibited no signs of impaired joint site determination at embryonic stage E12.5 (16). Here we demonstrate that knee sections from E15.5 and E18.5 RBPjκPrx1 mutants exhibit normal joint cavitation and morphogenesis similar to wild-type (WT) controls (Fig. S1A). ABH/OG staining reveals that RBPjκPrx1 mutants exhibit normal chondrogenesis of the epiphyseal, articular, and meniscal cartilages. Additionally, morphological analyses of alcian blue and alizarin red stained skeletal preparations at E18.5 further confirmed that all joints of RBPjκPrx1 skeletons were comparable to controls (16).
Formation of the secondary ossification center (SOC) is important for establishing proper joint architecture and separation of the articular and growth plate cartilages. While the molecular details of this process are unclear, it is known that many of the same molecules governing chondrocyte maturation, cartilage matrix turnover, vascular invasion, and bone formation are critical regulators of SOC formation. Accordingly, histological and molecular analyses of RBPjκPrx1 mutant knees at two-weeks of age revealed that compared to control joints, loss of Notch signaling leads to a delay in SOC formation characterized by persistent hypertrophic cartilage in the epiphyseal region (Fig. S1B). In situ hybridization (ISH) for Mmp13, a requisite molecule in chondrocyte terminal maturation and cartilage matrix catabolism, revealed normal expression in terminal hypertrophic chondrocytes and subchondral osteoblasts of controls, while RBPjκPrx1 mutants exhibited impaired Mmp13 expression (Fig. S1B). Previously, we demonstrated that RBPjκ-dependent Notch signaling is required for growth plate chondrocyte terminal maturation, such that loss of RBPjκ-dependent Notch signaling leads to persistent hypertrophic cartilage and suppressed Mmp13 expression throughout growth plate cartilage development (18). Interestingly, although RBPjκ Prx1 mutants exhibit significant delays in SOC formation, the superficial and intermediate layers of the articular cartilage appear normal. Molecular analyses confirm that Prg4 maintains expression in both RBPjκPrx1 mutants and controls (Fig. S1B). These data demonstrate that RBPjκPrx1 mutant mice exhibit a delay in SOC formation, but articular cartilage development is largely unaffected.
Loss of Notch signaling in joint tissues results in an early and progressive OA-like pathology
Since delays in SOC development were observed in early postnatal RBPjκPrx1 mutant mice, we further evaluated the knee joints of RBPjκPrx1 mutants and controls ranging in age from 2 to 8-months. Micro-computed tomography (MicroCT) analyses revealed deformations of subchondral bone architecture of RBPjκPrx1 mutant tibias at 2-months of age. Tibial plateaus were flattened and broader as compared to controls (red arrows; Fig. S2). By 4-months of age and beyond, mineralized osteophytes surrounded the knee joints of RBPjκPrx1 mutants (green arrows; Fig. S2), with expansion and mineralization of the menisci by 6 to 8-months of age (blue arrows; Fig. S2). These morphological changes are consistent with tissue phenotypes observed in both human and murine OA.
Joint integrity was assessed histologically in 2 and 8-month old RBPjκPrx1 mutant and control mice (Fig. 1). By 2-months of age, hallmark features of early onset OA were observed in RBPjκPrx1 mutants including: 1) joint cartilage fibrosis or degeneration, 2) osteophyte formation, 3) subchondral bone sclerosis, and 4) synovial tissue expansion (Fig. 1A). ABH/OG staining demonstrated a localized loss of proteoglycan content in RBPjκPrx1 mutant knees consistent with articular cartilage degeneration. All OA-related changes observed in RBPjκPrx1 mutant mice by 2-months were exacerbated by 8-months (Fig. 1B). ABH/OG stained sections at this time point exhibited further joint cartilage degeneration including fibrillation and clefting on the femoral condyle, tibial plateau, and menisci. In regions of severe damage, loss of GAGs and/or proteoglycans was evidenced by depletion of ABH/OG staining in the extracellular and pericellular matrix of the joint cartilages (Fig. 1B). Interestingly, some regions in RBPjκPrx1 mutants retained ABH/OG staining and showed limited signs of damage, although upon closer examination the cells and matrix demonstrated changes consistent with the loss of superficial zone chondrocytes. ABH/OG stained sections revealed a tide-mark closer to the articular cartilage surface with fewer superficial zone cells (Fig. 1B). Histomophometry performed on ABH/OG stained knee sections from 2 and 8-month old RBPjκPrx1 mutant and controls established that upper zone articular cartilage area and thickness was significantly decreased (>75% for both) in RBPjκPrx1 mutants, while the deep zone cartilage area and thickness was increased at most time-points (Fig. S3A). Using an established OARSI scoring system (19), the severity of cartilage damage was assessed. The results were consistent with the histomorphometric analysis (Fig. S3B). These data indicate that RBPjκ-dependent Notch signals are important in maintaining superficial/intermediate zone chondrocytes throughout adult life.
Figure 1. Loss of RBPjκ-dependent Notch signaling results in histological and ECM changes consistent with joint cartilage degeneration and an OA-like pathology.

ABH/OG staining of 2-month old (A) and 8-month old (B) WT and RBPjκPrx1 mutant knee sections. Osteophyte formation (black arrows), meniscal degeneration (red arrow), synovial tissue expansion (black asterisks), and subchondral bone sclerosis (yellow asterisks) are shown in the RBPjκPrx1 mutants. At 2-months, 20X magnification of the articular cartilage (green box) shows severe fibrosis of RBPjκPrx1 mutant cartilage. At 8-months, 20X magnification of the articular cartilage (blue and green boxes) shows degeneration of the articular cartilage (blue box) and altered localization of the tide mark in RBPjκPrx1 mutant cartilage (green box). Orange dashed line indicates the tide mark. S, synovial tissue; M, meniscus; SB, subchondral bone; AC, articular cartilage. (N>5 for all time points). (C) Polarized light microscopy of 2 and 8-month old WT and RBPjκPrx1 mutant knee sections. White arrowheads denote a loss of normal Type II Collagen fibers. White asterisks denote Type I Collagen fibers indicating cartilage and meniscal fibrosis or de-differentiation.
To determine whether loss of RBPjκ-dependent Notch signaling affected collagen fiber composition or arrangement within the ECM of the joint cartilages, we analyzed ABH/OG stained knee sections with polarized light microscopy. The birefringence from a normal articular cartilage matrix revealed fluorescent fibers (mostly green) perpendicular to the surface of the articular cartilage in upper and deeper zones with parallel fibers at the surface (Fig. 1C). In the subchondral bone, the birefringence of the bony and fibrous matrix presented as fluorescent fibers (mostly white and pink) aligned in various orientations (Fig. 1C). At 2 and 8-months of age, polarized light microscopy revealed a loss of normal joint cartilage matrix structure in RBPjκPrx1 mutant sections, such that the birefringence of the collagen fibers in the articular cartilage matrix was nearly absent. Interestingly, in regions of cartilage fibrosis, the ECM birefringence was more consistent with that of a bony or fibrous matrix (white asterisks; Fig. 1C).
To identify specific changes in the expression of ECM-related molecules we performed immunohistochemistry (IHC) or in situ hybridization (ISH) for Type I Collagen (COL1A1), Type II Collagen (COL2A1), Type X Collagen (COL10A1), Matrix metalloprotease 13 (MMP13), Aggrecanase neoepitope (NITEGE), and Lubricin (PRG4) (Figs. 2, S4, and data not shown). Consistent with the ECM changes observed using polarized light microscopy, IHC analysis for COL1A1 demonstrated that fibrotic regions possessed enhanced COL1A1 expression (data not shown), suggesting chondrocyte de-differentiation in these regions. Expression of COL2A1 was significantly reduced in both the extracellular and pericellular matrix of the articular cartilage in RBPjκPrx1 mutant sections as early as 2-months, especially in regions of cartilage fibrosis (Fig. 2A). COL10A1 expression, which is normally localized to the pericellular matrix of deep zone chondrocytes and often up-regulated in the context of OA, was up-regulated in both the pericellular and extracellular matrix of RBPjκPrx1 mutant articular cartilage by 8-months (Fig. 2B). Consistent with the loss of superficial zone chondrocytes, we detected a progressive loss of Prg4 expression in joint cartilages and synovial fibroblasts (Fig. S4A). Furthermore, we observed increased expression of the catabolic markers, MMP13 and NITEGE, by 8-months of age within areas of infiltrating fibrotic tissue that impinges on the articular cartilage, as well as, the articular and meniscal chondrocytes undergoing degeneration (Fig. 2B).
Figure 2. Loss of RBPjκ-dependent Notch signaling results in ECM and molecular changes consistent with an early and progressive OA-like pathology.

(A) COL2A1 IHC of 2-month old WT and RBPjκPrx1 mutant knee sections. Red arrows indicate pericellular staining and green arrows indicate ECM staining. (B) COL10A1 IHC of 8-month old WT and RBPjκPrx1 mutant knee sections. Blue arrows indicate pericellular and ECM regions of the superficial zone. MMP13 and NITEGE IHC of 8-month old WT and RBPjκPrx1 mutant knee sections. (N>3 for all experiments). Yellow arrows denote MMP13 and NITEGE positive degenerating joint cartilages.
To determine the recombination efficiency of the Prx1Cre-mediated gene deletion and to assess the changes in the ECM-related gene expression during early stages of postnatal joint development, we isolated RNA from articular chondrocytes of RBPjκPrx1 mutants and controls at P2 prior to morphological joint degeneration. Real-time qPCR was performed for Rbpjκ, Sox9, Col2a1, Agc1, Mmp13, Adamts4 and Adamts5 (Fig. 3). These results demonstrated that Rbpjκ expression was reduced by 92% in P2 RBPjκPrx1 mutants as compared to controls, suggesting an efficient deletion of Rbpjκ (Fig. 3). The expression level of anabolic genes such as Sox9, Col2a1 and Agc1 were also reduced (by 51%, 23% and 35% respectively) in P2 RBPjκPrx1 mutants. Interestingly, the expression level of the catabolic genes Mmp13, Adamts4, and Adamts5 were dramatically decreased (by 70%, 68% and 45 % respectively), suggesting a dual role for Notch singling in regulating ECM related molecules (Fig. 3).
Figure 3. Impaired Notch signaling within articular chondrocytes results in ECM-related gene expression changes.
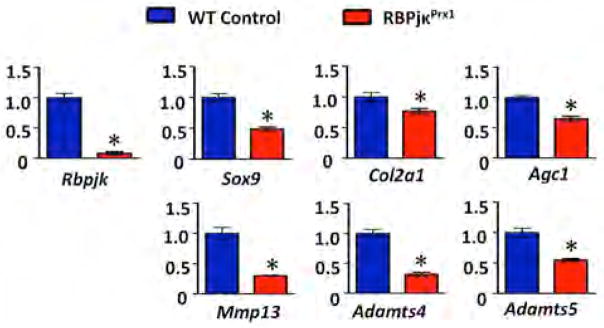
Real-time qPCR comparing gene expression in the articular chondrocytes isolated from P2 WT and RBPjκPrx1 mutants. Gene expression analyses were performed for Rbpjk, Sox9, Col2a1, Agc1, Mmp13, Adamts4 and Adamts5. Bars represent means +/− SD (n=3). All samples are normalized to Beta-actin and then normalized to the controls. “*” denotes statistical significance with p-value less than 0.05. All assays were performed in triplicate.
Each of the morphological and molecular changes identified in the ECM of aging RBPjκPrx1 mutants are consistent with a severe, early, and progressive OA-like pathology, which is likely caused by the loss of RBPjκ-dependent Notch signaling during early postnatal articular cartilage/joint maintenance but not during embryonic joint development. Furthermore, these data also suggest that the severe and early joint degeneration is likely caused by loss of Notch signaling in various joint cells that contribute to altered synovial cell behavior, an inferior joint cartilage ECM, and progressive catabolic joint changes that are enhanced with age.
Loss of Notch signaling within postnatal chondrocytes results in a progressive OA-like pathology
To determine whether Notch signaling within postnatal chondrocytes is directly required for joint cartilage maintenance, we generated a loss-of-function mouse model in which floxed alleles for Rbpjκ were conditionally deleted specifically within chondrocytes using the TM inducible Col2CreERT2 transgene. Both Col2CreERT2;Rbpjκf/f (RBPjκCol2TM) mutants and Cre negative control mice (WT) were administered TM via daily i.p. injections from P25–29. Joint integrity was analyzed via ABH/OG stained histology at 2 and 8-months of age. By 2-months of age (1-month post TM injections), no major structural changes were observed in the joints of RBPjκCol2TM mutants as compared to controls (Fig. 4A). By 8-months of age (7-months post TM injections), hallmark features of a progressive OA-like pathology were observed in RBPjκCol2TM mutants including: 1) articular cartilage and meniscus degeneration, 2) mineralization of joint cartilages, and 3) altered subchondral bone structure (Fig. 4B). ABH/OG staining demonstrated a general loss of GAG and/or proteoglycan content in RBPjκCol2TM mutant knees consistent with articular cartilage degeneration. In regions of severe degeneration, ABH/OG staining revealed a depletion of normal articular chondrocytes and the appearance of a fibrotic or bone-like tissue (yellow asterisk, Fig. 4B). In regions with more limited degeneration, a loss of upper zone chondrocytes (distance from articular surface to tidemark; yellow arrow) and an increase in chondrocytes of the deep zone with a hypertrophic-like morphology were observed (Fig. 4B). Histomophometry performed on ABH/OG stained knee sections established that upper zone articular cartilage area and thickness was significantly decreased (>50% for both) in RBPjκCol2TM mutants, while the deep zone articular cartilage thickness was increased by nearly 75% (Fig. S5A). OARSI scoring results were consistent with the histomorphometric analysis showing significant cartilage degeneration only in RBPjκCol2TM mutants by 8-months of age as compared to controls (Fig. S5B).
Figure 4. Cartilage-specific reductions in RBPjκ-dependent Notch signaling results in ECM changes consistent with a progressive OA-like pathology.
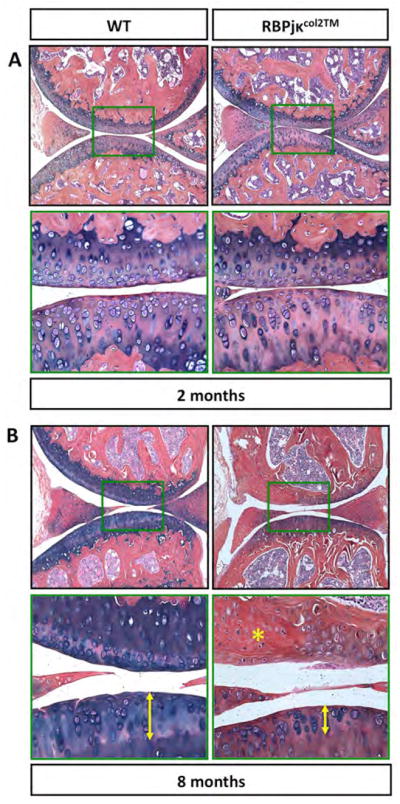
ABH/OG staining of 2 (A) and 8-month old (B) WT and RBPjκCol2TM mutant knee sections. 10X magnification of total knee joint and 40X magnification of a centralized domain of articular cartilage (green box). Yellow asterisk indicates articular cartilage degeneration/fibrosis. Yellow arrows indicate thickness of upper zone articular cartilage above the tide-mark. (N>5 for all time points).
To further characterize specific changes in the expression of ECM-related molecules in the RBPjκCol2TM mutant joint cartilages, we performed IHC analyses for COL2A1, COL10A1, MMP13, NITEGE, and PRG4 (Figs. 5, S4, and data not shown). Knee sections from 2-month old RBPjκCol2TM mutants showed early signs of reduced COL2A1 expression in both the extracellular and pericellular matrix of the articular cartilage (Fig. 5A). RBPjκCol2TM mutants also exhibited an inappropriate increase in COL10A1 expression within upper zone chondrocytes (Fig. 5A). By 8-months of age, RBPjκCol2TM mutants showed significant depletion of COL2A1, especially in regions of severe fibrosis. Additionally, enhanced COL10A1 expression was evident throughout all zones of the articular cartilage (Fig. 5B). Consistent with the loss of superficial zone chondrocytes in RBPjκCol2TM mutants, we detected an early and progressive loss of PRG4 expression in joint cartilages (Fig. S4B). Within areas of fibrosis and degeneration, we also observed an increase in both MMP13 and NITEGE (Fig 5B). Surprisingly, we also detected significant levels of MMP13 within the subchondral bone matrix of the RBPjκCol2TM mutants, which is rarely observed in controls (data not shown). All of the morphological and molecular changes identified in RBPjκCol2TM mutants are consistent with a progressive OA-like pathology, which is caused by the cartilage-specific loss of RBPjκ-dependent Notch signaling during postnatal articular cartilage/joint maintenance.
Figure 5. Cartilage-specific reductions in RBPjκ-dependent Notch signaling results in ECM and molecular changes consistent with a progressive OA-like pathology.

(A) COL2A1 and COL10A1 IHC of 2-month old WT and RBPjκCol2TM mutant knee sections (40X magnification). (B) COL2A1, COL10A1, MMP13, and NITEGE IHC of 8-month old WT and RBPjκCol2TM mutant knee sections (40X magnification). (N>3 for all experiments). Red arrows indicate positively or negatively stained degenerating cartilage.
To determine the recombination efficiency following TM induced Cre-mediated gene deletion and to assess early changes in ECM-related gene expression, we isolated articular chondrocytes and collected RNA from P30 RBPjκCol2TM mutants and controls following 5-days of TM administration. Real-time qPCR was performed for Rbpjκ, Hes1, Hey1, Sox9, Col2a1, Agc1, Prg4, Col10a1, Mmp13, Adamts4 and Adamts5. Gene expression results demonstrated that Rbpjκ expression was only reduced by ~31% in RBPjκCol2TM mutants, while Hes1 and Hey1 expression were reduced by ~50%, suggesting that Col2CreERT2-mediated recombination of Rbpjκ floxed alleles in articular cartilage was incomplete (Fig. 6A). Even with limited recombination, expression changes were observed in ECM-related molecules. The expression level of Sox9 and Col2a1 were reduced by ~55%, and Agc1 was reduced by ~40%. Prg4 expression did not show a significant decrease in RBPjκCol2TM mutants at this early time-point, suggesting that it is not a direct target and that loss of Prg4 expression in 2 and 8-month old RBPjκCol2TM mutants is progressive (Fig. S4B). The expression level of Mmp13 was not changed immediately following gene recombination, while the expression of Adamts4 and Adamts5, which are often up-regulated in OA-like disease, were significantly reduced (63% and 19% respectively) in RBPjκCol2TM mutant articular chondrocytes at this early time-point (Fig. 6A).
Figure 6. Impaired Notch signaling within articular chondrocytes results in rapid chondrocyte gene expression changes.

(A) Real-time qPCR comparing gene expression in the articular chondrocytes isolated from P30 WT and RBPjκCol2TM mutants following 5 days of TM administration (P25–29) and (B) WT cultured articular chondrocytes following two days of DAPT or DMSO treatment. Gene expression analyses were performed for Rbpjk, Hes1, Hey1, Sox9, Col2a1, Agc1, Prg4, Col10a1, Mmp13, Adamts4 and Adamts5. Bars represent means +/− SD (n=3). All samples are normalized to Beta-actin and then normalized to the controls. “*” denotes statistical significance with p-value less than 0.05. All assays were performed in triplicate.
We next verified these early gene expression changes following Notch inhibition by using an in vitro articular chondrocyte culture model adapted from (15). Articular chondrocytes isolated from P19–21 WT mice were cultured for 2 days in the presence and absence of the Notch inhibitor, DAPT. These data were consistent with our in vivo data in which Hes1, Sox9, and Col2a1 expression was significantly down-regulated immediately following Notch inhibition (Fig. 6B). The expression of Adamts4 and Adamts5 were not detectable in these cells. Surprisingly, we also observed a decrease in Prg4 expression, suggesting a reduction in superficial-like articular chondrocytes within our cultures (Fig. 6B). Taken together, all of these data demonstrate that chondrocyte-specific Notch signaling is at least partially required for the maintenance of the joint cartilages, and that Notch signaling is important in regulating either directly or indirectly the expression of critical ECM-related molecules.
Discussion
In this report, we have provided the first genetic evidence that although RBPjκ-dependent Notch signaling is dispensable for embryonic joint development, it is required for proper formation of the SOC and articular cartilage and joint maintenance. This study establishes that loss of RBPjκ-dependent Notch signals within joint tissues of mesenchymal origin results in a severe, early, and progressive OA-like pathology. We also demonstrate that partial loss of Notch signaling specifically within postnatal chondrocytes, following formation of the SOC, results in a progressive OA-like pathology. Collectively, these data indicate that Notch signaling is required throughout postnatal life within the articular and meniscal chondrocytes, and potentially other joint tissues, to maintain joint integrity. These data collectively implicate permanent alterations in Notch activity in the pathogenesis of OA and joint disease.
Notch signaling has been established as a critical pathway in skeletal development and disease in both mice (16, 18, 20–29) and humans (30–34). Recently, we have identified the Notch signaling pathway as a central regulator of cartilage development (16, 18, 25), suggesting its potential additional involvement in joint cartilage maintenance and degenerative diseases like OA. While human OA cartilage samples display an increase in the frequency of cells expressing NOTCH1 and other Notch pathway proteins (5, 35), the function of Notch in articular cartilage in normal or diseased states remains unclear. Several lines of evidence have demonstrated that at the molecular level, sustained Notch signaling in mesenchymal progenitors and growth plate chondrocytes suppresses Sox9, Col2a1, and Agc1 expression, while promoting Col10a1 and Mmp13 expression (18, 22, 25, 26, 28, 36). Inhibition of Notch signaling in growth plate and articular chondrocytes has also been shown to suppress Mmp13 expression both in vitro and in vivo (18, 20, 25) (Fig. 6), demonstrating specific regulation of an important cartilage catabolic factor. Alternatively, transient NOTCH1 activation was shown to induce Sox9 and chondrogenic gene expression (23, 24). These data suggest that Notch signaling may regulate both anabolic and catabolic responses in chondrocytes. Notch signaling has recently been shown to mediate differential gene expression and cellular responses based on the amplitude and duration of the signal (37). Therefore it is possible that transient, physiological levels of Notch signaling in chondrocytes favor a balanced anabolic and catabolic cartilage maintenance response, while sustained or high levels of Notch activity elicits a pathological or primarily catabolic response. Consistent with this hypothesis, it has recently been shown that Notch signals are dramatically up-regulated within the articular cartilage and inflammatory setting of injury induced knee arthritis of the mouse. This study further demonstrated that loss or reductions in cartilage-specific Notch signaling are capable of reducing MMP13 expression within murine joints and delaying cartilage degeneration over the short-term (8-weeks) (38). Our data supports the concept that short-term Notch inhibition results in a decrease of both anabolic and catabolic factors within articular chondrocytes, although we further demonstrate that long-term (8+ months) and/or widespread loss of RBPjκ-dependent Notch signaling in joint tissues disrupts normal physiology of the cartilage, ultimately resulting in an age-related and progressive joint disease. Therefore, physiological Notch signaling within chondrocytes and joint related tissues is essential for joint cartilage maintenance, although transient modulation of these signals may provide a means for modifying the joint cartilages in both normal and pathological situations.
The progressive joint degeneration observed in the cartilage-specific RBPjκCol2TM mutants was quite different than the early, progressive joint degeneration phenotype observed in RBPjκPrx1 mutant mice. There exist several possibilities to explain these different phenotypes. First, the TM-inducible RBPjκCol2TM mutant mice demonstrated relatively poor postnatal recombination of the Rbpjκ floxed alleles, and therefore exhibit an incomplete impairment in Notch signaling as compared to the more complete recombination observed in RBPjκPrx1 mutant cartilages (18). Second, RBPjκPrx1 mutants exhibit a loss of Notch signaling during early limb development within mesenchymal lineage cells that not only give rise to articular and meniscal chondrocytes, but also synovial fibroblasts, subchondral osteoblasts, and ligamentous cells. The early removal of Rbpjκ alleles from multiple joint tissues resulted in delayed SOC formation altering the shape of the postnatal joint and likely leading to altered biomechanics. Furthermore, the RBPjκPrx1 mutants display a severe synovial hyperplasia not observed in RBPjκCol2TM mutants or most other OA-like genetic models. However, a strikingly similar joint phenotype is observed in the Prg4−/− mice, where synovial fibroblasts proliferate extensively and adhere directly to the joint cartilages resulting in severe fibrosis and joint failure (39). Based on our data, we speculate that Notch signaling may be an important and potentially indirect regulator of Prg4 expression, and that the progressive loss of Prg4 in each of these joint tissues exacerbates and alters the RBPjκPrx1 mutant joint phenotype. Furthermore, data from both our in vivo genetic models and our in vitro articular chondrocyte culture model suggest that loss of Notch signaling primarily results in decreased anabolic regulation of articular cartilage ECM molecules and that likely predisposes the joint cartilages to catabolic degeneration over time. In conclusion, our findings have identified the RBPjκ-dependent Notch signaling pathway as a novel and essential regulator of several ECM-related factors important for maintaining normal joint cartilage structure and function. This critical role in maintaining joint cartilage integrity implicates the Notch pathway as a key molecular participant in the pathogenesis of OA and raises the possibility that manipulation of this pathway has therapeutic implications for this disease and other cartilage degenerative disorders.
Supplementary Material
(A) ABH/OG staining of E15.5 and E18.5 WT and RBPjκPrx1 knee sections. (B) ABH/OG staining and in situ hybridization (ISH) of WT and RBPjκPrx1 knee sections at two weeks of age. ISH signal for Mmp13 and Prg4 expression is shown in red. M, meniscus; AC, articular cartilage; SOC, secondary ossification center; HC, hypertrophic cartilage. (N>3 for all experiments).
MicroCT analyses of 2, 4, 6, and 8-month old WT and RBPjκPrx1 mutant knees. Flattening of tibial plateau (red arrows), synovial tissue expansion and mineralization (blue arrows), and osteophyte formation (green arrows) are highlighted in RBPjκPrx1 mutants. (N>3 for all time points).
(A) Histomorphometric analyses of articular cartilage area and thickness from 2-month and 8-month old ABH/OG stained WT and RBPjκPrx1 knee sections. The tide-mark was used to determine upper and deep zone articular cartilage regions. At least 3 slides were measured per animal. (B) OARSI scoring for RBPjκPrx1 mutants and controls at 2- and 8-months of age. “*” Statistical significance with p-value less than 0.05. Data presented as the means +/− SE (N>5 for all time points).
(A) Prg4 ISH of 2 and 8-month old WT and RBPjκPrx1 mutant knee sections. Red arrows indicate diminishing or absent Prg4 expression in the articular cartilage and synovium of the RBPjκPrx1 mutants. (B) PRG4 IHC of 2 and 8-month old WT and RBPjκCol2TM mutant knee sections.
(A) Histomorphometric analyses of articular cartilage area and thickness from 2-month and 8-month old ABH/OG stained WT and RBPjκCol2TM knee sections. The tide-mark was used to determine upper zone (superficial and intermediate) and deep zone articular cartilage regions. (B) OARSI scoring for RBPjκCol2TM mutants and controls at 2- and 8-months of age. “*” Statistical significance with p-value less than 0.05. Data presented as the means +/− SE (N>5 for all time points).
Acknowledgments
This work was supported in part by the following United States National Institute of Health grants: R01 grants (AR057022 and AR063071 to MJH), R21 grant (AR059733 to MJH), a P50 Center of Research Translation grant (AR054041 to RJO), T32 training grants (AR053459 and GM007356 to AK, trainee), and a P30 Core Center grant (AR061307). We thank Drs. Tasuku Honjo (Kyoto University) and Di Chen (Rush University) for providing important mouse strains.
We would also like to gratefully acknowledge the technical expertise and assistance of Ryan Tierney, Sarah Mack, Kathy Maltby, and Ashish Thomas within the Center for Musculoskeletal Research Histology, Biochemistry, and Molecular Imaging Core.
Footnotes
Disclosure: There exist no conflicts of interest and we have nothing to disclose.
References
- 1.Felson DT, Lawrence RC, Dieppe PA, Hirsch R, Helmick CG, Jordan JM, et al. Osteoarthritis: new insights. Part 1: the disease and its risk factors. Annals of internal medicine. 2000;133(8):635–46. doi: 10.7326/0003-4819-133-8-200010170-00016. [DOI] [PubMed] [Google Scholar]
- 2.Sandell LJ. Etiology of osteoarthritis: genetics and synovial joint development. Nat Rev Rheumatol. 2012;8(2):77–89. doi: 10.1038/nrrheum.2011.199. [DOI] [PubMed] [Google Scholar]
- 3.Kopan R, Ilagan MX. The canonical Notch signaling pathway: unfolding the activation mechanism. Cell. 2009;137(2):216–33. doi: 10.1016/j.cell.2009.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dowthwaite GP, Bishop JC, Redman SN, Khan IM, Rooney P, Evans DJ, et al. The surface of articular cartilage contains a progenitor cell population. Journal of cell science. 2004;117(Pt 6):889–97. doi: 10.1242/jcs.00912. [DOI] [PubMed] [Google Scholar]
- 5.Grogan SP, Miyaki S, Asahara H, D’Lima DD, Lotz MK. Mesenchymal progenitor cell markers in human articular cartilage: normal distribution and changes in osteoarthritis. Arthritis research & therapy. 2009;11(3):R85. doi: 10.1186/ar2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hayes AJ, Dowthwaite GP, Webster SV, Archer CW. The distribution of Notch receptors and their ligands during articular cartilage development. Journal of anatomy. 2003;202(6):495–502. doi: 10.1046/j.1469-7580.2003.00185.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yabe Y, Matsumoto T, Tsurumoto T, Shindo H. Immunohistological localization of Notch receptors and their ligands Delta and Jagged in synovial tissues of rheumatoid arthritis. Journal of orthopaedic science : official journal of the Japanese Orthopaedic Association. 2005;10(6):589–94. doi: 10.1007/s00776-005-0943-3. [DOI] [PubMed] [Google Scholar]
- 8.Chen M, Lichtler AC, Sheu TJ, Xie C, Zhang X, O’Keefe RJ, et al. Generation of a transgenic mouse model with chondrocyte-specific and tamoxifen-inducible expression of Cre recombinase. Genesis. 2007;45(1):44–50. doi: 10.1002/dvg.20261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Han H, Tanigaki K, Yamamoto N, Kuroda K, Yoshimoto M, Nakahata T, et al. Inducible gene knockout of transcription factor recombination signal binding protein-J reveals its essential role in T versus B lineage decision. Int Immunol. 2002;14(6):637–45. doi: 10.1093/intimm/dxf030. [DOI] [PubMed] [Google Scholar]
- 10.Logan M, Martin JF, Nagy A, Lobe C, Olson EN, Tabin CJ. Expression of Cre Recombinase in the developing mouse limb bud driven by a Prxl enhancer. Genesis. 2002;33(2):77–80. doi: 10.1002/gene.10092. [DOI] [PubMed] [Google Scholar]
- 11.Hilton MJ, Tu X, Long F. Tamoxifen-inducible gene deletion reveals a distinct cell type associated with trabecular bone, and direct regulation of PTHrP expression and chondrocyte morphology by Ihh in growth region cartilage. Dev Biol. 2007;308(1):93–105. doi: 10.1016/j.ydbio.2007.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hilton MJ, Tu X, Cook J, Hu H, Long F. Ihh controls cartilage development by antagonizing Gli3, but requires additional effectors to regulate osteoblast and vascular development. Development. 2005;132(19):4339–51. doi: 10.1242/dev.02025. [DOI] [PubMed] [Google Scholar]
- 13.Sampson ER, Hilton MJ, Tian Y, Chen D, Schwarz EM, Mooney RA, et al. Teriparatide as a chondroregenerative therapy for injury-induced osteoarthritis. Sci Transl Med. 2011;3(101):101ra93. doi: 10.1126/scitranslmed.3002214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sampson ER, Beck CA, Ketz J, Canary KL, Hilton MJ, Awad H, et al. Establishment of an index with increased sensitivity for assessing murine arthritis. Journal of orthopaedic research : official publication of the Orthopaedic Research Society. 2011;29(8):1145–51. doi: 10.1002/jor.21368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gosset M, Berenbaum F, Thirion S, Jacques C. Primary culture and phenotyping of murine chondrocytes. Nature protocols. 2008;3(8):1253–60. doi: 10.1038/nprot.2008.95. [DOI] [PubMed] [Google Scholar]
- 16.Dong Y, Jesse AM, Kohn A, Gunnell LM, Honjo T, Zuscik MJ, et al. RBPjkappa-dependent Notch signaling regulates mesenchymal progenitor cell proliferation and differentiation during skeletal development. Development. 2010;137(9):1461–71. doi: 10.1242/dev.042911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pacifici M, Koyama E, Iwamoto M. Mechanisms of synovial joint and articular cartilage formation: recent advances, but many lingering mysteries. Birth Defects Res C Embryo Today. 2005;75(3):237–48. doi: 10.1002/bdrc.20050. [DOI] [PubMed] [Google Scholar]
- 18.Kohn A, Dong Y, Mirando AJ, Jesse AM, Honjo T, Zuscik MJ, et al. Cartilage-specific RBPjkappa-dependent and -independent Notch signals regulate cartilage and bone development. Development. 2012;139(6):1198–212. doi: 10.1242/dev.070649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Glasson SS, Chambers MG, Van Den Berg WB, Little CB. The OARSI histopathology initiative - recommendations for histological assessments of osteoarthritis in the mouse. Osteoarthritis Cartilage. 2010;18 (Suppl 3):S17–23. doi: 10.1016/j.joca.2010.05.025. [DOI] [PubMed] [Google Scholar]
- 20.Blaise R, Mahjoub M, Salvat C, Barbe U, Brou C, Corvol MT, et al. Involvement of the Notch pathway in the regulation of matrix metalloproteinase 13 and the dedifferentiation of articular chondrocytes in murine cartilage. Arthritis and rheumatism. 2009;60(2):428–39. doi: 10.1002/art.24250. [DOI] [PubMed] [Google Scholar]
- 21.Engin F, Yao Z, Yang T, Zhou G, Bertin T, Jiang MM, et al. Dimorphic effects of Notch signaling in bone homeostasis. Nature medicine. 2008;14(3):299–305. doi: 10.1038/nm1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grogan SP, Olee T, Hiraoka K, Lotz MK. Repression of chondrogenesis through binding of notch signaling proteins HES-1 and HEY-1 to N-box domains in the COL2A1 enhancer site. Arthritis and rheumatism. 2008;58(9):2754–63. doi: 10.1002/art.23730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Haller R, Schwanbeck R, Martini S, Bernoth K, Kramer J, Just U, et al. Notch1 signaling regulates chondrogenic lineage determination through Sox9 activation. Cell death and differentiation. 2011 doi: 10.1038/cdd.2011.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hardingham TE, Oldershaw RA, Tew SR. Cartilage, SOX9 and Notch signals in chondrogenesis. Journal of anatomy. 2006;209(4):469–80. doi: 10.1111/j.1469-7580.2006.00630.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hilton MJ, Tu X, Wu X, Bai S, Zhao H, Kobayashi T, et al. Notch signaling maintains bone marrow mesenchymal progenitors by suppressing osteoblast differentiation. Nature medicine. 2008;14(3):306–14. doi: 10.1038/nm1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mead TJ, Yutzey KE. Notch pathway regulation of chondrocyte differentiation and proliferation during appendicular and axial skeleton development. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(34):14420–5. doi: 10.1073/pnas.0902306106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tao J, Chen S, Yang T, Dawson B, Munivez E, Bertin T, et al. Osteosclerosis owing to Notch gain of function is solely Rbpj-dependent. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 2010;25(10):2175–83. doi: 10.1002/jbmr.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Watanabe N, Tezuka Y, Matsuno K, Miyatani S, Morimura N, Yasuda M, et al. Suppression of differentiation and proliferation of early chondrogenic cells by Notch. Journal of bone and mineral metabolism. 2003;21(6):344–52. doi: 10.1007/s00774-003-0428-4. [DOI] [PubMed] [Google Scholar]
- 29.Zanotti S, Smerdel-Ramoya A, Stadmeyer L, Durant D, Radtke F, Canalis E. Notch inhibits osteoblast differentiation and causes osteopenia. Endocrinology. 2008;149(8):3890–9. doi: 10.1210/en.2008-0140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ando K, Kanazawa S, Tetsuka T, Ohta S, Jiang X, Tada T, et al. Induction of Notch signaling by tumor necrosis factor in rheumatoid synovial fibroblasts. Oncogene. 2003;22(49):7796–803. doi: 10.1038/sj.onc.1206965. [DOI] [PubMed] [Google Scholar]
- 31.Chapman G, Sparrow DB, Kremmer E, Dunwoodie SL. Notch inhibition by the ligand DELTA-LIKE 3 defines the mechanism of abnormal vertebral segmentation in spondylocostal dysostosis. Human molecular genetics. 2011;20(5):905–16. doi: 10.1093/hmg/ddq529. [DOI] [PubMed] [Google Scholar]
- 32.Guegan K, Stals K, Day M, Turnpenny P, Ellard S. JAG1 mutations are found in approximately one third of patients presenting with only one or two clinical features of Alagille syndrome. Clinical genetics. 2011 doi: 10.1111/j.1399-0004.2011.01749.x. [DOI] [PubMed] [Google Scholar]
- 33.Majewski J, Schwartzentruber JA, Caqueret A, Patry L, Marcadier J, Fryns JP, et al. Mutations in NOTCH2 in families with Hajdu-Cheney syndrome. Human mutation. 2011;32(10):1114–7. doi: 10.1002/humu.21546. [DOI] [PubMed] [Google Scholar]
- 34.Stahl EA, Raychaudhuri S, Remmers EF, Xie G, Eyre S, Thomson BP, et al. Genome-wide association study meta-analysis identifies seven new rheumatoid arthritis risk loci. Nature genetics. 2010;42(6):508–14. doi: 10.1038/ng.582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Karlsson C, Brantsing C, Egell S, Lindahl A. Notch1, Jagged1, and HES5 are abundantly expressed in osteoarthritis. Cells Tissues Organs. 2008;188(3):287–98. doi: 10.1159/000121610. [DOI] [PubMed] [Google Scholar]
- 36.Chen S, Tao J, Bae Y, Jiang MM, Bertin T, Chen Y, et al. Notch gain of function inhibits chondrocyte differentiation via Rbpj-dependent suppression of Sox9. J Bone Miner Res. 2012 doi: 10.1002/jbmr.1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ninov N, Borius M, Stainier DY. Different levels of Notch signaling regulate quiescence, renewal and differentiation in pancreatic endocrine progenitors. Development. 2012;139(9):1557–67. doi: 10.1242/dev.076000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hosaka Y, Saito T, Sugita S, Hikata T, Kobayashi H, Fukai A, et al. Notch signaling in chondrocytes modulates endochondral ossification and osteoarthritis development. Proc Natl Acad Sci U S A. 2013;110(5):1875–80. doi: 10.1073/pnas.1207458110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rhee DK, Marcelino J, Baker M, Gong Y, Smits P, Lefebvre V, et al. The secreted glycoprotein lubricin protects cartilage surfaces and inhibits synovial cell overgrowth. J Clin Invest. 2005;115(3):622–31. doi: 10.1172/JCI200522263. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) ABH/OG staining of E15.5 and E18.5 WT and RBPjκPrx1 knee sections. (B) ABH/OG staining and in situ hybridization (ISH) of WT and RBPjκPrx1 knee sections at two weeks of age. ISH signal for Mmp13 and Prg4 expression is shown in red. M, meniscus; AC, articular cartilage; SOC, secondary ossification center; HC, hypertrophic cartilage. (N>3 for all experiments).
MicroCT analyses of 2, 4, 6, and 8-month old WT and RBPjκPrx1 mutant knees. Flattening of tibial plateau (red arrows), synovial tissue expansion and mineralization (blue arrows), and osteophyte formation (green arrows) are highlighted in RBPjκPrx1 mutants. (N>3 for all time points).
(A) Histomorphometric analyses of articular cartilage area and thickness from 2-month and 8-month old ABH/OG stained WT and RBPjκPrx1 knee sections. The tide-mark was used to determine upper and deep zone articular cartilage regions. At least 3 slides were measured per animal. (B) OARSI scoring for RBPjκPrx1 mutants and controls at 2- and 8-months of age. “*” Statistical significance with p-value less than 0.05. Data presented as the means +/− SE (N>5 for all time points).
(A) Prg4 ISH of 2 and 8-month old WT and RBPjκPrx1 mutant knee sections. Red arrows indicate diminishing or absent Prg4 expression in the articular cartilage and synovium of the RBPjκPrx1 mutants. (B) PRG4 IHC of 2 and 8-month old WT and RBPjκCol2TM mutant knee sections.
(A) Histomorphometric analyses of articular cartilage area and thickness from 2-month and 8-month old ABH/OG stained WT and RBPjκCol2TM knee sections. The tide-mark was used to determine upper zone (superficial and intermediate) and deep zone articular cartilage regions. (B) OARSI scoring for RBPjκCol2TM mutants and controls at 2- and 8-months of age. “*” Statistical significance with p-value less than 0.05. Data presented as the means +/− SE (N>5 for all time points).