

Abstract
Diabetes-induced hyperglycemia increases the extracellular concentration of methylglyoxal. Methylglyoxal-derived hydroimidazolones (MG-H) form advanced glycation end products (AGEs) that accumulate in the serum of diabetic patients. The binding of hydroimidozolones to the receptor for AGEs (RAGE) results in long-term complications of diabetes typified by vascular and neuronal injury. Here we show that binding of methylglyoxal-modified albumin to RAGE results in signal transduction. Chemically synthesized peptides containing hydroimidozolones bind specifically to the V domain of RAGE with nanomolar affinity. The solution structure of an MG-H1–V domain complex revealed that the hydroimidazolone moiety forms multiple contacts with a positively charged surface on the V domain. The high affinity and specificity of hydroimidozolones binding to the V domain of RAGE suggest that they are the primary AGE structures that give rise to AGEs–RAGE pathologies.
Nonenzymatic protein glycation results in the formation of advanced glycation end products (AGEs), which comprise a structurally diverse class of post-translational protein modifications.1,2 AGEs have been linked to complications of diabetes, chronic inflammation, Alzheimer’s disease, and cancer.3−6 AGEs mediate their effects primarily through a receptor-dependent pathway by binding to a specific cell surface receptor, the receptor for AGEs (RAGE).7,8 RAGE is a member of the immunoglobulin (Ig) superfamily of cell surface receptors and consists of three extracellular domains, V, C1, and C2, a transmembrane helix, and a short cytoplasmic tail.9 RAGE is located in the major histocompatibility complex class III (MHC III) region, suggesting its involvement in immune responses.10,11
Methylglyoxal (2-oxoaldehyde) is a reactive α-oxaldehyde metabolite and precursor of AGEs.12 Glycation by methylglyoxal affects mainly arginine and results in a loss of positive charge via the formation of hydroimidazolones. Methylglyoxal-derived hydroimidazolones (MG-H) form three structural isomers that are physiological ligands of RAGE:13−17 MG-H1 [Nδ-(5-hydro-5-methyl-4-imidazolon-2-yl)ornithine], MG-H2 [5-(2-amino-5-hydro-5-methyl-4-imidazolon-1-yl)norvaline], and MG-H3 [5-(2-amino-4-hydro-4-methyl-5-imidazolon-1-yl)norvaline]. The concentration of methylglyoxal in human tissues and blood plasma can reach relatively high levels, approximately 1–5 μM.18 Among reported arginine-derived AGEs, MG-Hs are believed to be the most prevalent in humans,19 and at least one MG-H adduct is present on 3–13% of the proteins found in the human body;20 furthermore, MG-H modifications comprise 1.3% by weight of the total protein in heat-treated foods.21
AGEs bind only to the V domain of RAGE.7,22 This binding does not accelerate clearance or degradation but rather begins a sustained period of cellular activation mediated by receptor-dependent signaling that leads to inflammation. It is proposed that RAGE activation is largely responsible for the pathogenicity associated with AGEs.23,24 Despite the fact that AGE–RAGE biology has been studied for more than 20 years, very little is known about the structural biology of AGE–RAGE complexes. This is mostly due to the extensive heterogeneity of AGEs created by glycation reactions: Glycation reactions are not largely dependent on sequence specificity, and lysine and arginine residues, which are particularly susceptible to glycation, are very common in proteins.
Recently, we and others have shown that major free AGE adducts of lysine, Nε-carboxymethyl-lysine (CML) and Nε-carboxyethyl-lysine (CEL), do not bind to RAGE7,25 and the binding of CML (CEL)-containing peptides to the V domain of RAGE is weak, in the micromolar range.25 The solution structure of the V domain–CEL peptide complex revealed a positively charged pocket on the surface of the V domain that recognizes not only the CEL side chain but also the peptide backbone, thus explaining the puzzling lack of sequence specificity for CML (CEL) ligands of RAGE.25
Here, we determined a solution structure of an MG-H1–V domain complex and structurally characterized an MG-H1-containing peptide–V domain complex. MG-H1 binds to a molecular surface on the V domain that is similar to that recognized by CML (CEL) peptides. In addition to specific electrostatic contacts, the MG-H1 imidazolone ring tightly fits into the V domain binding site. The structure suggests that MG-Hs, as the most prevalent AGEs in humans, may contribute to ligand-induced RAGE signaling.
Experimental Procedures
Reagents and Chemicals
Restriction enzymes and Taq polymerase were from NEB. MG-H isomers, MG-H1, MG-H2, MG-H3, and G-H1 were from PolyPeptide. Dulbecco’s modified Eagle’s medium (DMEM), penicillin/streptomycin, and fetal bovine serum (FBS) were purchased from Gibco-Life Technologies. The 4 to 12% bis-tris gels were from Novex-Life Technologies. Lipofectamine was from Invitrogen-Life Technologies, RAGE si-RNA from Ambion, and scramble si-RNA from Thermo Scientific. Phosphorylated and nonphosphorylated Janus N-terminal kinase (p-JNK and JNK, respectively), glyceraldehyde-3-phosphate dehydrogenase (GAPDH), and anti-mouse IgG (HRP) antibodies were purchased from Cell Signaling. The RAGE antibody was from Millipore. The anti-RAGE (A-9) antibody was from Santa Cruz Biotechnology, and HRP-conjugated anti-rabbit IgG was from Promega. All other chemicals used were reagent grade or better.
Solid Phase MG-H1 Peptide Synthesis
Peptides were synthesized on a SYRO2000 multiple synthesizer (MultiSynTech GmbH, Witten, Germany) employing 9-fluorenylmethoxycarbonyl/tert-butyl (Fmoc/tBu) chemistry on Wang resin.25,26 MG-H1 was introduced as an Nα-allyloxycarbonyl (Alloc)-protected ornithine derivative [Fmoc-Orn(Alloc)-OH]. After the peptide synthesis had been completed, the Alloc group was cleaved with tetrakis(triphenylphosphine)palladium(0) in the presence of N-methylmorpholine27 and MG-H1 was synthesized as described previously.28 Peptides were cleaved with TFA containing a 12.5% (v/v) scavenger mixture (1:2:2:2 ethanedithiol/m-cresol/thioanisole/water mixture) at room temperature for 2 h. The peptides were precipitated with cold diethyl ether and purified by reverse phase high-performance liquid chromatography (RP-HPLC) using a Jupiter C18 column (21.2 mm internal diameter, 250 mm length, 15 μm particle size, 30 nm pore size) (Phenomenex Inc., Torrance, CA). The purities of the peptides were confirmed by RP-HPLC and the molecular weights by matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF MS; 4700 proteomic analyzer from Applied Biosystems GmbH, Darmstadt, Germany) using an α-cyano-4-hydroxycinnamic acid matrix.
Plasmid Construction
Human RAGE cDNA library clone BC020669 was obtained from Open Biosystems and used as a template for polymerase chain reaction (PCR) amplifications. DNA encoding the V domain (amino acids 24–125) was amplified via PCR using Taq polymerase and oligonucleotides 5′-TTTCATATGGCTCAAAACATCACAGCCCGGATTGG-3′ and 5′-GTCTAAGGACCCTTCGGTCTTACTCAGCTGTTT-3′ containing flanking 5′-NdeI and 3′-SalI restriction sites. The restriction-digested PCR products were ligated into expression vector pET28a (Novagen), which confers kanamycin resistance. The resulting plasmid, pET28-V, expresses a C-terminal His-tagged V domain of RAGE.
Site-Directed Mutagenesis of the V Domain
To singly mutate the V domain of RAGE, the QuikChange II XL Site-Directed Mutagenesis Kit (Strategene) was used. Following mutagenic PCR, pET28-V was restriction digested with DpnI for 1 h and transformed into Escherichia coli strain DH10B. Mutated plasmids were isolated and purified using a Mini-Prep Kit (Qiagen). DNA sequencing identified plasmids pET28-R98A-V and pET28-Q100A-V, which encode the appropriate mutant V domain.
Labeling, Expression, and Purification of Wild-Type and Mutant V Domains
To uniformly label the V domain, pET28-V, pET28-R98A-V, or pET28-Q100A-V was transformed into E. coli strain BL21(DE3) Codon+ (Novagen). For uniform15N labeling or uniform 13C and 15N labeling, cells were grown at 37 °C in minimal medium (M9) containing 35 mg/L kanamycin, 4 g/L unlabeled dextrose or [U-13C]dextrose as the sole carbon source, and 1 g/L [U-15N]ammonium chloride as the sole nitrogen source. Cells were grown to an A600 of 0.7 at 37 °C, induced with 0.5 mM isopropyl 1-thio-β-d-galactopyranoside (IPTG), and grown overnight. Cells were harvested and resuspended in 20 mM Hepes-Na buffer (pH 7.0) containing 8 M urea and heat lysed at 100 °C for 10 min. The lysate was centrifuged, and the supernatant was loaded onto a nickel-nitrilo-triacetic acid-agarose (Ni-NTA) column (Qiagen). The column was washed with 20 mM Hepes-Na buffer (pH 7.0), and the protein was allowed to renature on the column before being eluted with 20 mM Hepes-Na (pH 7.0) containing 500 mM imidazole. Fractions containing the eluted protein were pooled and dialyzed into nuclear magnetic resonance (NMR) buffer [10 mM sodium phosphate (pH 6.5), 100 mM NaCl, and 0.02% (w/v) NaN3]. The C-terminal His tag of the V domain was cleaved by thrombin (Novagen) at room temperature for 1 h before gel filtration chromatography on an SE-75 column (Amersham Biosciences). The fractions containing the eluted protein were concentrated by using Ultra-Centricones (Millipore). The purity was estimated to be >95% by Coomassie-stained sodium dodecyl sulfate–polyacrylamide gel electrophoresis.
NMR Experiments
MG-H1-containing peptide residues were assigned by using two-dimensional 1H{1H} TOCSY and 1H{1H} ROESY experiments,29 which provide through-bond and through-space proton connectivities. Protein samples of [U-15N]V domain, with concentrations ranging from 60 to 300 μM, were dissolved in NMR buffer, 10 μM potassium phosphate (pH 6.5), 100 mM NaCl, and 0.02% (w/v) NaN3, in a 90% H2O/10% D2O mixture, and unlabeled MG-H1 or an MG-H1-containing peptide, MG-H1-PEP, was added to a 2-fold molar excess. To obtain backbone resonance assignments of the [U-15N,13C]V domain–MG-H1 complex, standard triple-resonance HN(CA)CO, HNCO, HN(CO)CA, HNCA, CBCA(CO)NH, and HNCACB29 spectra were recorded at 298 K using an Avance Bruker spectrometer operating at a 1H frequency of 700 MHz equipped with a single Z-axis gradient cryoprobe. To obtain the side chain resonance assignments of the V domain bound to MG-H1, 1H{15N} HSQC and CC(CO)NH, HCC(CO)NH, and three-dimensional (3D) 1H{15N} NOESY-HSQC experiments29 were performed. To assign intra- and intermolecular nuclear Overhauser effects (nOes), 3D 1H{15N} and 1H{13C}20 NOESY-HSQC experiments29 and 3D 15N-edited 13C-filtered NOESY-HSQC29 experiments were performed on samples containing 500 μM free [U-13C,15N]V domain and 500 μM [U-13C,15N]V domain with 1 mM unlabeled MG-H1. All spectra were processed using TOPSPIN version 2.1 (Bruker), and assignments were made using CARA.30
Structure Calculation
Structural calculations were conducted with Cyana 2.131 using 986 intramolecular and five intermolecular distance restraints derived from 13C-edited NOESY and 15N-edited and 15N-edited 13C-filtered NOESY spectra, 150 pairs of backbone torsion angle restraints derived from TALOS,32 38 restraints for hydrogen bonds, and the restraints from one disulfide bond between Cys38 and Cys99. nOes were converted to upper limit distances using the CALIBA module in CYANA.31 The reference volume determined by CALIBA was increased 2 times before conversion to loosen the distance restraints. All upper limit distances for intermolecular nOes were set to 6 Å. The geometry of the unnatural amino acid (R)-epimer of MG-H1 was added to the CYANA library. These experimental restraints are summarized in Table S1 of the Supporting Information. To perform CYANA calculations, a single polypeptide chain was constructed for the V domain and MG-H1 molecules.
For refinement, the CYANA-generated distance and angle restraints were converted into CNS format in CCPN.33 The structure for the nonstandard amino acid MG-H1 was generated and energy-minimized in PRODRG2.34 A total of 1000 structures were calculated, and the 200 lowest-energy structures were subjected to water refinement and further analysis by PROCHECK_NMR;35 80.5% of the V domain residues were in the most favorable regions of the Ramachandran plot, 18.2% in additional allowed regions, and 1.3% in generously allowed regions. There were no residues in the disallowed regions. The structural statistics of the 20 best structures are listed in Table S1 of the Supporting Information.
Fluorescence Titration
Measurements were performed on a Fluorolog-3 fluorescence spectrophotometer (HORIBA Jobin Yvon) at 25 °C in a 1 mL stirred cuvette. For fluorescence titration experiments, the V domain was dissolved in 10 mM phosphate buffer (pH 6.5) and 100 mM NaCl at a concentration of 100 nM, and the concentration of MG-Hs or MG-H1-containing peptides was increased from 5 nM to 10 μM in 11 steps using 1 mM stock solutions. Titrations in the absence of the V domain and in the absence of MG-H ligands were performed as references. The tryptophan fluorescence was measured using an excitation wavelength of 295 nm. The fluorescence emission signal was subtracted from the signal of the reference titrations, and the differences adjusted by the dilution factor were plotted against the final concentration of added MG-H molecules or MG-H1-containing peptides. Curve fitting (OriginLab) was performed to find the best values for Kd using a single-site binding isotherm approximation.36
Enzyme-Linked Immunosorbent Assay (ELISA)
The binding affinity of the V domain for methylglyoxal-modified bovine serum albumin (MGO-BSA) was determined using an ELISA. MGO-BSA was prepared according to the method of Ahmed et al.37 Ninety-six-well plates (Falcon ProBind, Becton Dickinson) were first coated with MGO-BSA at a level of 500 ng/well in 0.1 M sodium carbonate buffer (pH 9.6) and incubated overnight at 4 °C. The wells were blocked with 1% BSA (Fraction V, Calbiochem) in wash buffer, 10 mM potassium phosphate (pH 6.5), and 100 mM NaCl and incubated at room temperature for 2 h. The purified V domain, at concentrations ranging up to 10 μM in wash buffer, was added to the wells and incubated at room temperature for 2 h. Anti-RAGE (A-9), a mouse monoclonal antibody that recognizes residues 23–43 at the N-terminus of RAGE, was diluted 1:100 in wash buffer containing 1% BSA, added to each well, and incubated overnight at 4 °C. The secondary antibody, anti-mouse IgG (HRP), was diluted 1:1000 in wash buffer containing 1% BSA, added to each well, and incubated at room temperature for 2 h. After the addition of a chromogenic 3,3′,5,5′-tetramethylbenzidine, TMB, substrate solution (Thermo Scientific), plates were read on a scanner. All washes were performed three to five times. Data were analyzed by using GraphPad Prism version 5.
MGO-BSA, the purified V domain, and MG-Hs (PolyPeptide) were used in ELISA-based competition experiments. Ninety-six-well plates were first coated with MGO-BSA at a level of 500 ng/well in 0.1 M sodium carbonate buffer (pH 9.6) and incubated overnight at 4 °C. The wells were blocked with wash buffer containing 1% BSA and incubated at room temperature for 2 h. MG-Hs, at concentrations of up to 1 mM, mixed with 1 μM V domain dissolved in wash buffer, were added to the wells and incubated at room temperature for 2 h. Anti-RAGE (A-9) was diluted 1:100 in wash buffer containing 1% BSA, added to each well, and incubated overnight at 4 °C. A secondary antibody, anti-mouse IgG (HRP) (Cell Signaling), was diluted 1:1000 in wash buffer containing 1% BSA, added to each well, and incubated for 2 h at room temperature. After the addition of a chromogenic TMB substrate solution, plates were read on a scanner. All washes were performed three to five times. Data were analyzed by using GraphPad Prism version 5.
Cell Lines and Materials
Human alveolar epithelial cells, A549, were maintained in DMEM supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin in a 5% CO2 incubator at 37 °C. For experiments, cells were plated at a density of 1 × 105 cells/mL in 60 mm dishes. Cells were serum-starved overnight after they had reached 70–80% confluence, treated the next day with 12 μg/mL BSA and MGO-BSA for 7 min, and used in Western blot analyses.
Western Blot Analyses
Cells were lysed, and total cellular lysates were immunoblotted and probed with p-JNK (Thr183/Tyr185)/JNK, RAGE, and GAPDH antibodies. Briefly, ∼40 μg of proteins from cell lysates were denatured and resolved on 4 to 12% bis-tris gels and transferred onto nitrocellulose membranes. Membranes were blocked by using 5% nonfat dry milk and incubated with primary antibodies, p-JNK/JNK, RAGE, and GAPDH. HRP-conjugated anti-rabbit IgG was used to identify sites of primary antibody binding. Membranes were stripped of bound primary antibodies and reprobed with the GAPDH antibody as a loading control.
Si-RNA Transfection
Si-RNA transfection was performed according to the manufacturer’s protocol. A549 cells were transfected with 150 pM RAGE si-RNA using lipofectamine. After incubation for 6 h, transfection medium was replaced with fresh DMEM containing 10% FBS. Transfected cells were serum-starved, incubated with 12 μg/mL BSA or MGO-BSA for 7 min, and used in Western blot analyses.
Results
To prove that methylglyoxal-modified proteins, which contain hydroimidozolones, are RAGE ligands, we monitored the phosphorylation of JNK, an effector of RAGE signaling, in response to RAGE activation by MGO-BSA in human alveolar epithelial cells, A549 (Figure 1). Incubating A549 cells with 200 nM MGO-BSA results in an increase in the level of phosphorylation of JNK as compared to that of A549 treated with unmodified BSA (Figure 1A). To verify that this effect is specific to RAGE signaling, we decreased the level of RAGE expression in A549 cells by using RAGE si-RNA. Phosphorylation of JNK in response to MGO-BSA was decreased in A549 cells transfected with RAGE si-RNA compared to that of A549 cells transfected with scrambled si-RNA (Figure 1B,C). MGO-BSA binds to the V domain (Figure 1D), and methylglyoxal-derived hydroimidazolones, MG-H1, MG-H2, and MG-H3, can successfully block binding of MGO-BSA to the V domain (Figure 1E). We concluded that methylglyoxal-modified proteins are indeed RAGE ligands.
Figure 1.

Methylglyoxal-modified proteins are RAGE ligands. (A) In the top panel, MGO-BSA-stimulated activation of RAGE increases JNK phosphorylation (p-JNK) relative to that with BSA alone in human alveolar epithelial cells, A549. The bottom panel shows the JNK loading control. (B) In the top panel, decreasing the level of RAGE expression by adding RAGE si-RNA reduces MGO-BSA-stimulated activation of pJNK relative to nonspecific scramble si-RNA in A549 cells. The bottom panel is the JNK loading control. (C) RAGE si-RNA specifically reduces RAGE overexpression in A549 cells relative to scramble si-RNA. The bottom panel is the GAPDH loading control. (D) MGO-BSA binds to the V domain. MG-H1, MG-H2, and MG-H3 (left to right, respectively) compete with MGO-BSA for the V domain in ELISAs. Kd, R2, and IC50 are the dissociation constant, Pearson’s correlation coefficient, and the half-maximal inhibitory concentration, respectively. (E) Before MG-H1.
The MG-Hs used in this work are a series of methylglyoxal-modified arginines, composed of a racemic mixture of (R)- and (S)-epimers of three isomers, MG-H1, MG-H2, and MG-H3, each of which contains an imidazolone ring with a methyl group at a different site (Figure 2).12 To investigate the interaction between MG-Hs and the V domain of RAGE, we synthesized short peptides containing MG-H1 (Figure S1 of the Supporting Information). The amino acid sequences of the peptides represent the primary glycation sites found in human serum albumin, a major plasma protein.
Figure 2.

Synthesis and structure of arginine-derived AGEs MG-H1 [Nδ-(5-hydro-5-methyl-4-imidazolon-2-yl)ornithine], MG-H2 [5-(2-amino-5-hydro-5-methyl-4-imidazolon-1-yl)norvaline], and MG-H3 [5-(2-amino-4-hydro-4-methyl-5-imidazolon-1-yl)norvaline]. Only (R)-epimers of hydroimidazolones are shown.
Free MG-Hs bind to the V domain of RAGE, unlike ε-amino group-modified lysines, CML and CEL.25 The binding affinities were estimated by monitoring changes in the native tryptophan fluorescence of the V domain upon titration with MG-Hs (Table 1 and Figure S2 of the Supporting Information). The three MG-H isomers bind to the V domain with similar affinities, indicating that RAGE does not discriminate between MG-Hs or specifically recognize MG-Hs based on the location of the methyl group. This was confirmed by examining the binding of G-H1 [Nδ-(5- hydro-4-imidazolon-2-yl)ornithine], which lacks a methyl group on the imidazolone ring; the affinity of G-H1 for the V domain is comparable to that of the MG-Hs (Table 1). MG-H1 peptides also bind to the V domain with an affinity similar to that of isolated MG-Hs, implying that the MG-H moiety is the key recognition element and defines the major molecular binding surface on the V domain.
Table 1. Binding Affinities of Hydroimidazolones and Hydroimidazolone-Containing Peptides for Wild-Type and Mutant V Domains.
| molecule | V domain Kd (nM)a | Q100A V domain Kd (nM)a | R98A V domain Kd (nM)a |
|---|---|---|---|
| MG-H1 | 40 ± 5 | >10000 | >10000 |
| MG-H2 | 44 ± 5 | – | – |
| MG-H3 | 31 ± 5 | – | – |
| G-H1 | 43 ± 7 | – | – |
| LSQ(MG-H1)FPKAE | 35 ± 4 | – | – |
| ALT(MG-H1)EYTG | 44 ± 6 | – | – |
The dissociation constant was obtained by fitting fluoresceence titration data with a single-site binding isotherm.
Chemical shift perturbation and changes in the intensities of NMR resonances upon ligand binding can be used to identify amino acid residues of the V domain that are affected by the binding of MG-H125 (Figure 3 and Figure S3 of the Supporting Information). Titrating the 15N-labeled V domain with free MG-H1 results in changes in the chemical shifts of residues I26, V35, E50, L53, N81, V89, Q100, and G106, and the most significant changes in intensity for residues S22, V35, K52, S74, R98, and V117 (Figure S3B of the Supporting Information). We also titrated the 15N-labeled V domain with a peptide in which MG-H1 is inserted between amino acids glutamine and phenylalanine, LSQ(MG-H1)FPKAE. Because of the extra amino acids, the peptide structure contributes an interaction surface area larger than that of free MG-H1. The changes in the chemical shifts of residues G31, C38, K52, T55, F85, D93, F97, R98, C99, M102, and K110 and changes in intensity for residues A41, G56, L64, K110, and Y113 delineate the interaction surface between the peptide and the V domain (Figure S3C of the Supporting Information). Two key residues, K52 and R98, as well as the surrounding area of the V domain, composed of residues E50, L53, C99, Q100, M102, and K110, are affected by both free MG-H1 and the MG-H1-containing peptide (Figure 3A,B).
Figure 3.

RAGE V domain does not discriminate between MG-H1 and MG-H1 embedded within the peptide sequence LSQ(MG-H1)FPKAE. (A) Overlay of 15N HSQC spectra of the free (black) and MG-H1 bound (red) V domain. (B) Overlay of 15N HSQC spectra of the free (black) and LSQ(MG-H1)FPKAE peptide-bound (red) V domain.
To elucidate the chemical nature of the interaction between the V domain and MG-Hs, we determined the solution structure of the V domain (residues 23–123 of the 356-residue RAGE) in complex with MG-H1. Only the (R)-epimer of MG-H1 can be fit into the MG-H1–V domain binding site, implying stereospecific recognition of MG-H1. The structured regions comprised residues 23–92 of the V domain and the MG-H1 ligand. On the basis of the constraints obtained from NMR experiments, 20 structures with the lowest target function values were superimposed (Figure 4 and Table S1 of the Supporting Information). The solution structure of the V domain–MG-H1 complex is similar to that of the free V domain38 and of the V domain within VC1.39 The root-mean-square (rms) deviations between the solution structure of the MG-H1-bound V domain and the free V domain (PDB entry 2E5E) or the V domain within VC1 (PDB entry 3CJJ) are 2.0 and 2.1 Å for the backbone and 2.0 and 2.9 Å for all heavy atoms (carbon atoms) in the ordered regions.6 At the binding site, the rms deviation between the solution structure of the MG-H1-bound V domain and the free V domain is 0.98 Å for the backbone and 2.39 Å for all heavy atoms (carbon atoms).
Figure 4.

Stereo overlay of the 20 lowest-energy MG-H1–V domain complex backbone traces. The Ramachandran plot shows 84.4% of the residues in the most favored regions, 14% of the residues in additional allowed regions, and only 1.6% of the residues in generously allowed regions. The rms deviations of the solution structure of the V domain (PDB entry 3CJJ) and MG-H1 are 0.84 and 0.72 Å for the backbone and 1.7 and 0.7 Å for all heavy atoms. The N- and C-termini of the V domain and MG-H1 are indicated. The figure was prepared by using SwissPDB.
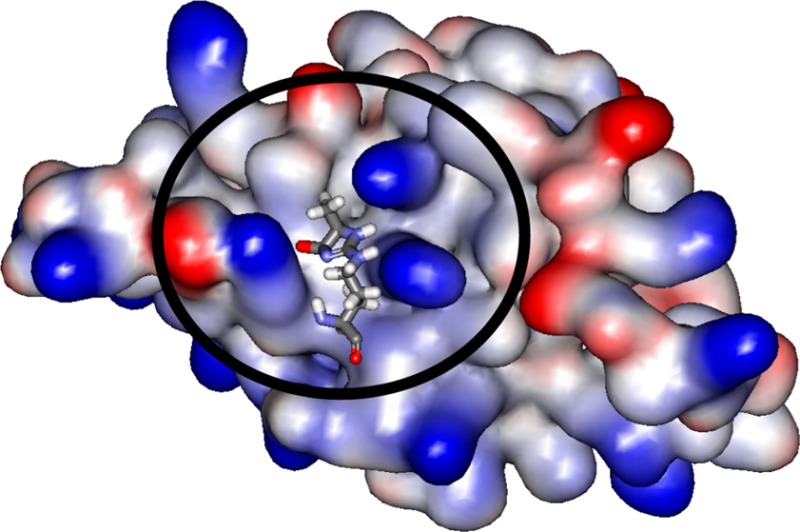
The solution structure of the MG-H1–V domain complex is shown in Figure 5. The MG-H1 binding site is small, spanning 80 Å2, and fits well into a groove formed by strands C, F, and G; side chain interactions provide additional stability for MG-H1 binding. Importantly, the methyl group of MG-H1 imidazolone is oriented away from the binding site, suggesting that this group does not contribute to the binding free energy. As a result, any (R)-epimer of MG-H can bind to the RAGE V domain with high affinity at the same binding site despite the different locations of the methyl group on the imidazolone ring.
Figure 5.

Solution structure of the MG-H1–V domain complex. (A) Structure of MG-H1 (balls and sticks) bound to the V domain (ribbon representation). Secondary structural elements are labeled following the immunoglobulin convention.47,48 (B) Residues surrounding the interaction surface between MG-H1 and the V domain. (C and D) Electrostatic potential surface of the V domain (C) and a close-up showing dipole–dipole forces (D). MG-H1 fits well into a groove composed of K52, R98, Q100, and K110 on the V domain. Positively and negatively charged surfaces are colored blue and red, respectively. The red arrow represents a dipole of the MG-H1 molecule, and yellow arrows represent environmental dipoles on the V domain surface. Both shape complementarity and dipole–dipole forces contribute to the stabilization of the MG-H1–V domain complex.40,49
We detected an extended network of nOes confidently placing the small MG-H1 into the groove by this cluster (Table S2 of the Supporting Information). The electrostatic potential surface of the V domain is shown in Figure 5C. Three positively charged amino acids, K52, R98, and K110, are localized on the surface of the V domain, with two nearby negatively charged residues, E50 and E108, suggesting strong charge–dipole and dipole–dipole interactions (Figure 5D). The amine hydrogen and the carbonyl group of MG-H also create a molecular dipole that aligns well with the surface charges in the MG-H binding site on the V domain40 (Figure 5D).
To further examine the role of residues that are involved in MG-H1 binding, we made single R98A and Q100A mutants of the V domain to perturb the protein–ligand electrostatic interaction. Indeed, the affinities of MG-H1 for the mutant V domains are greater than 10 μM, much weaker than for the wild-type V domain (Table 1 and Figure S2 of the Supporting Information). The results are consistent with R98 and Q100 being critical for MG-H1 binding.
AGEs recognize the V domain through a specific binding pattern, as do a large number of RAGE activation ligands.22 Despite the difference in conformation between MG-H1 and CML (CEL)-PEP, the two ligands share a similar binding surface. Because it is the MG-H1 moiety specifically that binds to the V domain, MG-H1-containing peptides or proteins, which have different secondary and tertiary structures, exhibit similar binding affinities and binding surfaces. As a result, MG-H1 is an essential structure for AGE-induced RAGE signal transduction.
Discussion
Many functionally and structurally different AGEs are observed under various physiological and pathological conditions, but the interactions with RAGE of only a few have been quantitatively studied. Methylglyoxal-derived hydroimidazolones, such as CML, CEL, and MGHs, are the predominant AGE modifications found in vivo.19 A previous study of the CEL-PEP–V domain complex revealed that CML (CEL)-modified proteins bind to RAGE even when the ligand modification site possesses very different primary, secondary, and tertiary structures.25 In these cases, the V domain makes molecular contacts with both the CEL (CML) moiety and the peptide backbone in the immediate vicinity of CEL (CML). Here we establish a model of a RAGE ligand–V domain complex in which the V domain specifically recognizes the MG-H1 moiety instead of the residues surrounding the modified site.
The positively charged cavity on the V domain, consisting of residues K52, R98, and K110, together with E50 and E108, presents a favorable polar environment for MG-H1 to bind (Figure 5D). The binding energy results from the tight fit between the imidazolone ring and the V domain binding site and also from charge–dipole and dipole–dipole interactions between the ligand and the protein.40,41 Because the peptide backbone of MG-H1-containing peptides is not involved in binding to the V domain, the binding does not depend on the primary, secondary, or tertiary structure of the MG-H1-modified protein. This explains why various MG-H-modified proteins found in tissue and plasma can bind to RAGE and is consistent with RAGE acting as a pattern recognition receptor.22,42 The geometry of MG-H1 is critical for molecular recognition, as only the (R)-epimer of MG-H1 can fit into the V domain binding site. The short bent conformation allows the MG-H1 structure to fit well inside a small area, and the methyl group does not interfere with the interaction between the MG-H isomer and the V domain.
An important question is whether the precursor of MG-H1, arginine, can fit into the MG-H1 binding groove and therefore possibly compete with MG-H1-containing ligands for the V domain, thus activating RAGE signaling. MG-H1 binds to the V domain in a compact and bent conformation that allows for a fit better than that afforded by the arginine side chain and facilitates van der Waals interactions that stabilize the MG-H1–V domain complex. Importantly, the affinity of arginine for the MG-H1 binding site on the V domain is expected to be very low because of its positive charge. Indeed, we did not detect any binding of arginine to the V domain.
MG-Hs contribute to various disease states such as diabetes,13,17 cancer,43 cardiovascular disease,14 and renal failure6 by interacting with RAGE. In contrast to CML (CEL)-modified proteins,22 which are present at submicromolar concentrations in human plasma and bind with micromolar affinity, MG-Hs and MG-H-modified proteins are present at micromolar concentrations and bind to the V domain with nanomolar affinity. Because RAGE forms dimers and higher-order oligomers,44,45 polyvalent engagement of MG-H proteins by RAGE results in tight binding and triggers RAGE-dependent signaling.22,25 Overall, these observations suggest an important role for MG-H modifications in the physiology and pathology of disease states.46
Supporting Information Available
Synthetic MG-H1 peptide, LSQ(MG-H1)FPKAE, that contains the correct chemical structure of methylglyoxal-derived hydroimidazolone isomer (Figure S1), binding of MG-H-containing peptides to the V domain (Figure S2), interaction surface between MG-H1 and MG-H1-containing peptides and the V domain (Figure S3), intermolecular nOEs establishing the MG-H1–V domain binding mode (Figure S4), NMR and refinement statistics for the MG-H1–V domain complex (Table S1), and intermolecular NOE distance constraints used to model the MG-H1–V domain complex (Table S2). This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
This work was supported, in whole or in part, by National Institutes of Health Grant R01 GM085006 (A.S.), the Juvenile Diabetes Research Foundation (A.S.), and the Department of Biotechnology, Government of India (V.R.).
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Brownlee M. (1995) Advanced protein glycosylation in diabetes and aging. Annu. Rev. Med. 46, 223–234. [DOI] [PubMed] [Google Scholar]
- Brownlee M. (2001) Biochemistry and molecular cell biology of diabetic complications. Nature 414, 813–820. [DOI] [PubMed] [Google Scholar]
- Valente T.; Gella A.; Fernandez-Busquets X.; Unzeta M.; Durany N. (2010) Immunohistochemical analysis of human brain suggests pathological synergism of Alzheimer’s disease and diabetes mellitus. Neurobiol. Dis. 37, 67–76. [DOI] [PubMed] [Google Scholar]
- Thornalley P. J. (1999) Clinical significance of glycation. Clin. Lab. 45, 263–273. [Google Scholar]
- Ishiguro H.; Nakaigawa N.; Miyoshi Y.; Fujinami K.; Kubota Y.; Uemura H. (2005) Receptor for advanced glycation end products (RAGE) and its ligand, amphoterin are overexpressed and associated with prostate cancer development. Prostate 64, 92–100. [DOI] [PubMed] [Google Scholar]
- Ramasamy R.; Yan S. F.; Schmidt A. M. (2012) Advanced glycation endproducts: From precursors to RAGE: Round and round we go. Amino Acids 42, 1151–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kislinger T.; Fu C.; Huber B.; Qu W.; Taguchi A.; Du Yan S.; Hofmann M.; Yan S. F.; Pischetsrieder M.; Stern D.; Schmidt A. M. (1999) Nε-(Carboxymethyl)lysine adducts of proteins are ligands for receptor for advanced glycation end products that activate cell signaling pathways and modulate gene expression. J. Biol. Chem. 274, 31740–31749. [DOI] [PubMed] [Google Scholar]
- Basta G.; Lazzerini G.; Massaro M.; Simoncini T.; Tanganelli P.; Fu C.; Kislinger T.; Stern D. M.; Schmidt A. M.; De Caterina R. (2002) Advanced glycation end products activate endothelium through signal-transduction receptor RAGE: A mechanism for amplification of inflammatory responses. Circulation 105, 816–822. [DOI] [PubMed] [Google Scholar]
- Neeper M.; Schmidt A. M.; Brett J.; Yan S. D.; Wang F.; Pan Y. C.; Elliston K.; Stern D.; Shaw A. (1992) Cloning and expression of a cell surface receptor for advanced glycosylation end products of proteins. J. Biol. Chem. 267, 14998–15004. [PubMed] [Google Scholar]
- Schmidt A. M.; Stern D. M. (2001) Receptor for age (RAGE) is a gene within the major histocompatibility class III region: Implications for host response mechanisms in homeostasis and chronic disease. Front. Biosci. 6, D1151–D1160. [DOI] [PubMed] [Google Scholar]
- Sugaya K.; Fukagawa T.; Matsumoto K.; Mita K.; Takahashi E.; Ando A.; Inoko H.; Ikemura T. (1994) Three genes in the human MHC class III region near the junction with the class II: Gene for receptor of advanced glycosylation end products, PBX2 homeobox gene and a notch homolog, human counterpart of mouse mammary tumor gene int-3. Genomics 23, 408–419. [DOI] [PubMed] [Google Scholar]
- Thornalley P. J. (2005) Dicarbonyl intermediates in the maillard reaction. Ann. N.Y. Acad. Sci. 1043, 111–117. [DOI] [PubMed] [Google Scholar]
- Berlanga J.; Cibrian D.; Guillen I.; Freyre F.; Alba J. S.; Lopez-Saura P.; Merino N.; Aldama A.; Quintela A. M.; Triana M. E.; Montequin J. F.; Ajamieh H.; Urquiza D.; Ahmed N.; Thornalley P. J. (2005) Methylglyoxal administration induces diabetes-like microvascular changes and perturbs the healing process of cutaneous wounds. Clin. Sci. 109, 83–95. [DOI] [PubMed] [Google Scholar]
- Bourajjaj M.; Stehouwer C. D.; van Hinsbergh V. W.; Schalkwijk C. G. (2003) Role of methylglyoxal adducts in the development of vascular complications in diabetes mellitus. Biochem. Soc. Trans. 31, 1400–1402. [DOI] [PubMed] [Google Scholar]
- Ma H.; Li S. Y.; Xu P.; Babcock S. A.; Dolence E. K.; Brownlee M.; Li J.; Ren J. (2009) Advanced glycation endproduct (AGE) accumulation and AGE receptor (RAGE) up-regulation contribute to the onset of diabetic cardiomyopathy. J. Cell. Mol. Med. 13, 1751–1764. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Li W.; Xu H.; Hu Y.; He P.; Ni Z.; Xu H.; Zhang Z.; Dai H. (2013) Edaravone Protected Human Brain Microvascular Endothelial Cells from Methylglyoxal-Induced Injury by Inhibiting AGEs/RAGE/Oxidative Stress. PLoS One 8, e76025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao D.; Brownlee M. (2010) Hyperglycemia-induced reactive oxygen species increase expression of the receptor for advanced glycation end products (RAGE) and RAGE ligands. Diabetes 59, 249–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornalley P. J. (2008) Protein and nucleotide damage by glyoxal and methylglyoxal in physiological systems: Role in ageing and disease. Drug Metab. Drug Interact. 23, 125–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornalley P. J.; Battah S.; Ahmed N.; Karachalias N.; Agalou S.; Babaei-Jadidi R.; Dawnay A. (2003) Quantitative screening of advanced glycation endproducts in cellular and extracellular proteins by tandem mass spectrometry. Biochem. J. 375, 581–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed N.; Dobler D.; Dean M.; Thornalley P. J. (2005) Peptide mapping identifies hotspot site of modification in human serum albumin by methylglyoxal involved in ligand binding and esterase activity. J. Biol. Chem. 280, 5724–5732. [DOI] [PubMed] [Google Scholar]
- Walter A. W.; Henle T.; Haessner R.; Klostermeyer H. (1994) Studies on the formation of lysinomethylalanine and histidinomethylalanine in milk products. Z. Lebensm.-Unters. Forsch. 199, 243–247. [DOI] [PubMed] [Google Scholar]
- Xie J.; Reverdatto S.; Frolov A.; Hoffmann R.; Burz D. S.; Shekhtman A. (2008) Structural basis for pattern recognition by the receptor for advanced glycation end products (RAGE). J. Biol. Chem. 283, 27255–27269. [DOI] [PubMed] [Google Scholar]
- Hofmann M. A.; Drury S.; Fu C.; Qu W.; Taguchi A.; Lu Y.; Avila C.; Kambham N.; Bierhaus A.; Nawroth P.; Neurath M. F.; Slattery T.; Beach D.; McClary J.; Nagashima M.; Morser J.; Stern D.; Schmidt A. M. (1999) RAGE mediates a novel proinflammatory axis: A central cell surface receptor for S100/calgranulin polypeptides. Cell 97, 889–901. [DOI] [PubMed] [Google Scholar]
- Schmidt A. M.; Hori O.; Chen J. X.; Li J. F.; Crandall J.; Zhang J.; Cao R.; Yan S. D.; Brett J.; Stern D. (1995) Advanced glycation endproducts interacting with their endothelial receptor induce expression of vascular cell adhesion molecule-1 (VCAM-1) in cultured human endothelial cells and in mice. A potential mechanism for the accelerated vasculopathy of diabetes. J. Clin. Invest. 96, 1395–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue J.; Rai V.; Singer D.; Chabierski S.; Xie J.; Reverdatto S.; Burz D. S.; Schmidt A. M.; Hoffmann R.; Shekhtman A. (2011) Advanced glycation end product recognition by the receptor for AGEs. Structure 19, 722–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knappe D.; Zahn M.; Sauer U.; Schiffer G.; Strater N.; Hoffmann R. (2011) Rational design of oncocin derivatives with superior protease stabilities and antibacterial activities based on the high-resolution structure of the oncocin-DnaK complex. ChemBioChem 12, 874–876. [DOI] [PubMed] [Google Scholar]
- Dangles O.; Guibe F.; Balavoine G.; Lavielle S.; Marquet A. (1987) Selective cleavage of the allyl and (allyloxy)carbonyl groups through palladium-catalyzed hydrostannolysis with tributyltin hydride. Application to the selective protection-deprotection of amino acid derivatives and in peptide synthesis. J. Org. Chem. 52, 4984–4993. [Google Scholar]
- Gruber P.; Hofmann T. (2005) Chemoselective synthesis of peptides containing major advanced glycation end-products of lysine and arginine. J. Pept. Res. 66, 111–124. [DOI] [PubMed] [Google Scholar]
- Cavanagh J., Fairbrother W. J., Palmer A. G., and Skelton N. J. (1996) Protein NMR Spectroscopy: Principles and Practice, Academic Press, San Diego. [Google Scholar]
- Masse J. E.; Keller R. (2005) AutoLink: Automated sequential resonance assignment of biopolymers from NMR data by relative-hypothesis-prioritization-based simulated logic. J. Magn. Reson. 174, 133–151. [DOI] [PubMed] [Google Scholar]
- Guntert P. (2004) Automated NMR structure calculation with CYANA. Methods Mol. Biol. 278, 353–378. [DOI] [PubMed] [Google Scholar]
- Cornilescu G.; Delaglio F.; Bax A. (1999) Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J. Biomol. NMR 13, 289–302. [DOI] [PubMed] [Google Scholar]
- Fogh R.; Ionides J.; Ulrich E.; Boucher W.; Vranken W.; Linge J. P.; Habeck M.; Rieping W.; Bhat T. N.; Westbrook J.; Henrick K.; Gilliland G.; Berman H.; Thornton J.; Nilges M.; Markley J.; Laue E. (2002) The CCPN project: An interim report on a data model for the NMR community. Nat. Struct. Biol. 9, 416–418. [DOI] [PubMed] [Google Scholar]
- Schuttelkopf A. W.; van Aalten D. M. (2004) PRODRG: A tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr. D60, 1355–1363. [DOI] [PubMed] [Google Scholar]
- Laskowski R. A.; Rullmannn J. A.; MacArthur M. W.; Kaptein R.; Thornton J. M. (1996) AQUA and PROCHECK-NMR: Programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR 8, 477–486. [DOI] [PubMed] [Google Scholar]
- Eftink M. R. (1997) Fluorescence methods for studying equilibrium macromolecule-ligand interactions. Methods Enzymol. 278, 221–257. [DOI] [PubMed] [Google Scholar]
- Westwood M. E.; Argirov O. K.; Abordo E. A.; Thornalley P. J. (1997) Methylglyoxal-modified arginine residues: A signal for receptor-mediated endocytosis and degradation of proteins by monocytic THP-1 cells. Biochim. Biophys. Acta 1356, 84–94. [DOI] [PubMed] [Google Scholar]
- Matsumoto S.; Yoshida T.; Murata H.; Harada S.; Fujita N.; Nakamura S.; Yamamoto Y.; Watanabe T.; Yonekura H.; Yamamoto H.; Ohkubo T.; Kobayashi Y. (2008) Solution structure of the variable-type domain of the receptor for advanced glycation end products: New insight into AGE-RAGE interaction. Biochemistry 47, 12299–12311. [DOI] [PubMed] [Google Scholar]
- Koch M.; Chitayat S.; Datillo B. M.; Schiefner A.; Diez J.; Chazin W.; Fritz G. (2010) Structural Basis for Ligand Recognition and Activation of RAGE. Structure 18, 1342–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthew J. B. (1985) Electrostatic effects in proteins. Annu. Rev. Biophys. Biophys. Chem. 14, 387–417. [DOI] [PubMed] [Google Scholar]
- Patrick G. L. (2005) An Introduction to Medicinal Chemistry, 3rd ed., Oxford University Press, Oxford, U.K. [Google Scholar]
- Chavakis T.; Bierhaus A.; Al-Fakhri N.; Schneider D.; Witte S.; Linn T.; Nagashima M.; Morser J.; Arnold B.; Preissner K. T.; Nawroth P. P. (2003) The pattern recognition receptor (RAGE) is a counterreceptor for leukocyte integrins: A novel pathway for inflammatory cell recruitment. J. Exp. Med. 198, 1507–1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stopper H.; Schinzel R.; Sebekova K.; Heidland A. (2003) Genotoxicity of advanced glycation end products in mammalian cells. Cancer Lett. 190, 151–156. [DOI] [PubMed] [Google Scholar]
- Xie J.; Burz D. S.; He W.; Bronstein I. B.; Lednev I.; Shekhtman A. (2007) Hexameric calgranulin C (S100A12) binds to the receptor for advanced glycated end products (RAGE) using symmetric hydrophobic target-binding patches. J. Biol. Chem. 282, 4218–4231. [DOI] [PubMed] [Google Scholar]
- Zong H.; Madden A.; Ward M.; Mooney M. H.; Elliott C. T.; Stitt A. W. (2010) Homodimerization is essential for the receptor for advanced glycation end products (RAGE)-mediated signal transduction. J. Biol. Chem. 285, 23137–23146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt A. M.; Hofmann M.; Taguchi A.; Yan S. D.; Stern D. M. (2000) RAGE: A multiligand receptor contributing to the cellular response in diabetic vasculopathy and inflammation. Semin. Thromb. Hemostasis 26, 485–493. [DOI] [PubMed] [Google Scholar]
- Bork P.; Holm L.; Sander C. (1994) The immunoglobulin fold. Structural classification, sequence patterns and common core. J. Mol. Biol. 242, 309–320. [DOI] [PubMed] [Google Scholar]
- Chothia C.; Jones E. Y. (1997) The molecular structure of cell adhesion molecules. Annu. Rev. Biochem. 66, 823–862. [DOI] [PubMed] [Google Scholar]
- Martin S. F.; Clements J. H. (2013) Correlating structure and energetics in protein-ligand interactions: Paradigms and paradoxes. Annu. Rev. Biochem. 82, 267–293. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.