

Abstract
1. The aim of the present study was to test the hypothesis that increasing kidney tissue concentrations of epoxyeicosatrienoic acids (EETs) by preventing their degradation to the biologically inactive dihydroxyeicosatrienoic acids (DHETEs) using blockade of soluble epoxide hydrolase (sEH) would attenuate the progression of chronic kidney disease (CKD).
2. Ren-2 transgenic rats (TGR) after 5/6 renal mass reduction (5/6 NX) served as a model of CKD associated with angiotensin (Ang) II-dependent hypertension. Soluble epoxide hydrolase was inhibited using cis-4-[4-(3-adamantan-1-yl-ureido)cyclohexyloxy]benzoic acid (c-AUCB; 3 mg/L drinking water) for 20 weeks after 5/6 NX. Sham-operated normotensive transgene-negative Hannover Sprague-Dawley (HanSD) rats served as controls.
3. When applied in TGR subjected to 5/6 NX, c-AUCB treatment improved survival rate, prevented the increase in blood pressure, retarded the progression of cardiac hypertrophy, reduced proteinuria and the degree of glomerular and tubulointerstitial injury and reduced glomerular volume. All these organ-protective actions were associated with normalization of the intrarenal EETs : DHETEs ratio, an index of the availability of biologically active EETs, to levels observed in sham-operated HanSD rats. There were no significant concurrent changes of increased intrarenal AngII content.
4. Together, these results show that 5/6 NX TGR exhibit a profound deficiency of intrarenal availability of active epoxygenase metabolites (EETs), which probably contributes to the progression of CKD in this model of AngII-dependent hypertension, and that restoration of intrarenal availability of EETs using long-term c-AUCB treatment exhibits substantial renoprotective actions.
Keywords: 5/6 nephrectomy, chronic kidney disease, cytochrome P450 enzymes, end-organ damage, epoxyeicosatrienoic acids, hypertension, renin–angiotensin system, soluble epoxide hydrolase
Introduction
Chronic kidney disease (CKD) represents a serious problem of current nephrology and its incidence has been increasing steadily.1 The recognition that CKD tends to progress to end-stage renal disease (ESRD), regardless of the initial cause, has initiated extensive research of the underlying mechanism(s) and of potential therapeutic strategies aimed at interrupting or at least slowing down the progression of CKD to ESRD.2–4 To this end, a model of 5/6 renal mass reduction (5/6 NX), consisting of unilateral nephrectomy combined with surgical removal of two-thirds of the contralateral kidney, has been widely used. Despite extensive investigation conducted over the past 40 years, the mechanism(s) responsible for the progression of CKD to ESRD remain poorly understood. However, it is clear that different renal diseases exhibit common pathomorphological signs, such as tubulointerstitial fibrosis and tubular atrophy, followed by glomerulosclerosis.4–7 The studies using the 5/6 NX model have demonstrated that hypertension and inappropriately activated renin–angiotensin system (RAS) are two critical determinants of the progression rate of CKD to ESRD.3,8–13 In addition, both experimental and clinical studies have shown that RAS blocking agents, such as angiotensin-converting enzyme inhibitors (ACEI) and angiotensin (Ang) II receptor blockers (ARBs), are highly effective inhibitors of the progression of CKD (renoprotective action).8,14–17 Therefore, an antihypertensive therapeutic regimen involving direct inhibition of the RAS is currently accepted as a gold standard in therapy.
However, the effectiveness of the renoprotective action of RAS inhibition is limited, especially in advanced CKD.18–20 This points to the need for more complex pharmacological strategies that would possibly also target control systems other than the RAS. Considerable attention has been focused on the role of active metabolites of arachidonic acid (ARA), those generated by cytochrome P450 (CYP)-dependent enzymes. Evidence indicates that ARA is metabolized by CYP epoxygenases to produce epoxyeicosatrienoic acids (EETs), which are important regulators of cardiovascular and renal function.21–23 It has been shown that increasing kidney tissue EETs by preventing their degradation to biologically inactive dihydroxyeicosatrienoic acids (DHETEs), as achieved by blockade of soluble epoxide hydrolase (sEH), exhibits antihypertensive, cardio- and renoprotective actions.24–32 Therefore, increasing the bioavailability of EETs in the kidney could be a new strategy to prevent the progression of CKD to ESRD. However, recent studies addressing this issue have yielded opposing results. A study that used overexpression of CYP2J2, an enzyme that produces EETs, demonstrated important renoprotective effects in 5/6 NX rats,33 whereas another study that inhibited sEH pharmacologically failed to elicit renoprotective actions and increased the rate of progression of CKD in 5/6 NX mice.34
Considering this controversy and in view of the established knowledge that hypertension and increased intrarenal RAS are critical determinants of hypertension-associated end-organ damage and progression of CKD, we examined whether long-term treatment with an sEH inhibitor would retard the progression of CKD in 5/6 NX Ren-2 renin transgenic rats (TGR). In addition, application of this well-defined monogenetic model of AngII-dependent hypertension with endogenous activation of RAS35 appeared well-suited for the purpose.
Results
Series 1: Role of RAS and CYP metabolites in the early phase after 5/6 NX
As shown in Fig. 1a, plasma AngII levels in sham-operated TGR were significantly higher than in sham-operated HanSD (62 ± 5 vs 16 ± 3 fmol/mL, respectively; P < 0.05) and 5/6 NX elicited in the early phase substantial increases in plasma AngII compared with sham-operated TGR (196 ± 15 vs 62 ± 5 fmol/mL, respectively; P < 0.05). Similarly, total kidney AngII concentrations in sham-operated TGR were significantly higher than those in sham-operated HanSD and, in the early phase, 5/6 NX caused a further increase in concentrations that were much higher than those in sham-operated TGR (369 ± 14 vs 87 ± 6 fmol/g, respectively; P < 0.05; Fig. 1b). As shown in Fig. 1c, the intrarenal availability of biologically active epoxygenase metabolites, expressed as the EETs : DHETEs ratio, was significantly lower in sham-operated TGR than in sham-operated HanSD rats and, already in the early phase, 5/6 NX elicited a further profound decrease in the EETs : DHETEs ratio compared with sham-operated TGR (0.72 ± 0.14 vs 1.48 ± 0.17, respectively; P < 0.05).
Fig. 1.
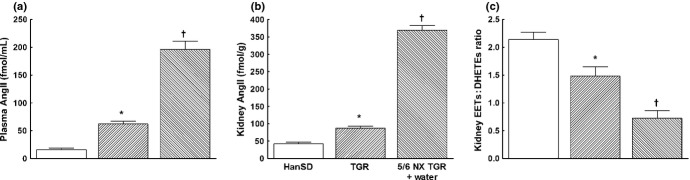
(a) Plasma and (b) kidney angiotensin (Ang) II concentrations and (c) kidney epoxyeicosatrienoic acids (EETs) : dihydroxyeicosatrienoic acids (DHETEs) ratio, measured 4 weeks (early phase) after 5/6 renal mass reduction (5/6 NX), in sham-operated Hannover Sprague-Dawley rats (HanSD; transgene negative), heterozygous Ren-2 transgenic rats (TGR) and untreated (i.e. vehicle (water) treated) 5/6 NX TGR rats. Data are the mean ± SEM. *P < 0.05 compared with sham-operated HanSD rats; †P < 0.05 compared with sham-operated TGR. ( ), HanSD; (
), HanSD; ( ), TGR; (
), TGR; ( ), 5/6 NX TGR + water.
), 5/6 NX TGR + water.
Densitometric analysis revealed no significant differences in CYP2C3 or sEH protein expression in the renal cortex between HanSD rats and TGR (data normalized against β-actin; Fig. 2). In the case of sEH, protein expression tended to be higher in TGR than HanSD rat, although the difference failed to reach statistical significance (Fig. 2b). In the early phase, 5/6 NX did not alter protein expression of CYP2C3, but significantly increased sEH expression.
Fig. 2.
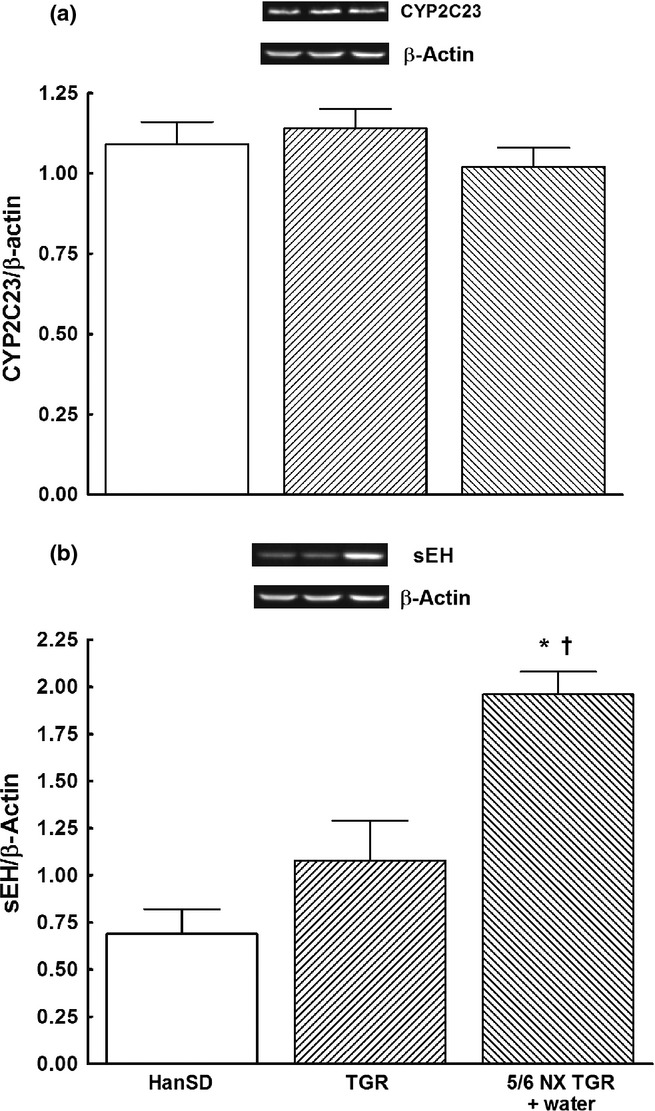
Expression of (a) CYP2C3 protein and (b) soluble epoxide hydrolase (sEH) in the kidney cortex, measured 4 weeks (early phase) after 5/6 renal mass reduction (5/6 NX), in sham-operated Hannover Sprague-Dawley rats (HanSD; transgene negative), heterozygous Ren-2 transgenic rats (TGR) and untreated (i.e. vehicle (water) treated) 5/6 NX TGR rats. Data are the mean ± SEM. *P < 0.05 compared with sham-operated HanSD rats; †P < 0.05 compared with sham-operated TGR at the same time point. ( ), HanSD; (
), HanSD; ( ), TGR; (
), TGR; ( ) 5/6 NX TGR + water.
) 5/6 NX TGR + water.
As shown in Fig. 3, there were no significant differences in CYP4A protein expression and 20-hydroxyeicosatetraenoic acid (20-HETE) concentrations in the renal cortex between HanSD rats and TGR, and 5/6 NX did not alter either value.
Fig. 3.
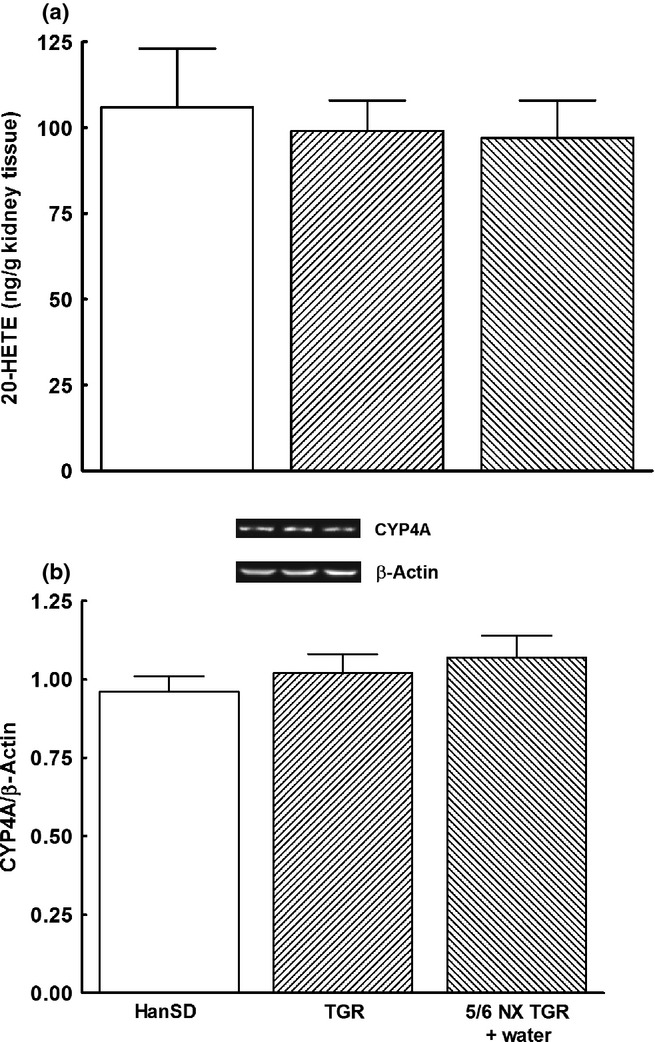
(a) Concentrations of 20-hydroxyeicosatrienoic acid (20-HETE) and (b) CYP4A protein expression in the kidney cortex, measured 4 weeks (early phase) after 5/6 renal mass reduction (5/6 NX), in sham-operated Hannover Sprague-Dawley rats (HanSD; transgene negative), heterozygous Ren-2 transgenic rats (TGR) and untreated (i.e. vehicle (water) treated) 5/6 NX TGR rats. Data are the mean ± SEM. ( ), HanSD; (
), HanSD; ( ), TGR; (
), TGR; ( ), 5/6 NX TGR + water.
), 5/6 NX TGR + water.
Series 2: Effects of RAS blockade and sEH inhibition on survival rate and signs of end-organ damage
All sham-operated TGR and HanSD rats survived until the end of the experiment. As shown in Fig. 4a, untreated 5/6 NX TGR began to die at Week 9 after 5/6 NX, with a final survival rate of 25%. Both therapeutic regimens (i.e. RAS blockade with a combination of 6 mg/L trandolapril and 100 mg/L losartan and sEH inhibition with 3 mg/L cis-4-[4-(3-adamantan-1-yl-ureido)cyclohexyloxy]benzoic acid (c-AUCB)), substantially improved post-5/6 NX survival rates in TGR; however, the former regimen was more effective (88% vs 72%, respectively; P < 0.05).
Fig. 4.
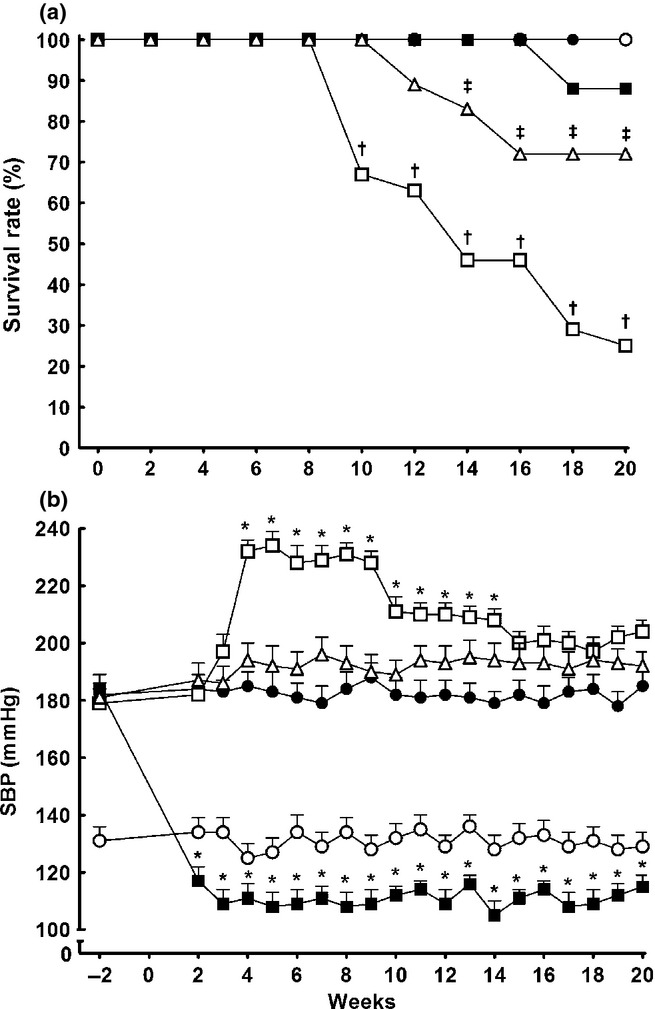
(a) Survival rates and (b) systolic blood pressure (SBP) in sham-operated Hannover Sprague-Dawley rats (HanSD; transgene negative; ○) and heterozygous Ren-2 transgenic rats (TGR; •), as well as in TGR rats after 5/6 renal mass reduction (5/6 NX) that were either untreated (□) or treated with: (i) combined renin–angiotensin system (RAS) blockade with trandolapril, an angiotensin-converting enzyme inhibitor, and losartan, an angiotensin receptor blocker (▪); or (ii) the soluble epoxide hydrolase inhibitor cis-4-[4-(3-adamantan-1-yl-ureido)cyclohexyloxy]benzoic acid (△). Data are the mean ± SEM. *P < 0.05 compared with baseline values; †P < 0.05 compared with sham-operated TGR at the same time point; †P < 0.05 compared with 5/6 NX TGR + RAS blockade at the same time point. ( ), HanSD; (
), HanSD; ( ), 5/6 NX TGR + water; (
), 5/6 NX TGR + water; ( ), 5/6 NX TGR + sEHi; (
), 5/6 NX TGR + sEHi; ( ), TGR; (
), TGR; ( ), 5/6 NX TGR + RAS blockade.
), 5/6 NX TGR + RAS blockade.
As shown in Fig. 4b, sham-operated HanSD rats remained normotensive and sham-operated TGR were considerably hypertensive, without any significant changes in systolic blood pressure (SBP) during the experiment. In TGR, 5/6 NX caused further significant increases in SBP above the initial SBP of 179 ± 5 mmHg, with the maximum reached at Week 8 after 5/6 NX (231 ± 4 mmHg; P < 0.05). Treatment with an sEH inhibitor prevented this increase and SBP remained at the same level as observed in sham-operated TGR. In contrast, RAS blockade not only prevented an increase in SBP in TGR after 5/6 NX, but rapidly reduced it, even below the levels observed in sham-operated HanSD rats (115 ± 4 vs 129 ± 4 mmHg, respectively; P < 0.05).
As shown in Fig. 5a, sham-operated HanSD rats exhibited minimal proteinuria throughout the experiment (7.2 ± 0.9 mg/24 h at the end of the experiment). Sham-operated TGR had pronounced proteinuria, more than threefold higher than that observed in sham-operated HanSD rats, throughout the experiment. Untreated 5/6 NX TGR exhibted a marked increase in proteinuria, reaching the maximum 8 weeks after 5/6 NX (161 ± 12 mg/24 h; P < 0.05 vs all other corresponding values). Blockade of the RAS prevented the increase in proteinuria that occurred after 5/6 NX in TGR and, until end of the experiment, the proteinuria was even lower than observed in sham-operated HanSD rats (2.7 ± 1.2 vs 7.2 ± 0.9 mg/24 h, respectively; P < 0.05). Treatment with the sEH inhibitor attenuated the increase in proteinuria in 5/6 NX TGR and, 16 weeks after 5/6 NX, the values observed were similar as those in sham-operated TGR; subsequently (at Week 20 after 5/6 NX) proteinuria was higher than observed in sham-operated TGR, but still significantly lower than that in untreated 5/6 NX TGR (35 ± 3 vs 67 ± 8 mg/24 h, respectively; P < 0.05).
Fig. 5.
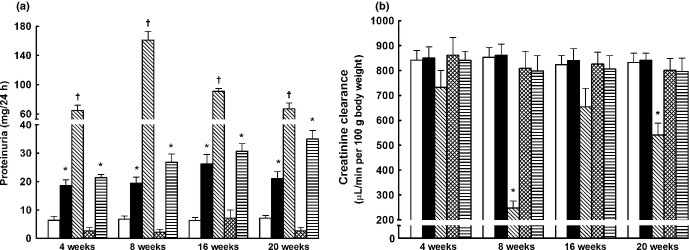
(a) Proteinuria and (b) creatinine clearance in sham-operated Hannover Sprague-Dawley rats (HanSD; transgene negative;  ) and heterozygous Ren-2 transgenic rats (TGR; ▪), as well as in TGR rats after 5/6 renal mass reduction (5/6 NX) that were either untreated (
) and heterozygous Ren-2 transgenic rats (TGR; ▪), as well as in TGR rats after 5/6 renal mass reduction (5/6 NX) that were either untreated ( ) or treated with: (i) combined renin–angiotensin system (RAS) blockade with trandolapril, an angiotensin-converting enzyme inhibitor, and losartan, an angiotensin receptor blocker (
) or treated with: (i) combined renin–angiotensin system (RAS) blockade with trandolapril, an angiotensin-converting enzyme inhibitor, and losartan, an angiotensin receptor blocker ( ); or (ii) the soluble epoxide hydrolase inhibitor cis-4-[4-(3-adamantan-1-yl-ureido)cyclohexyloxy]benzoic acid (
); or (ii) the soluble epoxide hydrolase inhibitor cis-4-[4-(3-adamantan-1-yl-ureido)cyclohexyloxy]benzoic acid ( ). Data are the mean ± SEM. *P < 0.05 compared with sham-operated HanSD rats at the same time point; †P < 0.05 compared with sham-operated TGR at the same time point. (
). Data are the mean ± SEM. *P < 0.05 compared with sham-operated HanSD rats at the same time point; †P < 0.05 compared with sham-operated TGR at the same time point. ( ), HanSD; (
), HanSD; ( ), 5/6 NX TGR + water; (
), 5/6 NX TGR + water; ( ), 5/6 NX TGR + sEHi; (▪), TGR; (
), 5/6 NX TGR + sEHi; (▪), TGR; ( ), 5/6 NX TGR + RAS blockade.
), 5/6 NX TGR + RAS blockade.
As shown in Fig. 5b, there were no significant differences in creatinine clearance between sham-operated HanSD and sham-operated TGR rats throughout the experimental period, indicating that despite severe hypertension, marked proteinuria and some degree of renal glomerular damage (see below) at the age of 8 months sham-operated TGR did not exhibit significant impairment of renal function. These findings are in accordance with previous studies showing that heterozygous TGR without any intervention (e.g. high salt or 5/6 NX) exhibit high resistance to the development of hypertension-induced renal damage.11,12 In contrast, untreated 5/6 NX TGR exhibited significantly lower creatinine clearance (especially 8 weeks after 5/6 NX, just before the first rats began to die) compared with sham-operated TGR. Both treatment regimens prevented decreases in creatinine clearance in 5/6 NX TGR.
As shown in Fig. 6a, sham-operated TGR clearly exhibited cardiac hypertrophy (measured as the ratio of left ventricular weight (LVW) : length (TL)) compared with sham-operated HanSD rats. In TGR, 5/6 NX induced a marked increase in LVW : TL above the values seen in sham-operated TGR (29.6 ± 1.1 vs 24.1 ± 1.2, respectively; P < 0.05). Renin–angiotensin system blockade not only prevented the increase in LVW : TL in 5/6 NX TGR, but even decreased it below values observed in sham-operated HanSD rats (14.4 ± 0.6 vs 16.1 ± 0.8, respectively; P < 0.05). Blockade of sEH prevented the increase in LVW : TL observed in TGR after 5/6 NX.
Fig. 6.
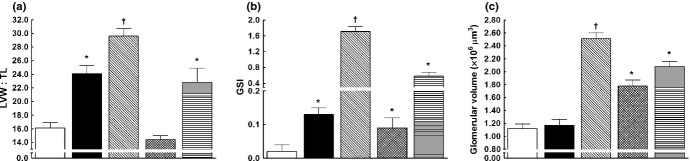
(a) Left ventricular weight (LVW) : tibia length ratio (mg/mm), (b) glomerulosclerosis index (GSI) and (c) glomerular volume in the late phase (i.e. 20 weeks after 5/6 renal mass reduction (5/6 NX)) in sham-operated Hannover Sprague-Dawley rats (HanSD; transgene negative;  ) and heterozygous Ren-2 transgenic rats (TGR; ▪), as well as in TGR rats after 5/6 renal mass reduction (5/6 NX) that were either untreated (
) and heterozygous Ren-2 transgenic rats (TGR; ▪), as well as in TGR rats after 5/6 renal mass reduction (5/6 NX) that were either untreated ( ) or treated with: (i) combined renin–angiotensin system (RAS) blockade with trandolapril, an angiotensin-converting enzyme inhibitor, and losartan, an angiotensin receptor blocker (
) or treated with: (i) combined renin–angiotensin system (RAS) blockade with trandolapril, an angiotensin-converting enzyme inhibitor, and losartan, an angiotensin receptor blocker ( ); or (ii) the soluble epoxide hydrolase inhibitor cis-4-[4-(3-adamantan-1-yl-ureido)cyclohexyloxy]benzoic acid (
); or (ii) the soluble epoxide hydrolase inhibitor cis-4-[4-(3-adamantan-1-yl-ureido)cyclohexyloxy]benzoic acid ( ). Data are the mean ± SEM. *P < 0.05 compared with sham-operated HanSD rats; †P < 0.05 compared with sham-operated TGR. (
). Data are the mean ± SEM. *P < 0.05 compared with sham-operated HanSD rats; †P < 0.05 compared with sham-operated TGR. ( ), HanSD; (
), HanSD; ( ), 5/6 NX TGR + water; (
), 5/6 NX TGR + water; ( ), 5/6 NX TGR + sEHi; (▪), TGR; (
), 5/6 NX TGR + sEHi; (▪), TGR; ( ), 5/6 NX TGR + RAS blockade.
), 5/6 NX TGR + RAS blockade.
Sham-operated HanSD rats showed only minimal glomerulosclerosis (measured as the glomerulosclerosis index (GSI); Fig. 6b). After 5/6 NX, TGR showed a substantial increase in GSI (1.71 ± 0.12 vs 0.13 ± 0.02 in sham-operated TGR; P < 0.05). Renin–angiotensin system blockade in 5/6 NX TGR normalized GSI to values observed in sham-operated TGR. Inhibition of sEH substantially attenuated the increase in GSI after 5/6 NX, but it remained markedly higher than in sham-operated TGR. The pattern of kidney tubulointerstitial injury closely resembled that of GSI in all groups (data not shown).
As shown in Fig. 6c, there was no significant difference between glomerular volume in sham-operated HanSD rats and sham-operated TGR. Untreated 5/6 NX TGR exhibited a substantial increase in glomerular volume compared with sham-operated TGR (2.51 ± 0.09 × 106 vs 1.17 ± 0.09 × 106 μm3, respectively; P < 0.05). Each of the two therapeutic regimens significantly reduced glomerular volume in 5/6 NX TGR, but RAS blockade was more effective than sEH inhibition (1.78 ± 0.09 × 106 vs 2.08 ± 0.08 × 106 μm3, respectively; P < 0.05).
Representative slices of renal tissue stained with periodic acid-Schiff (PAS) of sham-operated HanSD rats, untreated 5/6 NX TGR, 5/6 NX TGR treated with RAS blockade and 5/6 NX TGR treated with sEH inhibition are shown in Fig. 7.
Fig. 7.

Representative renal parenchyma images in (a) sham-operated Hannover Sprague-Dawley (transgene negative) rats, (c) 5/6 nephrectomized (5/6 NX) heterozygous Ren-2 transgenic rats (TGR) treated with the vehicle (water), (b) 5/6 NX TGR treated with combined renin–angiotensin system (RAS) blockade with trandolapril, an angiotensin-converting enzyme inhibitor, and losartan, an angiotensin receptor blocker and (d) 5/6 NX TGR treated with the soluble epoxide hydrolase inhibitor cis-4-[4-(3-adamantan-1-yl-ureido)cyclohexyloxy]benzoic acid. Bars, 100 μm.
As shown in Fig. 8a, plasma AngII levels were significantly higher in sham-operated TGR than in sham-operated HanSD rats and 5/6 NX caused, also in the late phase, further increases in plasma AngII levels in TGR compared with sham-operated TGR (71 ± 6 vs 36 ± 4 fmol/mL, respectively; P < 0.05). Long-term sEH inhibition did not significantly change plasma AngII levels in 5/6 NX TGR. In contrast, RAS blockade caused significant decrease in plasma AngII levels in 5/6 NX TGR to values observed in sham-operated TGR.
Fig. 8.
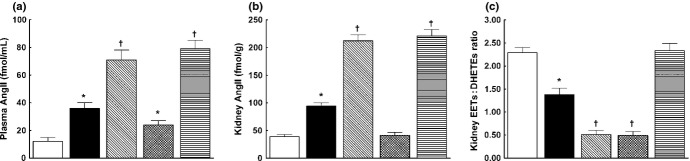
(a) Plasma and (b) kidney angiotensin (Ang) II concentrations and (c) kidney epoxyeicosatrienoic acids (EETs) : dihydroxyeicosatrienoic acids (DHETEs) ratio, measured 20 weeks (late phase) after 5/6 renal mass reduction (5/6 NX), in sham-operated Hannover Sprague-Dawley rats (HanSD; transgene negative;  ), heterozygous Ren-2 transgenic rats (TGR; ▪), in vehicle (water)-treated 5/6 NX TGR (
), heterozygous Ren-2 transgenic rats (TGR; ▪), in vehicle (water)-treated 5/6 NX TGR ( ), in 5/6 NX TGR treated with combined renin–angiotensin system (RAS) blockade with trandolapril, an angiotensin-converting enzyme inhibitor, and losartan, an angiotensin receptor blocker (
), in 5/6 NX TGR treated with combined renin–angiotensin system (RAS) blockade with trandolapril, an angiotensin-converting enzyme inhibitor, and losartan, an angiotensin receptor blocker ( ) and (d) 5/6 NX TGR treated with the soluble epoxide hydrolase inhibitor cis-4-[4-(3-adamantan-1-yl-ureido)cyclohexyloxy]benzoic acid (
) and (d) 5/6 NX TGR treated with the soluble epoxide hydrolase inhibitor cis-4-[4-(3-adamantan-1-yl-ureido)cyclohexyloxy]benzoic acid ( ). Data are the mean ± SEM. *P < 0.05 compared with sham-operated HanSD rats; †P < 0.05 compared with sham-operated TGR. (
). Data are the mean ± SEM. *P < 0.05 compared with sham-operated HanSD rats; †P < 0.05 compared with sham-operated TGR. ( ), HanSD; (
), HanSD; ( ), 5/6 NX TGR + sEHi; (▪), 5/6 NX TGR + RAS blockade.
), 5/6 NX TGR + sEHi; (▪), 5/6 NX TGR + RAS blockade.
Similarly, kidney AngII concentrations were significantly higher in sham-operated TGR than in sham-operated HanSD rats. After 5/6 NX, kidney AngII concenetrations in TGR further increased to levels substantially higher than in sham-operated TGR (212 ± 11 vs 94 ± 6 fmol/g, respectively; P < 0.05). In 5/6 NX TGR, RAS blockade decreased kidney AngII to values that were not significantly different from those in sham-operated HanSD rats (41 ± 6 vs 39 ± 4 fmol/g, respectively). In contrast, sEH blockade did not alter kidney AngII concentrations in 5/6 NX TGR (Fig. 8b). As shown in Fig. 8c, the intrarenal availability of biologically active epoxygenase metabolites, expressed as the EETs : DHETEs ratio, was significantly lower in sham-operated TGR than in sham-operated HanSD rats. The EETs : DHETEs ratio was much lower in untreated 5/6 NX TGR than in sham-operated TGR (0.51 ± 0.09 vs 1.28 ± 0.14, respectively; P < 0.05). Renin–angiotensin system blockade in 5/6 NX TGR did not significantly change the ratio; in contrast, sEH inhibition significantly increased the EETs : DHETEs ratio to levels observed in sham-operated HanSD rats.
Discussion
The present study evaluated the effectiveness of chronic EET activity, achieved by blockade of soluble sEH using c-AUCB, on the progression of CKD induced by 5/6 NX in TGR rats, a unique monogenetic model of RAS-dependent hypertension. This was compared with the effectiveness of a thorough two-level RAS blockade, which is currently accepted as the gold standard therapy used to retard the progression of CKD to ESRD.8,11,12,14–17,36
The first important finding of the present study was that the 5/6 NX in TGR not only increased circulating and kidney concentrations of AngII, as reported previously,12 but it aslo elicited a profound decrease in the intrarenal availability of biologically active epoxygenase metabolites, as evidenced by the decrease in the EETs : DHETEs ratio in the early and late phase after 5/6 NX. Because the renal generation of EETs in TGR after 5/6 NX is apparently in normal range, as indicated by unaltered protein expression of the CYP2C23 enzyme, the reduced intrarenal availability of biologically active epoxygenase metabolites is likely the result of increased conversion of EETs to DHETEs, as indicated by the distinctly increased renal expression of sEH protein in 5/6 NX TGR. Furthermore, our data show that intrarenal formation of 20-HETE was unaltered in 5/6 NX TGR compared with both sham-operated HanSD rats and sham-operated TGR. This finding is of particular importance because it is now well recognized that abnormalities in the CYP hydroxylase pathway play an important role in the pathophysiology of hypertension and hypertension-associated end-organ damage in various experimental models.37,38 In addition, sEH inhibition with c-AUCB in TGR subjected to 5/6 NX substantially improved the rats’ survival rate, prevented blood pressure increases, retarded the progression of cardiac hypertrophy, reduced proteinuria and the degree of glomerular and tubulointerstitial injury, prevented decreases in the clearance of endogenous creatinine and reduced glomerular volume. All these organ-protective actions were associated with normalization of the intrarenal EETs : DHETEs ratio to levels observed in sham-operated HanSD rats, but no significant change in increased intrarenal AngII. Remarkably, our finding that the dose of c-AUCB used in the present study (3 mg/L) substantially increased tissue concentrations of EETs without altering intrarenal AngII levels in the remnant kidney indicates that all the beneficial effects of long-term treatment with sEH inhibition in 5/6 NX TGR should be ascribed to increased intrarenal EETs bioavailability.
Together, the findings support the notion that the deficiency in the intrarenal availability of biologically active epoxygenase metabolites contributes to the progression of CKD and development of ESRD in 5/6 NX TGR. This view is in accordance with previous studies reporting that net intrarenal deficiency of EETs contributes to the pathophysiology of certain forms of AngII-dependent hypertension.22,24–32 It is of particular note that c-AUCB treatment with a dose that increased the intrarenal EETs : DHETEs ratio but did not alter the elevated circulating and renal RAS activity was still able to attenuate the development of AngII-dependent malignant hypertension and of hypertension-induced end-organ damage.29,31,32
Nevertheless, it is important to note that in a recent study with 5/6 NX mice, Jung et al.34 demonstrated that long-term c-AUCB treatment at an almost identical dosage to that used in the present study did not prevent the progression of CKD (no effect on SBP, no improvement in survival rate and no attenuation of renal glomerular and tubulointerstitial injury) and tended to accelerate rather than retard (vide augmented albuminuria) the course of CKD. There is no satisfactory explanation for these results, which contradict those of the present study, as well as earlier findings showing beneficial effects of sEH inhibitor treatment on the progression of end-organ damage.25,26,29,31 However, it should be noted that in the study of Jung et al.34 the 5/6 NX mice exhibited suppression of sEH expression and elevated EETs levels, the opposite to findings in AngII-dependent hypertension models in rats, in which increased expression and activity of sEH with consequent reduction of biologically active EETs were found.29,31,39,40 Apparently, blockade of sEH is antihypertensive and renoprotective only under conditions of increased sEH activity and reduced EETs bioavailability.22–24 The reasons for the suppression of sEH expression in 5/6 NX mice remain unclear; a species difference between rats and mice possibly involving different regulation of sEH expression and/or activity should be considered.
What are the mechanisms underlying the renoprotective actions of sEH inhhibitor treatment in 5/6 NX TGR? It will be recalled that the first theory explaining the mechanism of the progression of CKD, truly a milestone in nephrological research, was formulated by Brenner.2 On the basis of micropuncture studies with remnant kidneys of 5/6 NX rats,5 Brenner2 proposed that glomerular hyperfiltration and hypertension represented a compensatory response to a reduction in nephron number that results in damage to the healthy remnant glomeruli. The compensation is, in fact, maladaptive and, within the vicious circle, glomerulosclerosis progresses, ultimately leading to ESRD. According to this ‘haemodynamic’ theory, all renoprotective measures applied in CKD should focus on the correction of abnormal glomerular haemodynamics.2,4,5 However, subsequent studies using a serial micropuncture technique in remnant kidneys of 5/6 NX rats revealed that, on the one hand, haemodynamic abnormalities that developed in the remnant glomeruli had no correlation with the degree of sclerosis subsequently developing in the same glomerulus41,42 and, on the other hand, an unequivocal correlation between glomerular hypertrophy and glomerulosclerosis.43,44 Based on these studies, it had to be recognized that glomerular hypertrophy is an additional independent risk factor for the development of glomerulosclerosis, which, in accordance with Laplace's law (tension = radius × pressure), leads to an increase in wall tension and glomerular capillary damage, as well as to podocyte injury and depletion, the latter also identified as one of the major causes of glomerulosclerosis.7,43–45 Considering this ‘hypertrophy’ theory, of particular interest is our finding that sEH inhibitor treatment significantly reduced the glomerular volume in 5/6 NX TGR.
When the findings of the present study are considered in the context of the earlier studies and theories, we suggest that the mechanism underlying the renoprotective effects of long-term sEH inhibitor treatment in 5/6 NX TGR is related to attenuation of maladaptive compensatory glomerular growth, which in these animals is mediated through antiproliferative effects of normalized intrarenal EETs levels. Nevertheless, it is known that renal fibrosis is an important factor in the progression of CKD,7,46 and it has been documented that increased renal expression of fibronectin and collagen is a characteristic feature of 5/6 NX rats.47 Therefore, the lack of measurement of expression of these factors is an obvious limitation of the present study. Another limitation becomes apparent with the current evidence that oxidative stress is an important contributor to the pathogenesis of renal fibrosis.48 Because TGR, an AngII-dependent model of hypertension, exhibits increased generation of superoxide (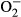 ),49 the role of oxidative stress in the progression of CKD in this model should also be considered and investigated. These two limitations inevitably narrow our insight into the complex mechanisms responsible for the renoprotective actions of sEH inhibitor treatment in 5/6 NX TGR.
),49 the role of oxidative stress in the progression of CKD in this model should also be considered and investigated. These two limitations inevitably narrow our insight into the complex mechanisms responsible for the renoprotective actions of sEH inhibitor treatment in 5/6 NX TGR.
Another important finding of the present study is that in 5/6 NX TGR, the antihypertensive regimen based on combined RAS blockade markedly improved survival rate and normalized blood pressure, cardiac hypertrophy and proteinuria; moreover, the treatment prevented the development of renal glomerular and tubulointerstitial injury and decreases in the clearance of endogenous creatinine. Furthermore, combined RAS blockade not only prevented the rise in kidney AngII concentration, but even decreased it to levels observed in sham-operated HanSD rats. All these findings are in good agreement with our recent study showing that hypertension and increased intrarenal RAS are two critical determinants of the rate of progression of CKD in 5/6 NX TGR,12 as well as with studies suggesting the crucial role of the intrarenal RAS in the pathophysiology of hypertension-associated end-organ damage.50–53 Nevertheless, the results of the present study indicate that the effectiveness of the renoprotective action of RAS blockade is limited during the late stage of CKD. Even though hypertension and intrarenal RAS activity were effectively controlled, some 5/6 NX TGR under RAS blockade started Week 18 (two of 17 rats), which was was preceded by a transient exacerbation of proteinuria. Together, these findings further support the notion that even if an antihypertensive regimen based on RAS blockade is a key component of the treatment strategy aimed at slowing-down the progression of CKD,8,11,12,14–17,36,54,55 there is a need to target vasoactive systems other than the RAS.
In view of our current encouraging findings indicating renoprotective effects of normalization of intrarenal EETs bioavailability on the progression of CKD, it seems logical and desirable to investigate whether combined sEH inhibition and RAS blockade may have additive beneficial effects in this model of CKD. However, it this regard it is important to emphasize that the renoprotective action of RAS blockade was much less successful in the late phase of CKD18–20 and, in agreement with the present results, we have found previously that in TGR the two-level RAS inhibition provides complete renoprotection when applied no later than 16 weeks after 5/6 NX.11,12 Therefore, it is reasonable to assume that potentially superior renoprotection of combined sEH inhibition and RAS blockade can possibly be demonstrated in very long-term studies only. Therefore, a relevant research project would require a follow-up period substantially longer than is routinely used in studies with 5/6 NX rats.11,12,55
In addition, we recently demonstrated in HanSD rats, which are originally normotensive and have normal plasma and kidney AngII levels, 5/6 NX led to the development of hypertension and marked activation of the intrarenal RAS in the remnant kidney and that these factors were dominant determinants of the rate of progression of CKD.12 Therefore, one could reasonably assume that 5/6 NX HanSD rats would also exhibit decreased EETs availability in the remnant kidney and that sEH inhibition could also exhibit renoprotective actions and retard the progression of CKD in initially normotensive HanSD rats subjected to renal mass reduction. However, the present study was designed to address first the role of intrarenal biologically active epoxygenase metabolites in the progression of CKD in hypertensive animals with already augmented RAS activity; the reason for this was that previous studies22–24,27–32 provided a good rationale for the use of sEH inhibition as a new therapeutic strategy to slow the progression of CKD in an AngII-dependent hypertensive model. Nevertheless, the findings of the present study highlight the need to establish whether the renoprotective effects of long-term treatment with an sEH inhibitor would also be demonstrable in 5/6 NX animals with originally normal blood pressure and without inappropriate activation of the RAS. This would make a desirable target of a future, hopefully more conclusive, study.
In conclusion, the results of the present study show that hypertensive TGR subjected to 5/6 renal mass reduction exhibit a profound decrease in the intrarenal EETs : DHETEs ratio, which strongly suggests that the deficiency of intrarenal active epoxygenase metabolites accelerates the progression of CKD in this AngII-dependent hypertensive model. In addition, long-term restoration of intrarenal biologically active EETs, achieved by pharmacological sEH inhibition, provided an important renoprotective effect. The finding that normalization of the intrarenal availability of active epoxygenase products substantially attenuates the progression of CKD in 5/6 NX TGR should be considered in attempts to develop new pharmacological strategies aimed at slowing down the progression of CKD to ESRD.
Methods
Ethical approval and animals
All studies were performed in accordance with the guidelines and practices established by the Animal Care and Use Committee of the Institute for Clinical and Experimental Medicine, Prague, which accord with the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes. All animals used in the present study were bred at the Center of Experimental Medicine of the Institute for Clinical and Experimental Medicine from stock animals supplied by the Max Delbrück Center for Molecular Medicine (Berlin, Germany), which is accredited by the Czech Association for Accreditation of Laboratory Animal Care. Heterozygous TGR were generated by breeding male homozygous TGR with female homozygous HanSD rats, as described previously;35 age-matched HanSD rats served as transgene-negative normotensive controls. Rats were kept on a 12 h light–dark cycle. Throughout the experiments, rats were fed a normal salt, normal protein diet (0.45% NaCl, 19–21% protein) produced by SEMED (Prague, Czech Republic) and had free access to tap water.
Chemicals
The sEH inhibitor cis-4-[4-(3-adamantan-1-yl-ureido)cyclohexyloxy]benzoic acid (c-AUCB) was prepared fresh three times a week and given in drinking water at 3 mg/L.27–32 The dose of c-AUCB used in the present study was selected on the basis of the results of a recent study in which this dose elicited substantial increases in tissue concentrations of EETs without altering RAS activity.29 We purposely chose a dose of c-AUCB that blocks only sEH activity without altering plasma and tissue AngII levels because the major aim of this treatment regimen was to evaluate the effects of pharmacologically induced EETs concentrations on the progression of CKD in 5/6 NX TGR. The activity of the RAS can be pharmacologically altered at various levels. We and others12,36,54,55 have demonstrated that pharmacological blockade using a combination of ACEI and ARB at high doses provides cardio- and renoprotection superior to that achieved with routine dosage. Thus, as in recent studies,11,12 for ‘RAS blockade’ in the present study we used a combination of trandolapril (6 mg/L in drinking water; Gopten; Abbot, Prague, Czech Republic) and losartan (100 mg/L in drinking water; Lozap; Zentiva, Prague, Czech Republic).
Experimental design
Series 1: Role of RAS and CYP metabolites in the early phase after 5/6 NX
The aim of this series of experiments was to evaluate the degree of activation of the RAS system and of the rate of synthesis within the two CYP-dependent enzymatic pathways, namely epoxygenase and ω-hydroxylase.
Male HanSD rats and TGR (9 weeks old) were anaesthetized with a mixture of tiletamine + zolazepam (8 mg/kg; Virbac, Carros, France) and xylazine (4 mg/kg, i.m.; Spofa, Prague, Czech Republic) and 5/6 NX was performed as described previously.11,12 Sham-operated HanSD rats served as healthy normotensive controls. It is well recognized that AngII is the major active product of the RAS and a reliable marker of the activity of the system.50 Therefore, at the end of the experiments, unaesthetised rats were decapitated and plasma and tissue AngII concentrations were determined by radioimmuassay. This approach was used because we have recently demonstrated that measured AngII levels are altered by anaesthesia.56,57 Moreover, we were able to compare the results of the present study with those of our earlier studies into the role of the RAS in the pathophysiology of hypertension.11,12,51–53,56,57 In addition, we determined EETs and DHETEs concentrations in the kidney cortex. Samples were extracted and the extracts separated by reverse-phase HPLC being being analysed by negative-mode electrospray ionization and tandem mass spectroscopy as described previously.27–32,58 Specifically, 8,9-EETs, 11,12-EETs and 14,15-EETs were measured separately and then presented as a total. These metabolites are the most active products formed in the CYP epoxygenase enzymatic pathway.22,23 The EETs : DHETES ratio was obtained using total concentrations of EETs and DHETEs. Western blot analysis of the protein expression of CYP2C23, the enzyme predominantly responsible for the formation of EETs in the kidney, and sEH, the enzyme responsible for the conversion of EETs to DHETEs, in the renal cortex was performed as described previously,29,58 with levels normalized against β-actin. In addition, 20-HETE and protein expression of CYP4A, the enzyme responsible for the formation of 20-HETE, were analysed in the renal cortex as described previously.28
The following experimental groups were investigated: (i) sham-operated HanSD + vehicle (water) treated (n = 7); (ii) sham-operated TGR + vehicle (water) treated (n = 7); and (iii) 5/6 NX TGR + vehicle (water) treated (n = 9).
Series 2: Effects of RAS blockade and sEH inhibition on survival rate and signs of end-organ damage
Male HanSD rats and TGR (8 weeks old) derived from several litters were randomly assigned to experimental groups to make sure that the animals from a single litter did not prevail in any group. To detect intergroup differences in SBP over time, SBP was measured in accordance with recommendations for blood pressure measurements in conscious animals by tail plethysmography using a tail-cuff apparatus (MC 4000 (Hatteras Instruments, Cary, NC, USA) and RTBP 1007 (Kent Scientific, Torrington, CT, USA)).59 Three days before starting actual measurements, rats were accustomed to the indirect tail-cuff SBP measurement procedure. This method, regularly used in our laboratory,11,12 was previously validated and a close correlation found between these values and those yielded by direct blood pressure measurements in conscious rats using an indwelling aortic catheter. Measurements of SBP were started 14 days before 5/6 NX and performed at 3 day intervals until the end of the experiment. On Day 0 (9 weeks of age), 5/6 NX was performed as described above. After 72 h recovery, rats were either started on treatment or left untreated. The following experimental groups were investigated: (i) sham-operated HanSD + water (initial n = 10); (ii) sham-operated TGR + water (initial n = 10); (iii) 5/6 NX TGR + water (initial n = 24); (iv) 5/6 NX TGR + RAS blockade (initial n = 17); and (v) 5/6 NX TGR + sEH inhibitor (initial n = 18).
The follow-up period was 20 weeks. At Weeks 4, 8, 16 and 20 after Day 0, after appropriate habituation training, rats were placed in individual metabolic cages and 24 h urine was collected for determination of protein and creatinine. Blood samples were taken from the tail vein and plasma creatinine concentrations were measured by the picric acid colorimetric method using a commercially available kit (Lachema, Brno, Czech Republic). This approach was validated previously and is regularly used in our studies.11,12 At the end of the experiments, unaesthetised rats were decapitated and AngII, EETs and DHETEs concentrations were assessed as described for Series 1.
Samples from the second half of the kidney were used to assess renal glomerular damage. Kidneys were fixed in 4% formaldehyde, dehydrated and embedded in paraffin. Sections were stained with haematoxylin–eosin and PAS and were examined and evaluated in a blinded manner. Fifty glomeruli in each kidney were examined on a semiquantitative scale as follows:60 Grade 0, all glomeruli normal; Grade 1, sclerotic area up to 25% (minimal sclerosis); Grade 2, sclerotic area 25–50% (moderate sclerosis); Grade 3, sclerotic area 50–75% (moderate-to-severe sclerosis); and Grade 4, sclerotic area 75–100% (severe sclerosis). The GSI was calculated as follows:
where nx is the number of glomeruli in Grade x of glomerulosclerosis.
Renal cortical tubulointerstitial injury was evaluated as described by Nakano et al.60 and as used in our recent studies11,12 to determine inflammatory cell infiltration, tubular dilatation, atrophy or interstitial fibrosis. Injury was graded semiquantitatively using the following scale of lesions: Grade 0, no abnormal findings; Grade 1, mild (< 25% of the cortex); Grade 2, moderate (25–50% of the cortex); and Grade 3, severe (> 50% of the cortex). Lesions were assessed in at least 30 random and non-overlapping fields in the renal cortex.
Morphometric evaluation of the glomerular volume was made in the same kidney sections that were examined for morphological changes using the method validated by Lane et al.60 and used in our recent studies11,12,61 using the Nikon NIS-Elements AR 3.1 morphometric program (Nikon, Tokyo, Japan).
Based on previous experience,51,52 we used the the ratio of LVW : to determine the degree of cardiac hypertrophy.
Statistical analysis
Statistical analyses of the data were performed using graphpad prism software (GraphPad Software, San Diego, CA, USA). To analyse changes in blood pressure within groups, we used anova for repeated measurements, followed by the Student–Newman–Keuls’ test. Statistical comparisons of other results ere made by Student's t-test or one-way anova. Unless noted otherwise, data are expressed as the mean ± SEM and n represents the number of animals. Two-sided P < 0.05 was considered significant. For clarity of presentation, statistical evaluation of only two comparisons are marked in the figures: (i) against values in sham-operated HanSD rats (the main control group); and (ii) for 5/6 NX TGR (the model of CKD) versus sham-operated TGR.
Acknowledgments
This study was supported by grant no. NT/14012-3 awarded by the Internal Grant Agency of the Ministry of Health of the Czech Republic to LČ. ZH was supported by a grant from the Ministry of Health within the project for the Development of Research Organization 00023001 (IKEM)–Institutional Support. The Center for Experimental Medicine (IKEM) received financial support from the European Commission within the Operational Program Prague–Competitiveness; project ‘CEVKOON’ (#CZ.2.16/3.1.00/22126). LČ was the recipient of a grant (MŠMT-Kontakt LH 11116) from the Ministry of Education, Youth and Sports of the Czech Republic for the support of international research cooperation, which funded the cooperation with KK and AN. JDI was supported by a grant from the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK; NIDDK38226). SHH was supported, in part, by National Institute of Environmental Health Science (NIEHS) grant ES02710, NIEHS Superfund grant P42 ES04699, National Heart, Lung and Blood Institute (NHLBI) grant HL059699 and the CounterACT Program, National Institutes of Health Office of the Director and the National Institute of Neurological Disorders and Stroke (no. U54 NS079202 awarded to BDH). BDH is a George and Judy Marcus Senior Fellow of the American Asthma Foundation.
Disclosure
The authors declare no conflicts of interest.
References
- 1.US Renal Data System. USEDS 2012 Annual Data Report. Atlas of Chronic Kidney Disease and End-Stage Renal Disease in the United States. Bethesda: National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases; 2012. [Google Scholar]
- 2.Brenner BM. Nephron adaptation to renal injury or ablation. Am. J. Physiol. 1985;249:F324–37. doi: 10.1152/ajprenal.1985.249.3.F324. [DOI] [PubMed] [Google Scholar]
- 3.Zoja C, Abbate M, Remuzzi G. Progression of chronic kidney disease: Insight from animal models. Curr. Opin. Nephrol. Hypertens. 2006;15:250–7. doi: 10.1097/01.mnh.0000222691.53970.83. [DOI] [PubMed] [Google Scholar]
- 4.Hostetter TH. Hyperfiltration and glomerulosclerosis. Semin. Nephrol. 2003;23:194–9. doi: 10.1053/anep.2003.50017. [DOI] [PubMed] [Google Scholar]
- 5.Hostetter TH, Olson JL, Rennke HG, Ventatachalam MA, Brenner BM. Hyperfiltration in remnant nephrons: A potentially adverse response to ablation. Am. J. Physiol. 1981;241:F85–93. doi: 10.1152/ajprenal.1981.241.1.F85. [DOI] [PubMed] [Google Scholar]
- 6.Shimamura T, Morrison AB. A progressive glomerulosclerosis occurring in partial five-sixths nephrectomized rats. Am. J. Pathol. 1975;79:95–106. [PMC free article] [PubMed] [Google Scholar]
- 7.Kriz W. Podocyte is the major culprit accounting for the progression of chronic renal disease. Micros. Res. Tech. 2002;57:189–95. doi: 10.1002/jemt.10072. [DOI] [PubMed] [Google Scholar]
- 8.Macconi D. Targeting the renin angiotensin system for remission/regression of chronic kidney disease. Histol. Histopathol. 2010;25:655–68. doi: 10.14670/HH-25.655. [DOI] [PubMed] [Google Scholar]
- 9.Kohan DE. Endothelin, hypertension and chronic kidney disease: New insight. Curr. Opin. Nephrol. Hypertens. 2010;19:134–9. doi: 10.1097/MNH.0b013e328335f91f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Briet M, Burns KD. Chronic kidney disease and vascular remodeling. Molecular mechanisms and clinical implications. Clin. Sci. 2012;123:399–416. doi: 10.1042/CS20120074. [DOI] [PubMed] [Google Scholar]
- 11.Vanecková I, Kujal P, Husková Z, et al. Effects of combined endothelin a receptor and renin–angiotensin system blockade on the course of end-organ damage in 5/6 nephrectomized Ren-2 hypertensive rats. Kidney Blood Press. Res. 2012;35:382–92. doi: 10.1159/000336823. [DOI] [PubMed] [Google Scholar]
- 12.Kujal P, Certíková Chábová V, Vernerova Z, et al. Similar renoprotection after renin–angiotensin-dependent and -independent antihypertensive therapy in 5/6-nephrectomized Ren-2 transgenic rats: Are there blood pressure-independent effects? Clin. Exp. Pharmacol. Physiol. 2010;37:1159–69. doi: 10.1111/j.1440-1681.2010.05453.x. [DOI] [PubMed] [Google Scholar]
- 13.Bidani AK, Polichnowski AJ, Loutzenhiser R, Griffin KA. Renal microvascular dysfunction, hypertension and CKD progression. Curr. Opin. Nephrol. Hypertens. 2013;22:1–9. doi: 10.1097/MNH.0b013e32835b36c1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ptinopolou AG, Pikilidou MI, Lasaridis N. The effect of antihypertensive drugs on chronic kidney disease: A comprehensive review. Hypertens. Res. 2013;36:91–101. doi: 10.1038/hr.2012.157. [DOI] [PubMed] [Google Scholar]
- 15.Turner JM, Bauer C, Abramowitz MK, Melamed ML, Hostetter TH. Treatment of chronic kidney disease. Kidney Int. 2012;81:351–62. doi: 10.1038/ki.2011.380. [DOI] [PubMed] [Google Scholar]
- 16.Rüster C, Wolf G. Renin–angiotensin–aldosterone system and progression of renal disease. J. Am. Soc. Nephrol. 2006;17:2985–91. doi: 10.1681/ASN.2006040356. [DOI] [PubMed] [Google Scholar]
- 17.Berl T. Renal protection by inhibition of the renin–angiotensin–aldosterone system. J. Renin Angiotensin Aldosterone Syst. 2009;10:1–8. doi: 10.1177/1470320309102747. [DOI] [PubMed] [Google Scholar]
- 18.Perico N, Amuchastegui SC, Colosio V, Sonzogni G, Bertani T, Remuzzi G. Evidence that an angiotensin-converting enzyme inhibitor has a different effect on glomerular injury according to the different phase of the disease at which the treatment is started. J. Am. Soc. Nephrol. 1994;5:1139–46. doi: 10.1681/ASN.V541139. [DOI] [PubMed] [Google Scholar]
- 19.Gordon J, Kopp JB. Off the beaten renin–angiotensin–aldosterone system pathway: New perspectives on antiproteinuric therapy. Adv. Chronic Kidney Dis. 2011;18:300–11. doi: 10.1053/j.ackd.2011.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lattanzio MR, Weir MR. Does blockade of the renin–angiotensin–aldosterone system slow progression of all forms of kidney disease? Curr. Hypertens. Rep. 2010;12:369–77. doi: 10.1007/s11906-010-0142-2. [DOI] [PubMed] [Google Scholar]
- 21.Campbell WB, Fleming I. Epoxyeicosatrienoic acids and endothelium-dependent response. Pfugers Arch. 2010;459:881–95. doi: 10.1007/s00424-010-0804-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Imig JD. Epoxides and soluble epoxide hydrolase in cardiovascular physiology. Physiol. Rev. 2012;92:101–30. doi: 10.1152/physrev.00021.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Capdevilla J, Wang W. Role of cytochrome P450 epoxygenase in regulating renal membrane transport and hypertension. Curr. Opin. Nephrol. Hypertens. 2013;22:163–9. doi: 10.1097/MNH.0b013e32835d911e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Elmarakby AA. Reno-protective mechanisms of epoxyeicosatrienoic acids in cardiovascular disease. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2012;302:R321–30. doi: 10.1152/ajpregu.00606.2011. [DOI] [PubMed] [Google Scholar]
- 25.Li J, Carroll MA, Chander PN, Falck JR, Sangras B, Stier CT. Soluble epoxide hydrolase inhibitor, AUDA, prevents early salt-sensitive hypertension. Front. Biosci. 2008;13:3480–7. doi: 10.2741/2942. [DOI] [PubMed] [Google Scholar]
- 26.Huang H, Morisseau C, Wang JF, et al. Increasing or stabilizing renal epoxyeicosatrienoic acid production attenuates abnormal renal function and hypertension in obese rats. Am. J. Physiol. Renal Physiol. 2007;293:F342–9. doi: 10.1152/ajprenal.00004.2007. [DOI] [PubMed] [Google Scholar]
- 27.Sporková A, Kopkan L, Varcabová A, et al. Role of cytochrome P450 metabolites in the regulation of renal function and blood pressure in 2-kidney, 1-clip hypertensive rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011;300:R1468–75. doi: 10.1152/ajpregu.00215.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Neckár J, Kopkan L, Husková Z, et al. Inhibition of soluble epoxide hydrolase by cis-4-[4-(3-adamantan-I-ylureido) cyclohexyl-oxy]benzoic acid exhibits antihypertensive and cardioprotective actions in transgenic rats with angiotensin II-dependent hypertension. Clin. Sci. 2012;122:513–25. doi: 10.1042/CS20110622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Honetschlägerová Z, Husková Z, Vanourkova Z, et al. Renal mechanisms contributing to the antihypertensive action of soluble epoxide hydrolase inhibition in Ren-2 transgenic rats with inducible hypertension. J. Physiol. 2011;589:207–19. doi: 10.1113/jphysiol.2010.199505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kopkan L, Husková Z, Sporková A, et al. Soluble epoxide hydrolase inhibition exhibits antihypertensive actions independently of nitric oxide in mice with renovascular hypertension. Kidney Blood Press. Res. 2012;35:595–607. doi: 10.1159/000339883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Honetschlägerová Z, Sporková A, Kopkan L, et al. Inhibition of soluble epoxide hydrolase improves the impaired pressure–natriuresis relationship and attenuates the development of hypertension and hypertension-associated end-organ damage in Cyp1a1-Ren-2 transgenic rats. J. Hypertens. 2011;29:1590–601. doi: 10.1097/HJH.0b013e328349062f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Honetschlägerová Z, Kitada K, Husková Z, et al. Antihypertensive and renoprotective actions of soluble epoxide hydrolase inhibition in ANG II-dependent malignant hypertension are abolished by pretreatment with l-NAME. J. Hypertens. 2013;31:321–32. doi: 10.1097/HJH.0b013e32835b50aa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhao G, Tu L, Li X, et al. Delivery of AAV2-CYP2J2 protects remnant kidney in the 5/6-nephrectomized rat via inhibition of apoptosis and fibrosis. Hum. Gene Ther. 2012;23:688–99. doi: 10.1089/hum.2011.135. [DOI] [PubMed] [Google Scholar]
- 34.Jung O, Jansen F, Mieth A, et al. Inhibition of the soluble epoxide hydrolase promotes albuminuria in mice with progressive renal disease. PLoS One. 2010;5:e11979. doi: 10.1371/journal.pone.0011979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mullins JJ, Peters J, Ganten D. Fulminant hypertension in transgenic rats harbouring the mouse Ren-2 gene. Nature. 1990;344:541–4. doi: 10.1038/344541a0. [DOI] [PubMed] [Google Scholar]
- 36.Ripley E. Complementary effects of angiotensin-converting enzyme inhibitors and angiotensin receptor blockers in slowing the progression of chronic kidney disease. Am. Heart J. 2009;157(Suppl):S7–16. doi: 10.1016/j.ahj.2009.04.008. [DOI] [PubMed] [Google Scholar]
- 37.Čertíková Chábová V, Walkowska A, Kompanowska-Jezierska E, et al. Combined inhibition of 20-hydroxyeicosatetraenoic acid formation and epoxyeicosatrienoic acids degradation attenuates hypertension and hypertension-induced end-organ damage in Ren-2 transgenic rats. Clin. Sci. 2010;118:617–32. doi: 10.1042/CS20090459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Williams JM, Murphy S, Burke M, Roman RJ. 20-Hydroxyeicosatetraenoic acid. A new target for the treatment of hypertension. J. Cardiovasc. Pharmacol. 2010;56:336–44. doi: 10.1097/FJC.0b013e3181f04b1c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ai D, Fu Y, Guo D, et al. Angiotensin II up-regulates soluble epoxide hydrolase in vascular endothelium in vitro and in vivo. Proc. Natl Acad. Sci. USA. 2007;104:9018–23. doi: 10.1073/pnas.0703229104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ai D, Shyy JYY, Zhu Y. Linking an insect enzyme to hypertension: Angiotensin II–epoxide hydrolase interactions. Kidney Int. 2010;77:88–92. doi: 10.1038/ki.2009.349. [DOI] [PubMed] [Google Scholar]
- 41.Yoshida Y, Fogo A, Shiraga H, Glick AD, Ichikawa I. Serial micropuncture analysis of single nephron function in subtotal renal ablation. Kidney Int. 1988;33:855–67. doi: 10.1038/ki.1988.77. [DOI] [PubMed] [Google Scholar]
- 42.Fogo A, Yoshida Y, Glick AD, Homma T, Ichikawa I. Serial micropuncture analysis of glomerular function in two rat models of glomerular sclerosis. J. Clin. Invest. 1988;82:322–30. doi: 10.1172/JCI113590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yoshida Y, Fogo A, Ichikawa I. Glomerular hemodynamic changes vs. hypertrophy in experimental glomerular sclerosis. Kidney Int. 1989;35:654–60. doi: 10.1038/ki.1989.35. [DOI] [PubMed] [Google Scholar]
- 44.Yoshida Y, Kawamura T, Ikoma M, Fogo A, Ichikawa I. Effects of antihypertensive drugs on glomerular morphology. Kidney Int. 1989;36:626–35. doi: 10.1038/ki.1989.239. [DOI] [PubMed] [Google Scholar]
- 45.Fukuda A, Wickman LT, Vekatareddy MP, et al. Angiotensin II-dependent persistent podocyte loss from destabilized glomeruli causes progression of end-stage kidney disease. Kidney Int. 2012;81:40–55. doi: 10.1038/ki.2011.306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Guarino M, Tosoni A, Nebuloni M. Direct contribution of epithelium to organ fibrosis: Epithelial–mesenchymal transition. Hum. Pathol. 2009;40:1365–76. doi: 10.1016/j.humpath.2009.02.020. [DOI] [PubMed] [Google Scholar]
- 47.Ni H, Chen J, Pan M, et al. FTY720 prevents progression of renal fibrosis by inhibiting renal microvasculature endothelial dysfunction in a rat model of chronic kidney disease. J. Mol. Histol. 2013;44:693–703. doi: 10.1007/s10735-013-9521-8. [DOI] [PubMed] [Google Scholar]
- 48.Barnes JL, Gorin Y. Myofibroblast differention during fibrosis. Role of NAD(P)H oxidases. Kidney Int. 2011;79:944–56. doi: 10.1038/ki.2010.516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kopkan L, Husková Z, Vanourková Z, et al. Reduction of oxidative stress does not attenuate the development of angiotensin II-dependent hypertension in Ren-2 transgenic rats. Vasc. Pharmacol. 2009;51:175–81. doi: 10.1016/j.vph.2009.06.001. [DOI] [PubMed] [Google Scholar]
- 50.Kobori H, Nangaku M, Navar LG, Nishiyama A. The intrarenal renin–angiotensin system: From physiology to the pathobiology of hypertension and kidney disease. Pharmacol. Rev. 2007;59:251–87. doi: 10.1124/pr.59.3.3. [DOI] [PubMed] [Google Scholar]
- 51.Husková Z, Vanourková Z, Erbanová M, et al. Inappropriately high circulating and intrarenal angiotensin II levels during dietary salt loading exacerbate hypertension in Cyp1a1-Ren-2 transgenic rats. J. Hypertens. 2010;28:495–509. doi: 10.1097/HJH.0b013e3283345d69. [DOI] [PubMed] [Google Scholar]
- 52.Vanourková Z, Kramer HJ, Husková Z, et al. AT1 receptor blockade is superior to conventional triple therapy in protecting against end-organ damage Cyp1a1-Ren-2 transgenic rats with inducible hypertension. J. Hypertens. 2006;24:2465–72. doi: 10.1097/01.hjh.0000251909.00923.22. [DOI] [PubMed] [Google Scholar]
- 53.Červenka L, Vanečková I, Husková Z, et al. Pivotal role of AT1A receptors in the development of two-kidney, one-clip hypertension: Study in AT1A receptor knockout mice. J. Hypertens. 2008;26:1379–89. doi: 10.1097/HJH.0b013e3282fe6eaa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cao Z, Bonnet F, Davis B, Allen TJ, Cooper ME. Additive and anti-albuminuric effects of angiotensin-converting enzyme inhibition and angiotensin receptor antagonism in diabetic spontaneously hypertensive rats. Clin. Sci. 2001;100:591–9. [PubMed] [Google Scholar]
- 55.Fujihara CK, Velho M, Malheiros DM, Zatz R. An extremely high dose of losartan affords superior renoprotection in the remnant model. Kidney Int. 2005;67:1913–24. doi: 10.1111/j.1523-1755.2005.00290.x. [DOI] [PubMed] [Google Scholar]
- 56.Huskova Z, Kramer HJ, Thumova M, et al. Effects of anesthesia on plasma and kidney ANG II levels in normotensive and ANG II-dependent hypertensive rats. Kidney Blood Press. Res. 2006;29:74–83. doi: 10.1159/000092981. [DOI] [PubMed] [Google Scholar]
- 57.Husková Z, Kramer HJ, Vanourková Z, Červenka L. Effects of changes in sodium balance on plasma and kidney angiotensin II levels in anesthetized and conscious Ren-2 transgenic rats. J. Hypertens. 2006;24:517–27. doi: 10.1097/01.hjh.0000209988.51606.c7. [DOI] [PubMed] [Google Scholar]
- 58.Varcabova S, Huskova Z, Kramer HJ, et al. Antihypertensive action of soluble epoxide hydrolase inhibition in Ren-2 transgenic rats is mediated by suppression of the intrarenal renin–angiotensin system. Clin. Exp. Pharmacol. Physiol. 2013;40:273–81. doi: 10.1111/1440-1681.12018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kurtz TW, Griffin KA, Bidani AK, Davisson RL, Hall JE. Recommendations for blood pressure measurements in humans and experimental animals. Part 2: Blood pressure measurements in experimental animals. Hypertension. 2005;45:299–310. doi: 10.1161/01.HYP.0000150857.39919.cb. [DOI] [PubMed] [Google Scholar]
- 60.Nakano Y, Hirano T, Uehara K, et al. New rat model induced by anti-glomerular basement membrane antibody shows severe glomerular adhesion in early stage and quickly progress to end-stage renal failure. Pathol. Int. 2008;58:361–70. doi: 10.1111/j.1440-1827.2008.02237.x. [DOI] [PubMed] [Google Scholar]
- 61.Lane PH, Steffes MW, Mauer MS. Estimation of glomerular volume: A comparison of four methods. Kidney Int. 1992;41:1085–9. doi: 10.1038/ki.1992.165. [DOI] [PubMed] [Google Scholar]