

Abstract
The ST6Gal-I sialyltransferase adds an α2–6-linked sialic acid to the N-glycans of certain receptors. ST6Gal-I mRNA has been reported to be upregulated in human cancer, but a prior lack of antibodies has limited immunochemical analysis of the ST6Gal-I protein. Here we show upregulated ST6Gal-I protein in several epithelial cancers, including many colon carcinomas. In normal colon, ST6Gal-I localized selectively to the base of crypts, where stem/progenitor cells are found, and the tissue staining patterns were similar to the established stem cell marker ALDH1. Similarly, ST6Gal-I expression was restricted to basal epidermal layers in skin, another stem/progenitor cell compartment. ST6Gal-I was highly expressed in induced pluripotent stem (iPS) cells, with no detectable expression in the fibroblasts from which iPS cells were derived. On the basis of these observations we investigated further an association of ST6Gal-I with cancer stem cells (CSCs). Selection of irinotecan resistance in colon carcinoma cells led to a greater proportion of CSCs compared with parental cells, as measured by the CSC markers CD133 and ALDH1 activity (Aldefluor). These chemoresistant cells exhibited a corresponding upregulation of ST6Gal-I expression. Conversely, shRNA-mediated attenuation of ST6Gal-I in colon carcinoma cells with elevated endogenous expression decreased the number of CD133/ALDH1-positive cells present in the cell population. Collectively, our results suggest that ST6Gal-I promotes tumorigenesis and may serve as a regulator of the stem cell phenotype in both normal and cancer cell populations.
Keywords: glycosylation, ST6Gal-I, colon carcinoma, stem cells, cancer stem cells
Introduction
Differences in the glycan profile of cancer cells as compared with normal cells are well documented. These changes are driven by various enzymes responsible for the addition and removal of sugars, such as glycosyltransferases and glycosidases. There is a selected subset of enzymes altered in cancer, suggesting a functional role for distinct glycans in the tumor phenotype. The ST6Gal-I sialyltransferase is an example of a glycosyltransferase commonly upregulated in cancer. This Golgi enzyme adds the negatively-charged sugar, sialic acid, in an α2–6 linkage to the termini of N-glycans. ST6Gal-I is overexpressed in many types of cancer including colon, breast, and ovarian, and upregulation correlates with increased metastatic potential and poor prognosis (reviewed in (1–3). ST6Gal-I is increased in cancer as a consequence of signaling by the ras oncogene (1–3)
The mechanistic role of ST6Gal-I in tumor progression remains poorly-understood. In vitro studies suggest that ST6Gal-I promotes cell migration and invasion (4, 5), and this enhanced migratory response is due, at least in part, to ST6Gal-I-mediated sialylation of the β1 integrin receptor (6–8). Animal models also implicate ST6Gal-I in tumor invasiveness. Bresalier et al. determined that metastatic murine cell lines were more highly sialylated than less metastatic parental lines, and neuraminidase treatment of the metastatic lines drastically decreased the amount of liver metastases after splenic injection (9). Also, Harvey et al. reported decreased metastasis to liver following splenic injections after blocking the transfer of sialic acid from its carrier, CMP-sialic acid (10).
In conjunction with cell migration, ST6Gal-I may regulate another important aspect of tumorigenicity, the ability to evade cell death. Work from our group revealed that the Fas death receptor is a substrate for ST6Gal-I, and that α2–6 sialylation of Fas reduces apoptotic signaling by hindering internalization of Fas after ligand-induced activation (11). We similarly reported that ST6Gal-I-mediated sialylation of the TNFR1 death receptor blocks TNFα-induced apoptosis (12). Baum’s group showed that sialylation of CD45 by ST6Gal-I prevents CD45 internalization, thereby protecting T-cells from apoptosis (13), and ST6Gal-I sialylation enhances PECAM surface retention, promoting survival of endothelial cells (14). These studies highlight the capacity of ST6Gal-I to modulate the function of specific receptors, particularly through regulation of cell surface retention. However, additional evidence has established ST6Gal-I as a key negative regulator of galectin-dependent apoptosis. Galectins are galactose-binding lectins that have many functions, including induction of cell death. The addition of α2–6 sialylation to galactosides prevents galectin binding and apoptotic activity (15). For example, our studies have shown that galectin-3 binds directly to the β1 integrin and stimulates apoptosis, but only when the β1 integrin lacks α2–6 sialylation (16). Finally, sialylation of EGFR by ST6Gal-I confers resistance to the EGFR-targeted chemotherapy reagent, gefitinib (17). These diverse findings suggest that ST6Gal-I acts as a critical regulator of tumor cell survival by inhibiting a multiplicity of cell death pathways.
While studies of specific receptors and signaling pathways have provided insight into the function of ST6Gal-I within a cellular context, a major gap in our knowledge is that ST6Gal-I expression in normal and tumor tissues has not been well-characterized. Due to a lack of effective anti-ST6Gal-I antibodies, prior investigations relied on measurements of ST6Gal-I mRNA levels, or tissue reactivity toward SNA, a lectin specific for α2–6 linked sialic acid. However, there are limitations associated with both of these approaches. The mRNA pool isolated from tumor tissue homogenates may include mRNA from noncancerous cells such as immune or stromal cells, and SNA reactivity is not completely restricted to ST6Gal-I-mediated α2–6 sialylation, since SNA can also recognize α2–6 sialic acids added to O-glycans by the ST6GalNAc family. To address this issue, immunohistochemical and immunoblot analyses of ST6Gal-I protein were performed in the current study using a newly-validated antibody. These studies revealed a dramatic upregulation of ST6Gal-I in tumor specimens compared with pairmatched uninvolved tissues. Surprisingly, the expression of ST6Gal-I in normal epithelium appeared to localize to the stem and/or progenitor cell compartment, and moreover, high ST6Gal-I levels corresponded with the expression of the cancer stem cell markers, CD133 and ALDH1. While many questions remain regarding ST6Gal-I function in cancer, these data suggest that ST6Gal-I activity may be involved in maintaining some aspect of stem-like cell behavior.
Methods
Cell Culture
HD3 colon carcinoma cells (18) were maintained in Dulbecco’s Modified Eagles medium (DMEM) low glucose (1 g/L) with 7% FBS and 1% antibiotic/antifungal solution containing streptomycin sulfate, penicillin G, and amphotericin B (Invitrogen, Grand Island, NY). The stable ST6Gal-I knockdown cell line was established as described (8). In brief, HD3 cells were transduced with lentivirus (Sigma, St. Louis, MO) expressing either shRNA against ST6Gal-I or an empty vector, and a pooled population of clones stably expressing shRNA was isolated by puromycin selection.
SW948 colon carcinoma cells were purchased from ATCC (Manassas, VA). Cells were maintained in DMEM: Liebovitz’ L-15 media in a 3:1 ratio with 10% denatured FBS and 2mM glutamine. To establish a chemoresistant subline, SW948 cells were treated with an initial dose of CPT-11 (Irinotecan hydrochloride, Pharmacia & Upjohn Co., Kalamazoo, MI) at 4 µg/mL, which is 2-fold the determined IC50 dose. Most cells were killed by day 10. Surviving cells were grown in drug-free media for 3 days, and then CPT-11 (4 µg/mL) was added back to the media for 5 days. After a 3-day recovery period in drug-free media, cells were capable of growth in CPT-11 containing media (4 µg/mL). Dosage was then increased stepwise for a period of 185 total days reaching a maximum of 20 µg/mL. Resistant cells were cultured in DMEM:L15 media containing 20 µg/mL CPT-11 and periodically screened for drug resistance. Cells maintained CPT-11 resistance even after growth in drug-free media out to 122 days.
Sample preparation and ST6Gal-I immunoblots
Colon tumor blot: Commercially available membrane containing three human colon tumor samples, one normal colon and one placental sample was purchased from Biochain Institute (Newark, CA). Tumor and pair-matched uninvolved colon specimens: Human tissues obtained from the Tissue Procurement Facility at UAB were snap-frozen in liquid nitrogen and stored at −80°C. Samples were homogenized using a polytron device in 50 mM Tris-HCl buffer (pH 7.4) with 1% Triton X-100 and protease inhibitors (Roche Applied Bioscience, Indianapolis, IN). Samples were centrifuged and supernatants used for immunoblotting. iPS, individual transcription factor-transduced, and human foreskin fibroblasts (HFF) cell lysates: Frozen lysates from cells including control HFFs, iPS cells derived from HFFs, or HFFs transduced with one of the following transcription factors, c-Myc, Klf4, Oct4, or Sox2, were obtained from Systems Biotechnologies. Colon carcinoma cell lines: HD3 and SW948 cells were lysed in 50mM Tris-HCl buffer containing 1% Triton X-100 and protease inhibitors. Lysates were centrifuged and supernatants collected for immunoblotting.
Samples were separated by SDS-PAGE, and transferred to PVDF membranes. Membranes were blocked in 5% dried non-fat milk (NFM) in Tris buffered saline containing 0.01% Tween-20 (TBST) at room temperature for one hour. The membranes were incubated overnight at 4°C with primary anti-ST6Gal-I antibody (catalogue # AF5924, R&D Systems, Minneapolis, MN), used at a concentration of 1 µg/mL and diluted into TBST containing 5% NFM. Membranes were washed in TBST and incubated with HRP conjugated secondary antibody (in 5% NFM/TBST) for one hour at room temperature. Blots were developed with Immobilon (Millipore, Billerica, MA). To control for protein loading, membranes were re-probed for GAPDH or β-actin (Cell Signaling Technologies, Danvers, MA). Densitometry was performed using Image J software.
SNA Precipitation and Fas immunoblots
Tissues were homogenized as described above, and 500µg of homogenate protein were incubated overnight at 4°C with 50 µL SNA-1 conjugated to agarose (EY Laboratories, San Mateo, CA) with rotation. α2–6 sialylated proteins complexed with SNA were collected by centrifugation, and washed. Sialylated proteins were released from complexes by boiling in SDS-PAGE sample buffer and immunoblotted for Fas (Santa Cruz Biotechnology, Inc, Santa Cruz, CA). To evaluate total Fas protein, Fas immunoblots were performed using aliquots of the initial tissue homogenates (not subjected to SNA precipitation).
Immunohistochemistry
Slides with paraffin embedded pair-matched tumor and uninvolved colon tissue were obtained from Biochain Institute (Newark, CA). Slides were rehydrated using xylene and a gradient of EtOH solutions including 100%, 95%, 80% and 70% EtOH in DiH2O for 5 minutes each. Frozen multi-tissue arrays were purchased from Biochain Institute (Newark, CA). Antigen retrieval was performed by boiling slides in citrate buffer (Vector Labs, Burlingame, CA) for 30 minutes. Slides were allowed to cool at room temperature for 60 minutes. Slides were blocked for 60 minutes in 10% normalized horse serum diluted in PBS. The following antibodies were then applied overnight at 4°C: 5 µg/mL ST6Gal-I (R&D Systems) or 2.5 µg/mL ALDH1 (BD Pharmingen, San Jose, CA), each diluted into blocking buffer. Slides were washed in PBS and secondary antibody was applied for 30 minutes at room temperature (Immpress, Vector Labs). Slides were developed with Immpact NovaRed (Vector Labs), and counter-stained with hematoxylin (Vector Labs). Slides were dehydrated through 70%, 85%, 95% and 100% EtOH, and xylene, and fixed with Permount (Vector Labs). Images were captured with ISCapture software.
Validation of ST6Gal-I antibody
Specificity of the ST6Gal-I antibody (R&D Systems #AF5924) was validated using two established cell lines (Supplementary Fig. S1). SW48 colon cancer cells and OV4 ovarian cancer cells have no endogenous ST6Gal-I, and ST6Gal-I expression was forced in these lines as reported (6, 7) (of note, SW48 and SW948 are distinct cell lines.) Immunoblotting (Fig S1A) was conducted as described above, and immunofluorescent staining (Fig. S1B) was performed using 1 µg/mL anti-ST6Gal-I antibody, followed by Alexa-conjugated secondary antibody (Life Technologies, Grand Island, NY). In addition, immunohistochemical staining was performed on formaldehyde-fixed OV4 cell cultures (Fig. S1C). To control for the effects of paraffinembedding and antigen retrieval, OV4 cells were detached and centrifuged, and the cell pellets were paraffin-embedded and sectioned. Antigen retrieval and immunohistochemical staining were performed on cell pellet sections (Fig. S1D) using the protocol described previously for tissue sections. Validation of tissue staining is shown in Supplementary Fig. S2. Frozen slides containing colon metastasis to liver were subjected to antigen retrieval and immunostaining. Samples were incubated with either primary or an isotype control antibody (Fig. S2A). As a final control, paraffin-embedded uninvolved colon and colon tumor specimens were exposed to secondary antibody alone (no primary) (Fig. S2B).
Flow Cytometry
Cells were detached from tissue culture flasks by brief trypsinization. 1×106 cells were analyzed for ALDH1 activity using the Aldefluor assay as recommended by the manufacturer (StemCell Technologies, Vancouver, BC). Samples from each cell line with inhibited Aldefluor staining were used as the gating control. CD133/1-PE antibody (AC133) was used according to the manufacturer’s protocol (Miltenyi Biotec, Auburn, CA). Results were gated for non-specific activity by isotype control (IgG1, Miltenyi Biotec). Additionally, for experiments measuring SNA reactivity, TRITC-conjugated SNA-1 (EY Laboratories) was used according to manufacturer instructions. Cells were analyzed by flow cytometry with a FACSCalibur (Becton-Dickinson, Franklin Lakes, NJ) at the UAB Rheumatic Diseases Core Center Analytic and Preparative Cytometry Facility. Statistical analysis of the flow cytometry results was accomplished using a z test for two proportions. p values less than 0.05 are considered significant.
Results
ST6Gal-I upregulation in human colon tumors
To address the lack of information regarding ST6Gal-I protein expression in human tissues, we screened several new commercial antibodies and identified one that reliably detects ST6Gal-I protein (Supplementary Figs. S1 and S2). Using this antibody, we evaluated ST6Gal- I levels in human colon cancer tissues using a commercial membrane blot containing three independent cases of human colon carcinoma, along with normal colon and normal placental specimens. As shown in Fig. 1A, higher ST6Gal-I expression was observed in the colon tumors as compared with normal colon and placenta. The upper band on the blots represents full-length ST6Gal-I, while the size of the lower band is consistent with the cleaved, secreted form of ST6Gal-I (19, 20).
Figure 1. ST6Gal-I is upregulated in human colon tumors and tumor-associated Fas receptors have elevated α2–6 sialylation.
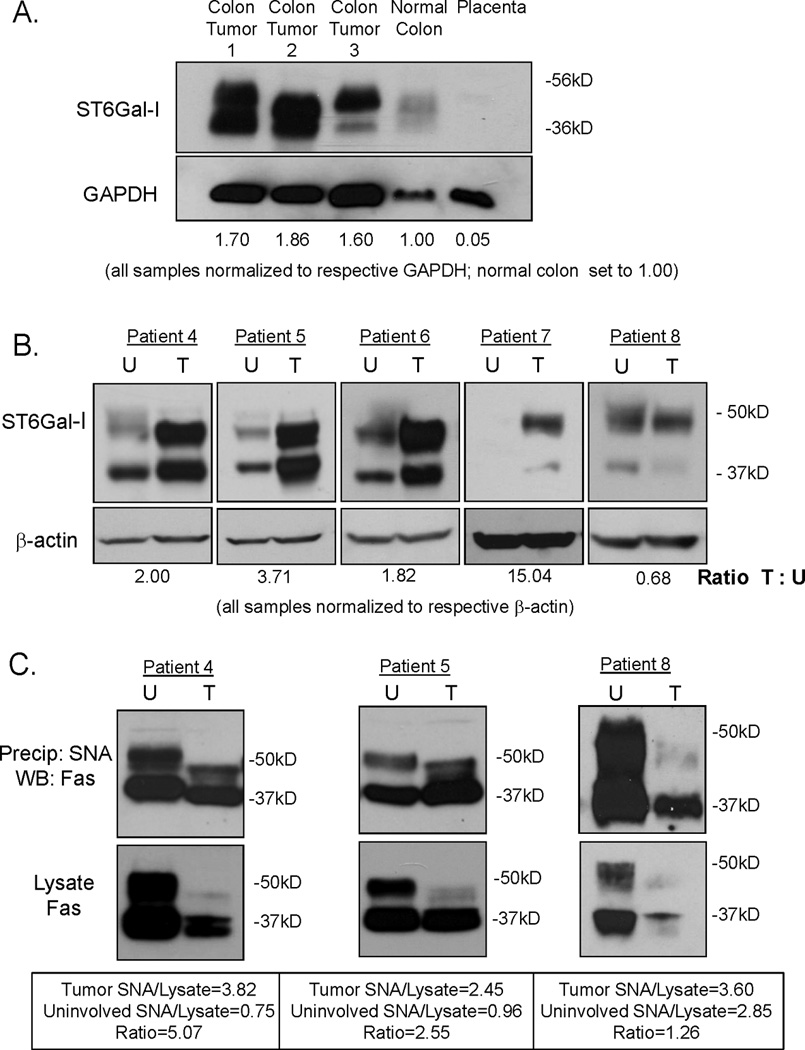
(A) A commercially purchased membrane for immunoblotting was probed for ST6Gal-I protein expression. Mature ST6Gal-I (upper band) is highly expressed in three separate colon tumors as compared to normal colon and placenta. The lower band is consistent with a cleaved, secreted form of ST6Gal-I (19). Densitometry was performed on the upper band. All samples were normalized to GAPDH and then compared to normal colon expression. (B) Tissue homogenates were prepared from colon tumors and pair-matched uninvolved colon specimens and immunoblotted for ST6Gal-I. ST6Gal-I was upregulated in 4/5 of colon tumors as compared to pair-matched uninvolved colon tissues. Densitometry was performed on the upper band. All samples were normalized to β-actin and then tumor samples were compared to pair-matched uninvolved colon. (C) α2–6 sialylated proteins were isolated using SNA1-agarose and immunoblotted for Fas. Total Fas levels were assessed by immunoblotting initial tissue homogenates (not subjected to SNA) for Fas. In all three patient samples, mature Fas (upper band) was downregulated in the tumor tissue relative to the respective uninvolved tissue. In patients with upregulated ST6Gal-I (patients 4 and 5), the proportion of sialylated Fas to total Fas was 5.07 and 2.55 respectively (densitometry was performed on the upper band). In the patient without upregulation of ST6Gal-I (patient 8), the proportion of sialylated Fas to total Fas was much lower at 1.26. U=uninvolved, T=tumor
We next examined ST6Gal-I expression in tumor and pair-matched uninvolved colon specimens obtained from the Tissue Procurement Shared Facility at UAB. Tissues were homogenized and immunoblotted for ST6Gal-I. Four of the five patient samples exhibited upregulated ST6Gal-I in the tumor compared with the cognate uninvolved specimens (Fig. 1B). Patient demographics can be found in Supplementary Table 1.
Elevated α2–6 sialylation of the Fas receptor in human colon carcinoma samples
To assess the functional consequence of ST6Gal-I upregulation in tumors, we measured levels of α2–6 sialylation on the Fas receptor. Utilizing patient samples for which sufficient tissue homogenate was available, tumor and pair-matched uninvolved colon tissue homogenates were incubated with agarose-conjugated SNA-1 lectin. The α2–6-sialylated proteins bound by SNA-agarose were isolated by centrifugation, resolved by SDS-PAGE and immunoblotted for Fas (Fig. 1C, top panels). To measure total Fas expression, samples of the original tissue homogenates (not subjected to SNA precipitation) were immunoblotted for Fas (Fig. 1C, lower panels). We found that total Fas expression was decreased in the tumors, consistent with other studies suggesting that Fas is downregulated in colon carcinoma as a mechanism for protection against Fas-mediated apoptosis (21). However, despite Fas downregulation, the proportion of α2–6 sialylated Fas in the tumors was distinctly higher than the proportion of α2–6 sialylated Fas in uninvolved colon tissues for those cases that exhibited ST6Gal-I upregulation (patients 4 and 5, Fig. 1C). Conversely, levels of α2–6 sialylated Fas were comparable in tumor and uninvolved tissues from the patient sample that did not exhibit ST6Gal-I upregulation (patient 8). Thus, ST6Gal-I overexpression in tumors acts to hypersialylate Fas despite an overall downregulation in Fas protein. Hypersialylation of Fas, which inhibits Fas receptor internalization and apoptotic signaling (11), may constitute a second line of defense through blocking the activity of Fas receptors remaining on the tumor cell surface. The determination that Fas has enhanced α2–6 sialylation in tumors is consistent with our prior studies showing that β1 integrins exhibit elevated α2–6 sialylation in colon tumors (6). These results indicate that upregulation of ST6Gal-I in tumors leads to elevated α2–6 sialylation of functionally-important ST6Gal-I targets.
ST6Gal-I upregulation and localization in colon tumors
Although ST6Gal-I upregulation in tumors was observed by immunoblotting, this approach does not address protein localization. Thus, paraffin-embedded tumor and uninvolved colon tissues from seven patients were stained by immunohistochemistry to visualize ST6Gal-I protein. Shown in Fig. 2A are results from a representative patient. In the uninvolved colonic mucosa (en face section), positive ST6Gal-I staining was observed in only a limited number of cells within the crypts. Longitudinal sections of the uninvolved colon tissue (Fig. 2B) revealed that the positively stained cells were localized to the base of the crypts. The staining was focal and located adjacent to the nucleus, typical of Golgi structure. However, the tumor samples demonstrated a dramatic upregulation in ST6Gal-I expression, with punctate-like staining apparent in the majority of the epithelial cancer cells. This type of punctate staining is characteristic of the disrupted Golgi architecture present in cancer cells (22, 23). All seven of the patients examined by immunohistochemistry exhibited the same type of staining pattern shown in Figs. 2A & B.
Figure 2. ST6Gal-I upregulation and localization in human colon tumors.
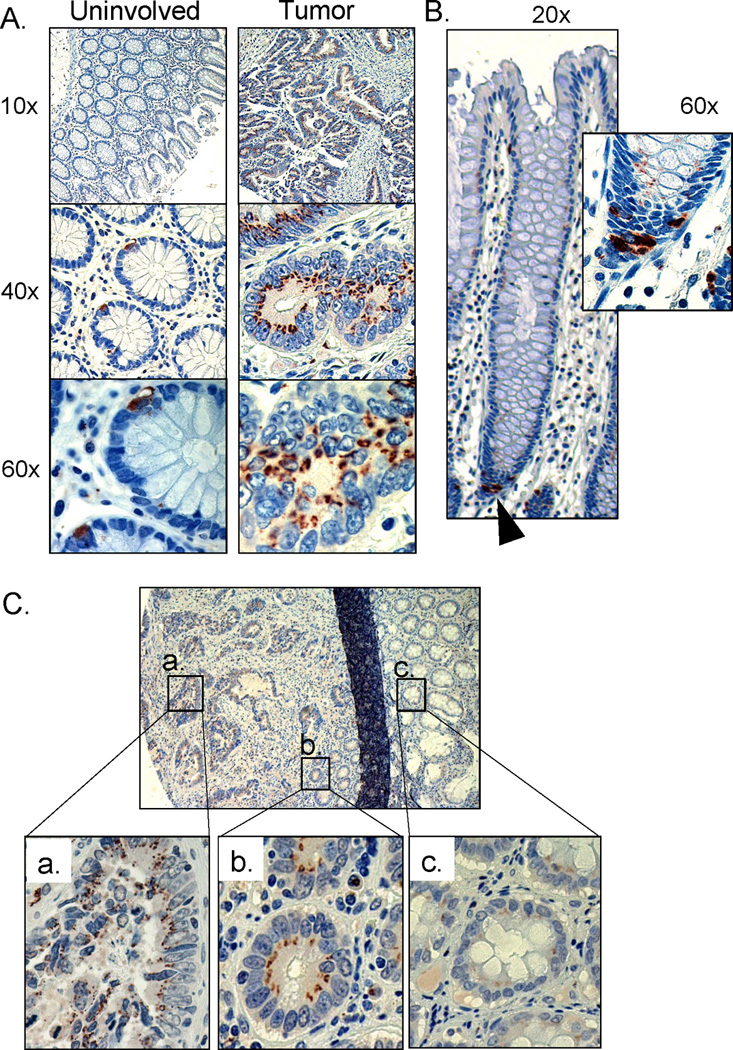
(A) Representative sample of pair-matched tissues stained for ST6Gal-I protein expression. Paraffin embedded specimens of uninvolved colon tissue and tumor tissues were immunohistologically stained for ST6Gal-I (brown), and counterstained with hematoxylin (blue). ST6Gal-I was highly upregulated in tumor tissue, whereas in uninvolved colon tissue, expression was restricted to a very few cells within each crypt structure. (B) Longitudinal view of a crypt from uninvolved tissue. ST6Gal-I staining was restricted to the base of the crypt (black arrow). Inset shows enlarged view with ST6Gal-I stain in cells at the base of the crypt. (C) ST6Gal-I staining in a patient sample showed gradient expression based on proximity to tumor. (a) upregulated expression of ST6Gal-I in malignant tissue, (b) aberrant expression in morphologically normal crypt structures directly adjacent to tumor and (c) low expression in crypts distal to the tumor.
Along with increased ST6Gal-I expression, we observed an interesting pattern within several tumor samples; ST6Gal-I levels were elevated in normal appearing crypts immediately adjacent to the tumor. Within the malignant region of the tissue section (Fig. 2Ca), the crypt structure was highly disrupted and ST6Gal-I was upregulated, as in Fig. 2A. However, in the morphologically normal-appearing crypts next to the tumor (Fig. 2Cb), ST6Gal-I staining was increased, and distributed in a punctate pattern, similar to staining in cancer cells. In the crypts more distal to the malignant tissue (Fig. 2Cc), ST6Gal-I expression was very low or undetectable, similar to the uninvolved pair-matched specimens. The upregulation of ST6Gal-I in crypts that appear morphologically intact is reminiscent of a “field effect” in which normalappearing epithelium is in fact the product of expansion of a genetically abnormal clone (24).
ST6Gal-I overexpression in multiple epithelial, but not non-epithelial, tumors
In addition to colon carcinoma, we examined ST6Gal-I protein expression in several other types of tumors. As shown in Fig. 3A, immunohistochemical staining performed on a multi-tissue array revealed ST6Gal-I upregulation in ovarian, stomach, pancreatic and prostate tumors compared with uninvolved tissues. In contrast, ST6Gal-I levels were low or undetectable in malignant and uninvolved tissues from brain and skeletal muscle (Fig. 3B).
Figure 3. ST6Gal-I is upregulated in several types of epithelial cancers, but not non-epithelial cancers.
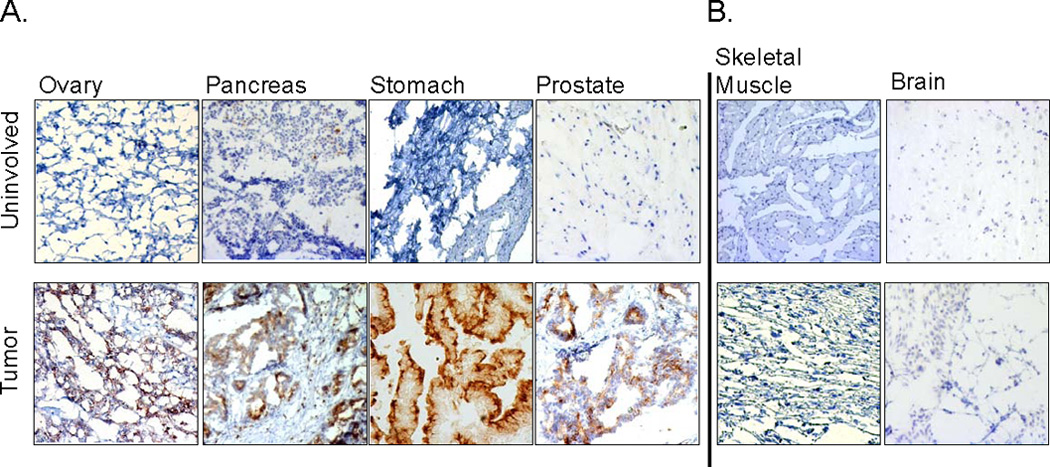
(A) Frozen epithelial tumors and pair-matched uninvolved tissues from ovary, pancreas, stomach, and prostate were stained for ST6Gal-I protein expression and counterstained with hematoxylin. ST6Gal-I upregulation was apparent in the tumor samples. (B) Frozen pair-matched tissues from skeletal muscle and brain exhibited low or undetectable levels of ST6Gal-I.
ST6Gal-I expression localizes to the stem or progenitor cell compartment in epithelia
The localization of ST6Gal-I within the base of crypts in non-malignant colon epithelium suggested that ST6Gal-I may be selectively expressed in the stem or progenitor compartment. It is well established that stem and progenitor cells reside in the base of the crypt of normal colon (25). Additionally, ST6Gal-I staining was very similar to what has been reported for the ALDH1 stem cell marker in normal colon (26). We therefore stained sections of normal human colon (cancer-free patients) for either ALDH1 or ST6Gal-I. As shown in Figs. 4A and B, both the ST6Gal-I and ALDH1 staining were in the base of the crypt, isolated to only a few cells within each crypt. No detectable staining of ST6Gal-I was observed in the differentiated colonocytes at the apical epithelial surface.
Figure 4. ST6Gal-I expression in stem/progenitor cell populations.
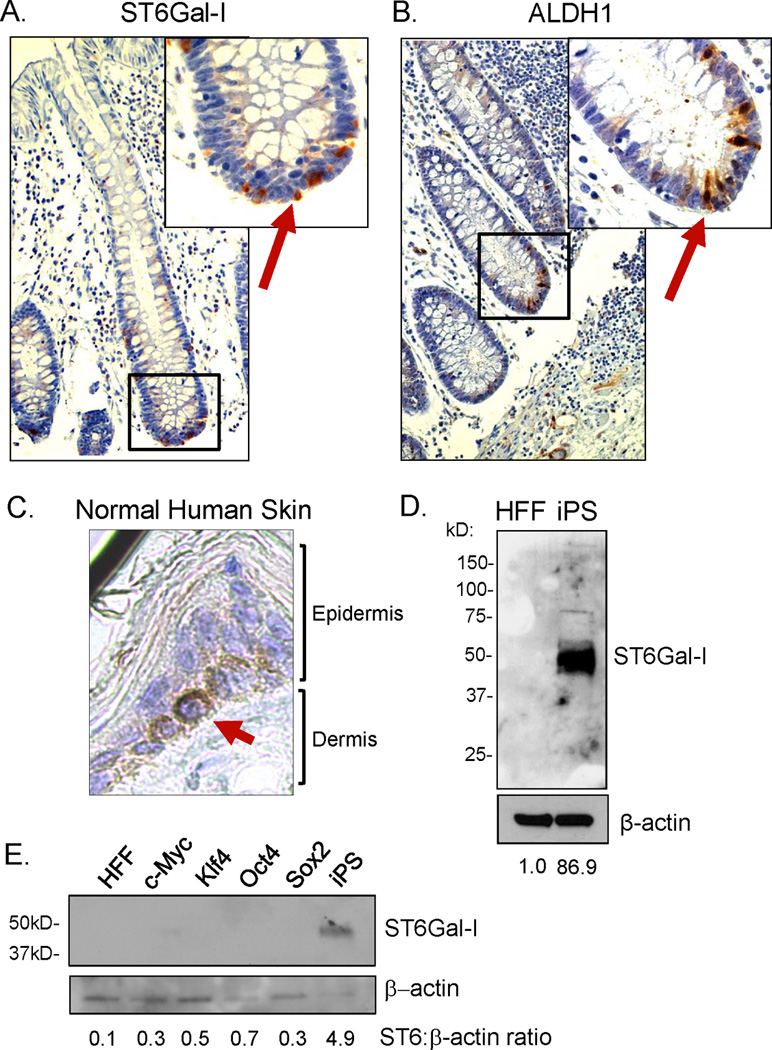
(A) Paraffin-embedded normal colon tissue (from a cancer-free patient) was stained for ST6Gal-I. ST6Gal-I expression was confined to the base of normal colon crypts, with no expression observed in the apical, differentiated epithelium. (B) Expression of the ALDH1 stem cell marker was localized to the base of the crypts in normal human colon, similar to ST6Gal-I. (C) Staining for ST6Gal-I in paraffin-embedded normal human skin tissue. ST6Gal-I expression was confined to the basal proliferative compartment of the epidermis in normal skin. (D) Immunoblot for ST6Gal-I expression in cell lysates obtained from human induced Pluripotent Stem cells (iPS) and the human foreskin fibroblast population (HFF) from which iPS cells were derived. There was no detectable ST6Gal-I expression in HFFs, whereas there was a dramatic upregulation of ST6Gal-I in the iPS cells. (E) Immunoblot for ST6Gal-I expression in HFFs, iPS cells, or HFFs transduced with only one of the individual Yamanaka factors: c-Myc; Klf4; Oct4; or Sox2. ST6Gal-I upregulation was observed in iPS cells (generated by simultaneous transduction of all four factors), but not in HFFs transduced with the single factors alone. Densitometry was completed by normalizing signal to the respective β-actin band and then comparing HFFs to iPS.
We next examined ST6Gal-I expression in the epidermis, which has clearly defined stem cell compartments (27). One of the compartments for epidermal stem/progenitor cells is the basal epidermal cell layer, immediately adjacent to the basement membrane. As basal epidermal cells differentiate, they migrate apically and lose the capacity for proliferation. As shown in Fig. 4C, ST6Gal-I expression was restricted to this basal layer, consistent with the concept that ST6Gal-I may be enriched in stem and/or progenitor cells.
ST6Gal-I is highly expressed in human induced pluripotent stem cells
To further explore a link between ST6Gal-I and stem cells, ST6Gal-I levels were evaluated in iPS cells, as well as in the human foreskin fibroblast (HFF) population from which iPS cells were derived. Immunoblots revealed that ST6Gal-I was highly expressed in iPS cells, with no detectable expression in HFFs (Fig. 4D). In addition, ST6Gal-I expression was assessed in HFFs transduced with only one of each of the four individual transcription factors used in combination to derive iPS cells (c-Myc, Klf4, Oct4, Sox2). As shown in Fig. 4E, ST6Gal-I upregulation was only observed in cells with simultaneous transduction of all four transcription factors (iPS cells), suggesting that ST6Gal-I upregulation may require genetic reprogramming. Consistent with these results, Hirabayashi’s group reported that ST6Gal-I mRNA is elevated in iPS cells relative to somatic cells, and then downregulated upon forced differentiation of iPS cells (28). Intriguingly, somatic cells exhibit both α2–3 and α2–6 sialylation, with α2–3 sialylation predominating, whereas surface sialylation becomes exclusively α2–6-linked following transformation of somatic cells into iPS cells (29). Although the biologic significance of this switch is currently unclear, these findings point to some important and distinct function for α2–6 sialylation in the stem cell phenotype.
ST6Gal-I expression correlates with stem cell enrichment in colon carcinoma cell lines
Based on the ST6Gal-I localization in normal and tumor tissues, we hypothesized that ST6Gal-I might be a marker for cancer stem cells (CSCs). ALDH1 is one of the well-studied markers for both normal and CSCs (26), and furthermore, immunohistochemical analyses revealed a similar staining pattern for ALDH1 and ST6Gal-I (Fig 4). Hence, we examined whether ST6Gal-I expression was associated with the level of stem-cell enrichment in colon cancer cell lines. Our group has generated the human colon carcinoma cell line, HD3, which overexpresses ST6Gal-I secondary to forced oncogenic ras expression (18). This line was previously transduced with shRNA to obtain a cell population with stable ST6Gal-I knockdown (8). Parental and ST6Gal-I knockdown cells were analyzed for CSC enrichment by flow cytometry using the ALDH1 activity assay, Aldefluor. As shown in Fig. 5A, in three independent experiments cells with high ST6Gal-I expression (HD3.par) exhibited significantly greater CSC enrichment than cells in which ST6Gal-I had been knocked down (HD3.sh). Fig. 5B shows a representative dot plot (Run #1, Fig. 5A). Additionally, cells were double-labeled with Aldefluor and TRITC-conjugated-SNA to detect cell surface α2–6 sialylation in order to examine the correlation between ST6Gal-I activity and stem cell enrichment (Fig 5C). Cells with ST6Gal-I knockdown exhibited a decrease in the fluorescent intensity of SNA labeling, indicating reduced α2–6 sialylation, and this was associated with diminished ALDH1 activity (note that there is variation in the level of α2–6 sialylation due to the polyclonal nature of the HD3.sh population). To more stringently assay for stem cell enrichment, cells were double-labeled for ALDH1 and an additional CSC marker, CD133. As shown in Fig. 5D, cells with high endogenous ST6Gal-I expression had significantly greater numbers of cells positive for CD133/ALDH1. This suggests that forced downregulation of ST6Gal-I significantly decreases the number of CSCs within cancer cell populations.
Figure 5. ST6Gal-I expression correlated with cancer stem cell enrichment.
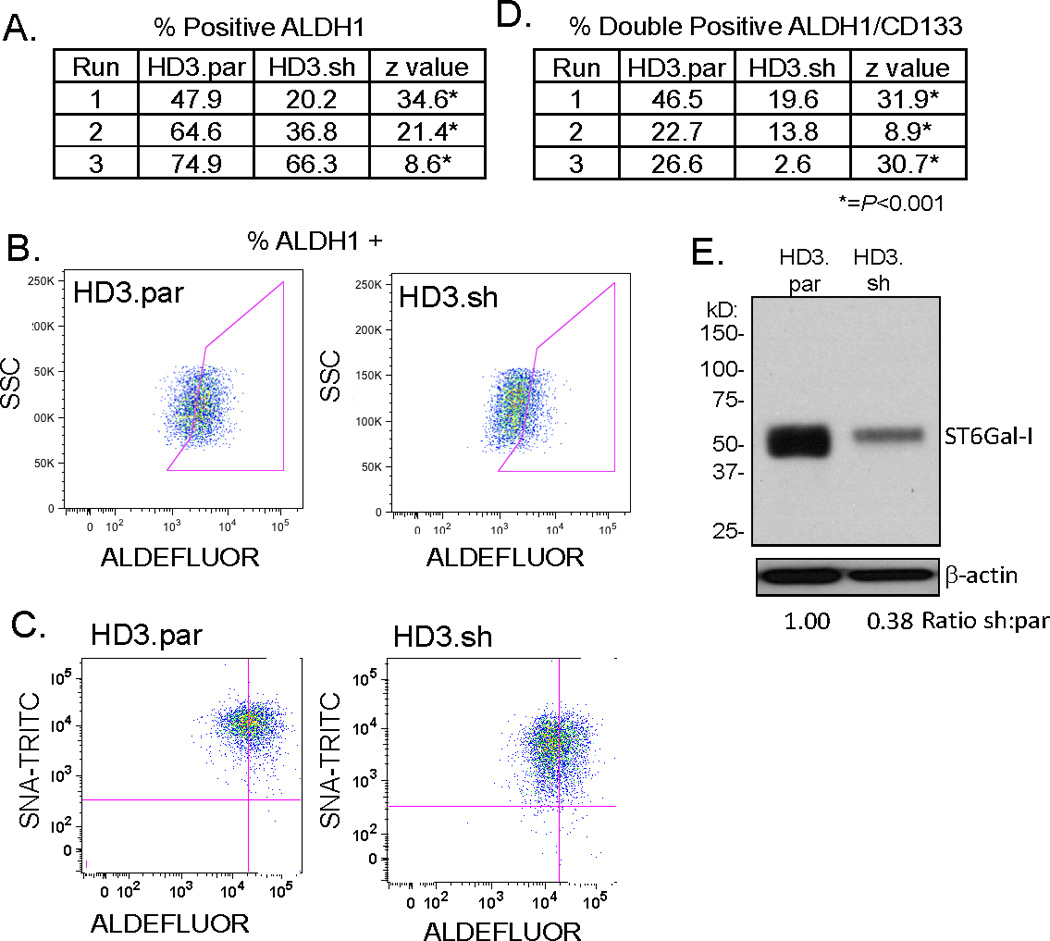
(A) Colon carcinoma cells, HD3.par and HD3.sh, were assayed for ALDH1 activity (Aldefluor) by flow cytometry. Enrichment of ALDH1 staining was significantly higher in HD3.par as compared to HD3.sh in three independent runs. (B) Representative dot plot (run #1, 5A) showing ALDH1 staining. (C) Aldefluor and SNA-TRITC double-labeling shows knockdown decreases α2–6 surface sialylation along with stem cell enrichment. (D) Double labeling for stem cell enrichment of HD3.par and HD3.sh cells with ALDH1 and CD133 by flow cytometry revealed that knockdown of ST6Gal-I lead to significantly decreased enrichment in three independent runs. (E) Immunoblot of HD3.par and HD3.sh cells showed that shRNA transduction reduced ST6Gal-I expression. Densitometry completed by normalizing to respective β-actin and then comparing HD3.sh to HD3.par. *=P <0.001.
One important characteristic of CSCs is the capacity to survive chemotherapy treatment. To study this cellular behavior, we established a cell line with acquired resistance to the camptothecin analog, Irinotecan (CPT-11), a drug used to treat colorectal carcinoma. SW948 colon carcinoma cells were treated serially with CPT-11 to obtain a stable cell line resistant to greater than 10-fold the IC50 dosage of parental cells. The parental (SW948.par) and CPT-11- resistant (SW948.CPT) lines were then assayed for ALDH1 activity. As shown in Fig. 6A, three independent experiments demonstrated significant enrichment of ALDH1 in the chemoresistant cells. Fig. 6B is a representative dot plot (Run #1, Fig 6A). Stem cell enrichment was further evaluated by double-labeling cells with anti-CD133 and Aldefluor, which revealed significantly greater numbers of CD133+/ALHD1+ cells in the SW948.CPT cells compared with SW948.par cells (Fig. 6C). We next evaluated ST6Gal-I expression in SW948.par and SW948.CPT cells by immunoblotting. Fig. 6D shows an acquired ST6Gal-I expression in the established chemoresistant cells. The chemoresistant cells also exhibit elevated ST6Gal-I activity indicated by increased intensity of SNA-TRITC labeling (Fig. 6E). Taken together, these data demonstrate a correlation between CSC enrichment and ST6Gal-I expression in two independent cell model systems. Forced ST6Gal-I downregulation decreases CSC number, whereas acquired chemoresistance yields higher CSC numbers with a corresponding increase in ST6Gal-I expression and activity.
Figure 6.
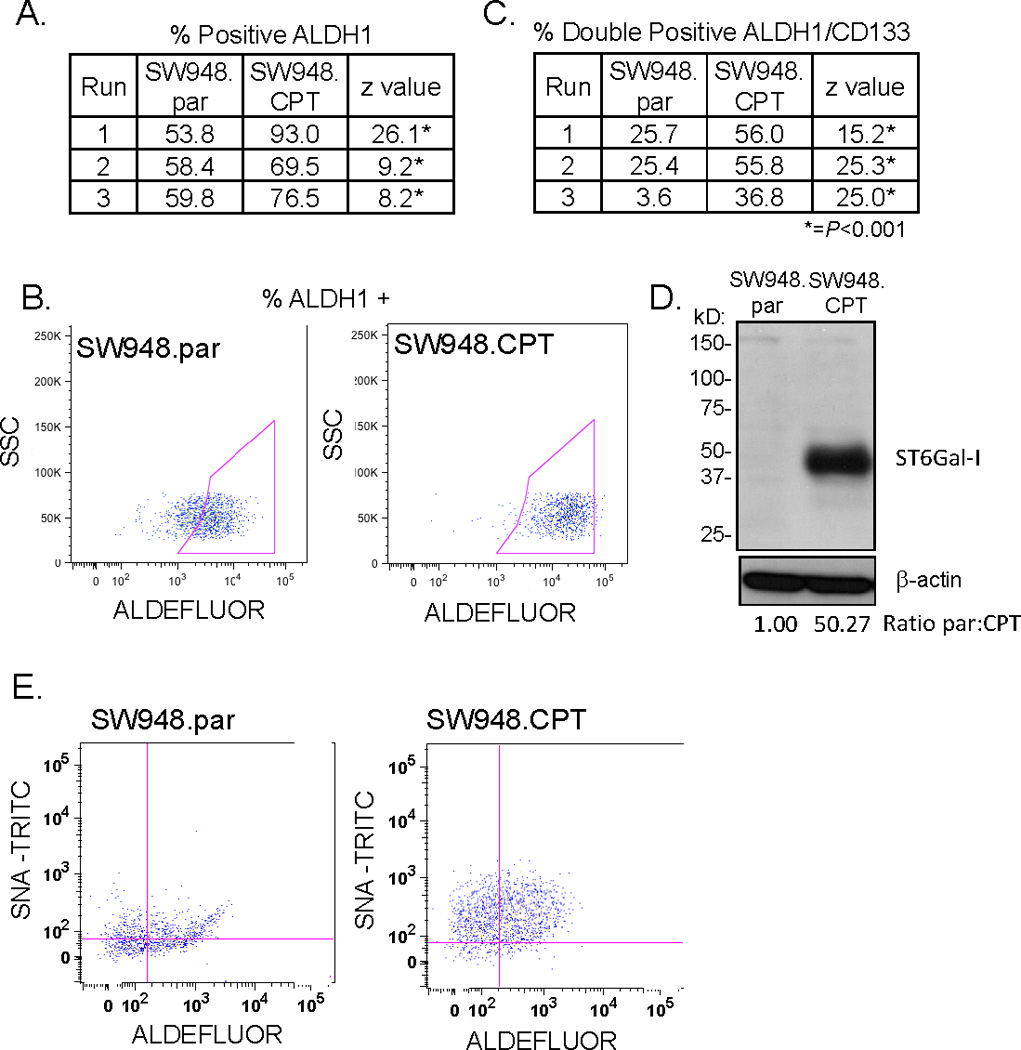
(A) ALDH1 activity was assayed by flow cytometry in colon carcinoma cell line SW948. SW948.CPT chemoresistant line had significant enrichment for ALDH1 staining in three independent runs as compared to SW948.par. (B) Representative dot plot of ALDH1 staining 28 (run #1, 6A). (C) Double-labeling of SW948.par and SW948.CPT with ALDH1 and CD133 showed significant increase in stem cell markers in the chemoresistant line (SW948.CPT) in three independent runs. (D) Immunoblot of SW948.par and SW948.CPT shows ST6Gal-I expression was upregulated in the SW948.CPT line. Densitometry completed by normalizing to respective β-actin and then comparing SW948.CPT to SW948.par. (E) Double-labeling with Aldefluor and SNA-TRITC demonstrates chemoresistant line has increased stem cell enrichment as well as increased surface α2–6 sialylation. *=P <0.001.
Discussion
Studies over the last two decades have reported increased ST6Gal-I mRNA in many human cancers (1, 2), and more recent gene expression profiling technologies confirm tumorassociated ST6Gal-I upregulation (30–32). Microarray performed on colon cancer cells isolated by laser capture microdissection revealed higher ST6Gal-I mRNA in tumors with high vs low risk of recurrence (and cells from both tumor types had higher ST6Gal-I than normal colonocytes) (33). Additional microarray studies indicate that ST6Gal-I is overexpressed in cervical (30), testicular (31) and pancreatic (32) cancers, and ST6Gal-I levels are higher in metastatic vs primary prostate cancer (34). As well, ST6Gal-I is one of the genes downregulated by the metastasis suppressor, BRMS1 (35). However few investigations have characterized ST6Gal-I protein expression in either cancer or normal tissues due to the prior lack of anti-ST6Gal-I antibodies. In one study utilizing a privately-generated antibody, ST6Gal-I was found to be upregulated in the majority of human colon tumors (36). In the present investigation, we screened multiple new commercial antibodies and identified a reagent with high specificity for ST6Gal-I. Using this antibody we observed extensive staining for ST6Gal-I in all of the human tumor tissues evaluated by immunohistochemistry, and markedly elevated ST6Gal-I expression in seven out of eight colon tumor samples examined by immunoblotting. Interestingly, the localization of ST6Gal-I in normal tissues was distinctly different from that of tumor tissues. Specifically, ST6Gal-I expression was found within a few cells in the base of the colonic crypts, with no detectable expression in the differentiated epithelial cells. Furthermore, ST6Gal-I expression was high in the basal, proliferative compartment of the epidermis, and high in iPS cells, but undetectable in the somatic cell population from which iPS cells were derived.
Given that ST6Gal-I expression in normal tissues appeared to associate with stem/progenitor cell populations, we evaluated whether ST6Gal-I levels might be elevated in CSCs. CSCs (alternately referred to as “tumor-initiating cells”), are posited to represent a subset of cells within the heterogeneous tumor that has a more aggressive and chemoresistant phenotype (37, 38). The level of CSC enrichment within a cancer cell population is identified by a variety of markers, including ALDH1 and CD133, which have been validated in colon carcinoma (26, 39, 40). CSCs are considered to be a driving force behind tumor recurrence due to the self-renewal properties of these cells, and resistance to chemotherapeutic drugs. This has been shown in a number of cancer types including breast, ovarian and colon carcinomas. In this study, we found that high ST6Gal-I expression consistently correlated with ALDH1 and CD133 expression, and forced ST6Gal-I downregulation reduced the percentage of CSCs within a heterogeneous cell population. As well, when SW948 colon cancer cells, which do not usually express ST6Gal-I, were treated serially with increasing concentrations of Irinotecan (CPT11), the stem-cell population was selectively protected, evidenced by an increase in ALDH1/CD133- positive cells, and correspondingly, ST6Gal-I expression and activity were markedly increased. Notably, microarray studies comparing gene expression in CD133+ vs CD133− colon cancer cells identified ST6Gal-I as one of the 39 genes with the highest selective expression in CD133+ cells, and ST6Gal-I was the only glycosylation-related gene in this pool (41). While further studies are needed, these results suggest that ST6Gal-I may represent a new marker for CSCs.
There are several hypotheses concerning the origin of CSCs. It is widely debated as to whether CSCs are derived from mutated normal stem cells, progenitor cells, or more differentiated cells (that subsequently revert to a less-differentiated phenotype). In colon tumorigenesis, it has been suggested that a tumor would more likely arise from a mutated stem or progenitor cell, due to the short half-life of differentiated colonocytes, as well as the clonal nature of crypt development, where the entire crypt is thought to be derived from a single stem cell or stem cell compartment located at the base of the crypt (25, 42). Interestingly, some of the fundamental evidence supporting the clonal crypt hypothesis was obtained from studies of a sialic acid variant, 9-O-acetylated sialic acid, which is generated by the enzyme, sialate-O-acetyltransferase (OAT). Loss of heterozygosity in stem cells of humans heterozygous for the OAT gene causes complete repopulation of the crypt by the progeny of the mutant stem cells (43). While the relationship between 9-O-acetylated sialic acids and ST6Gal-I activity is unclear, these studies are consistent with the concept that specific types of sialylation may be very important in maintaining some aspect of the stem cell phenotype. This hypothesis is further supported by the recent finding that sialic acids on iPS cells are exclusively α2–6-linked, in contrast to somatic cells which express a mixture of α2–3 and α2–6 sialylation, with α2–3 predominating (29).
ST6Gal-I-mediated receptor sialylation has been previously correlated with an undifferentiated or immature cell state, particularly in certain immune cell types. We reported that ST6Gal-I expression is decreased as monocytic cells differentiate down the macrophage lineage (19, 44). Others have shown that ST6Gal-I activity is initially important for monocytederived dendritic cell generation, but that maturation of dendritic cells is associated with a loss in ST6Gal-I (45). As well, removal of sialic acids via neuraminidase treatment stimulated dendritic cell differentiation, and dendritic cells from ST6Gal-I null mice have a more mature status than cells from wild type mice (46). ST6Gal-I is also markedly downregulated upon activation of murine CD4+ and CD8+ T lymphocytes (47). Fewer studies have addressed ST6Gal-I expression in epithelial cell differentiation, however SNA labeling of epidermis is inversely correlated with cell differentiation status (48). Finally, Varki and colleagues investigated the role of ST6Gal-I in the PyMT spontaneous mammary tumorigenesis model, and found that tumors from ST6Gal-I null mice were more differentiated than tumors from wild type mice (49).
The functional contribution of ST6Gal-I to an immature or undifferentiated cell phenotype has yet to be elucidated, however resistance to apoptosis may play a prominent role. Accumulating evidence points to ST6Gal-I as a major inhibitor of cell death pathways initiated by Fas, TNFR1 and galectins (2, 11, 12). Lee’s group also demonstrated that ST6Gal-I confers radiation-resistance in colon cancer cell lines (50). In the aggregate, these studies are consistent with the general concept that ST6Gal-I activity might underlie the survival or self-renewal characteristics of stem/progenitor cells, and/or selected cancer cell populations. A corollary hypothesis is that downregulation of ST6Gal-I in differentiated cells may sensitize cells to multiple apoptotic stimuli, thus limiting cell lifespan. Clearly there is a need for further investigation of ST6Gal-I function, however the current study provides important new insight into the localization of ST6Gal-I expression in normal and tumor epithelium, and also implicates ST6Gal-I as a potential new marker for CSCs.
Supplementary Material
Acknowledgements
The authors are grateful for technical support from Dr. Yuanyuan Xu, and assistance from Enid Keyser and the Rheumatic Diseases Core Center-Analytic and Preparative Cytometry Facility (funded by NIH P30 AR48311).
Financial Support: AFS: AHA 10PRE2390018; AILJ: DOD W81XWH-11-1-0151; MJS: NSF DGE-0950047; SLB: NCI CA84248 & DOD OC100141
Footnotes
Authors have no conflict of interest to disclose.
References
- 1.Schultz MJ, Swindall AF, Bellis SL. Regulation of the metastatic cell phenotype by sialylated glycans. Cancer Metastasis Rev. 2012;31:501–518. doi: 10.1007/s10555-012-9359-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhuo Y, Bellis SL. Emerging role of alpha2,6-sialic acid as a negative regulator of galectin binding and function. J Biol Chem. 2010;286:5935–5941. doi: 10.1074/jbc.R110.191429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dall'Olio F, Chiricolo M. Sialyltransferases in cancer. Glycoconj J. 2001;18:841–850. doi: 10.1023/a:1022288022969. [DOI] [PubMed] [Google Scholar]
- 4.Zhu Y, Srivatana U, Ullah A, Gagneja H, Berenson CS, Lance P. Suppression of a sialyltransferase by antisense DNA reduces invasiveness of human colon cancer cells in vitro. Biochim Biophys Acta. 2001;1536:148–160. doi: 10.1016/s0925-4439(01)00044-8. [DOI] [PubMed] [Google Scholar]
- 5.Lin S, Kemmner W, Grigull S, Schlag PM. Cell surface alpha 2,6 sialylation affects adhesion of breast carcinoma cells. Exp Cell Res. 2002;276:101–110. doi: 10.1006/excr.2002.5521. [DOI] [PubMed] [Google Scholar]
- 6.Seales EC, Jurado GA, Brunson BA, Wakefield JK, Frost AR, Bellis SL. Hypersialylation of beta1 integrins, observed in colon adenocarcinoma, may contribute to cancer progression by up-regulating cell motility. Cancer Res. 2005;65:4645–4652. doi: 10.1158/0008-5472.CAN-04-3117. [DOI] [PubMed] [Google Scholar]
- 7.Christie DR, Shaikh FM, Lucas JA, 4th, Lucas JA, 3rd, Bellis SL. ST6Gal-I expression in ovarian cancer cells promotes an invasive phenotype by altering integrin glycosylation and function. J Ovarian Res. 2008;1:3–10. doi: 10.1186/1757-2215-1-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shaikh FM, Seales EC, Clem WC, Hennessy KM, Zhuo Y, Bellis SL. Tumor cell migration and invasion are regulated by expression of variant integrin glycoforms. Exp Cell Res. 2008;314:2941–2950. doi: 10.1016/j.yexcr.2008.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bresalier RS, Rockwell RW, Dahiya R, Duh Q-Y, Kim YS. Cell surface sialoprotein alterations in metastatic murine colon cancer cell lines selected in an animal model for colon cancer metastasis. Cancer Res. 1990;50:1299–1307. [PubMed] [Google Scholar]
- 10.Harvey BE, Toth CA, Wagner HE, Steele GD, Jr, Thomas P. Sialytransferase activity and hepatic tumor growth in a nude mouse model of colorectal cancer metastases. Cancer Res. 1992;52:1775–1779. [PubMed] [Google Scholar]
- 11.Swindall AF, Bellis SL. Sialylation of the Fas death receptor by ST6Gal-I provides protection against Fas-mediated apoptosis in colon carcinoma cells. J Biol Chem. 2011;286:22982–22990. doi: 10.1074/jbc.M110.211375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu Z, Swindall AF, Kesterson RA, Schoeb TR, Bullard DC, Bellis SL. ST6Gal-I regulates macrophage apoptosis via alpha2–6 sialylation of the TNFR1 death receptor. J Biol Chem. 2011;286:39654–39662. doi: 10.1074/jbc.M111.276063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Amano M, Galvan M, He J, Baum LG. The ST6Gal I sialyltransferase selectively modifies N-glycans on CD45 to negatively regulate galectin-1-induced CD45 clustering, phosphatase modulation, and T cell death. J Biol Chem. 2003;278:7469–7475. doi: 10.1074/jbc.M209595200. [DOI] [PubMed] [Google Scholar]
- 14.Kitazume S, Imamaki R, Ogawa K, Komi Y, Futakawa S, Kojima S, et al. Alpha2,6-sialic acid on platelet endothelial cell adhesion molecule (PECAM) regulates its homophilic interactions and downstream antiapoptotic signaling. J Biol Chem. 2010;285:6515–6521. doi: 10.1074/jbc.M109.073106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hirabayashi J, Hashidate T, Arata Y, Nishi N, Nakamura T, Hirashima M, et al. Oligosaccharide specificity of galectins: a search by frontal affinity chromatography. Biochim Biophys Acta. 2002;1572:232–254. doi: 10.1016/s0304-4165(02)00311-2. [DOI] [PubMed] [Google Scholar]
- 16.Zhuo Y, Chammas R, Bellis SL. Sialylation of beta1 integrins blocks cell adhesion to galectin-3 and protects cells against galectin-3-induced apoptosis. J Biol Chem. 2008;283:22177–22185. doi: 10.1074/jbc.M8000015200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Park JJ, Yi JY, Jin YB, Lee YJ, Lee JS, Lee YS, et al. Sialylation of epidermal growth factor receptor regulates receptor activity and chemosensitivity to gefitinib in colon cancer cells. Biochem Pharmacol. 2012;83:849–857. doi: 10.1016/j.bcp.2012.01.007. [DOI] [PubMed] [Google Scholar]
- 18.Seales EC, Jurado GA, Singhal A, Bellis SL. Ras oncogene directs expression of a differentially sialylated, functionally altered beta1 integrin. Oncogene. 2003;22:7137–7145. doi: 10.1038/sj.onc.1206834. [DOI] [PubMed] [Google Scholar]
- 19.Woodard-Grice AV, McBrayer AC, Wakefield JK, Zhuo Y, Bellis SL. Proteolytic shedding of ST6Gal-I by BACE1 regulates the glycosylation and function of alpha4beta1 integrins. J Biol Chem. 2008;283:26364–26373. doi: 10.1074/jbc.M800836200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kitazume S, Nakagawa K, Oka R, Tachida Y, Ogawa K, Luo Y, et al. In vivo cleavage of alpha2,6-sialyltransferase by Alzheimer beta-secretase. J Biol Chem. 2005;280:8589–8595. doi: 10.1074/jbc.M409417200. [DOI] [PubMed] [Google Scholar]
- 21.Moller P, Koretz K, Leithauser F, Bruderlein S, Henne C, Quentmeier A, et al. Expression of APO-1 (CD95), a member of the NGF/TNF receptor superfamily, in normal and neoplastic colon epithelium. Int J Cancer. 1994;57:371–377. doi: 10.1002/ijc.2910570314. [DOI] [PubMed] [Google Scholar]
- 22.Kellokumpu S, Sormunen R, Kellokumpu I. Abnormal glycosylation and altered Golgi structure in colorectal cancer: dependence on intra-Golgi pH. FEBS letters. 2002;516:217–224. doi: 10.1016/s0014-5793(02)02535-8. [DOI] [PubMed] [Google Scholar]
- 23.Weller SG, Capitani M, Cao H, Micaroni M, Luini A, Sallese M, et al. Src kinase regulates the integrity and function of the Golgi apparatus via activation of dynamin 2. Proc Natl Acad Sci U S A. 107:5863–5868. doi: 10.1073/pnas.0915123107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chai H, Brown RE. Field effect in cancer-an update. Ann Clin Lab Sci. 2009;39:331–337. [PubMed] [Google Scholar]
- 25.Humphries A, Wright NA. Colonic crypt organization and tumorigenesis. Nat Rev Cancer. 2008;8:415–424. doi: 10.1038/nrc2392. [DOI] [PubMed] [Google Scholar]
- 26.Huang EH, Hynes MJ, Zhang T, Ginestier C, Dontu G, Appelman H, et al. Aldehyde dehydrogenase 1 is a marker for normal and malignant human colonic stem cells (SC) and tracks SC overpopulation during colon tumorigenesis. Cancer Res. 2009;69:3382–3389. doi: 10.1158/0008-5472.CAN-08-4418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Boehnke K, Falkowska-Hansen B, Stark HJ, Boukamp P. Stem cells of the human epidermis and their niche: composition and function in epidermal regeneration and carcinogenesis. Carcinogenesis. 2012;33:1247–1258. doi: 10.1093/carcin/bgs136. [DOI] [PubMed] [Google Scholar]
- 28.Tateno H, Toyota M, Saito S, Onuma Y, Ito Y, Hiemore K, et al. Glycome diagnosis of human induced pluripotent stem cells using lectin microarray. J Biol Chem. 2011;286:20345–20353. doi: 10.1074/jbc.M111.231274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hasehira K, Tateno H, Onuma Y, Ito Y, Asashima M, Hirabayashi J. Structural and quantitative evidence for dynamic glycome shift upon production of human induced pluripotent stem cells. Mol Cell Proteomics. 2012 Sep 27; doi: 10.1074/mcp.M112.020586. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Choi YW, Bae SM, Kim YW, Lee HN, Park TC, Ro DY, et al. Gene expression profiles in squamous cell cervical carcinoma using array-based comparative genomic hybridization analysis. Int J Gynecol Cancer. 2007;17:687–696. doi: 10.1111/j.1525-1438.2007.00834.x. [DOI] [PubMed] [Google Scholar]
- 31.Almstrup K, Hoei-Hansen CE, Wirkner U, Blake J, Schwager C, Ansorge W, et al. Embryonic stem cell-like features of testicular carcinoma in situ revealed by genomewide gene expression profiling. Cancer Res. 2004;64:4736–4743. doi: 10.1158/0008-5472.CAN-04-0679. [DOI] [PubMed] [Google Scholar]
- 32.Grutzmann R, Pilarsky C, Staub E, Scmitt AO, Foerder M, Specht T, et al. Systematic isolation of genes differentially expressed in normal and cancerous tissue of the pancreas. Pancreatology. 2003;3:169–178. doi: 10.1159/000070087. [DOI] [PubMed] [Google Scholar]
- 33.Kemmner W, Roefzaad C, Haensch W, Schlag PM. Glycosyltransferase expression in human colonic tissue examined by oligonucleotide arrays. Biochim Biophys Acta. 2003;1621:272–279. doi: 10.1016/s0304-4165(03)00079-5. [DOI] [PubMed] [Google Scholar]
- 34.LaTulippe E, Satagopan J, Smith A, Scher H, Scardino P, Reuter V, et al. Comprehensive gene expression analysis of prostate cancer reveals distinct transcriptional programs associated with metastatic disease. Cancer Res. 2002;62:4499–4506. [PubMed] [Google Scholar]
- 35.Champine PJ, Michaelson J, Weimer BC, Welch DR, DeWald DB. Microarray analysis reveals potential mechanisms of BRMS1-mediated metastasis suppression. Clin Exp Metastasis. 2007;24:551–565. doi: 10.1007/s10585-007-9092-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lise M, Belluco C, Perera SP, Patel R, Thomas P, Ganguly A. Clinical correlations of alpha2,6-sialyltransferase expression in colorectal cancer patients. Hybridoma. 2000;19:281–286. doi: 10.1089/027245700429828. [DOI] [PubMed] [Google Scholar]
- 37.Rosen JM, Jordan CT. The increasing complexity of the cancer stem cell paradigm. Science. 2009;324:1670–1673. doi: 10.1126/science.1171837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dalerba P, Cho RW, Clarke MF. Cancer stem cells: models and concepts. Ann Rev Med. 2007;58:267–284. doi: 10.1146/annurev.med.58.062105.204854. [DOI] [PubMed] [Google Scholar]
- 39.O'Brien CA, Pollett A, Gallinger S, Dick JE. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature. 2007;445:106–110. doi: 10.1038/nature05372. [DOI] [PubMed] [Google Scholar]
- 40.Ricci-Vitiani L, Lombardi DG, Pilozzi E, Biffoni M, Todaro M, Peschle C, et al. Identification and expansion of human colon-cancer-initiating cells. Nature. 2007;445:111–115. doi: 10.1038/nature05384. [DOI] [PubMed] [Google Scholar]
- 41.Ieta K, Tanaka F, Haraguchi N, Kita Y, Sakashita H, Mimori K, et al. Biological and genetic characteristics of tumor-initiating cells in colon cancer. Ann Surg Oncol. 2008;15:638–648. doi: 10.1245/s10434-007-9605-3. [DOI] [PubMed] [Google Scholar]
- 42.Preston SL, Wong WM, Chan AO, Poulsom R, Jeffery R, Goodlad RA, et al. Bottom-up histogenesis of colorectal adenomas: origin in the monocryptal adenoma and initial expansion by crypt fission. Cancer Res. 2003;63:3819–3825. [PubMed] [Google Scholar]
- 43.Campbell F, Williams GT, Appleton MA, Dixon MF, Harris M, Williams ED. Postirradiation somatic mutation and clonal stabilisation time in the human colon. Gut. 1996;39:569–573. doi: 10.1136/gut.39.4.569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Seales EC, Shaikh FM, Woodard-Grice AV, Aggarwal P, McBrayer AC, Hennessy KM, et al. A protein kinase C/Ras/ERK signaling pathway activates myeloid fibronectin receptors by altering beta1 integrin sialylation. J Biol Chem. 2005;280:37610–37615. doi: 10.1074/jbc.M508476200. [DOI] [PubMed] [Google Scholar]
- 45.Videira PA, Amado IF, Crespo HJ, Alguero MC, Dall'Olio F, Cabral MG, et al. Surface alpha 2–3- and alpha 2–6-sialylation of human monocytes and derived dendritic cells and its influence on endocytosis. Glycoconj J. 2008;25:259–268. doi: 10.1007/s10719-007-9092-6. [DOI] [PubMed] [Google Scholar]
- 46.Crespo HJ, Cabral MG, Teixeira AV, Lau JT, Trindade H, Videira PA. Effect of sialic acid loss on dendritic cell maturation. Immunology. 2009;128:e621–e631. doi: 10.1111/j.1365-2567.2009.03047.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Comelli EM, Sutton-Smith M, Yan Q, Amado M, Panico M, Gilmartin T, et al. Activation of murine CD4+ and CD8+ T lymphocytes leads to dramatic remodeling of N-linked glycans. J Immunol. 2006;177:2431–2440. doi: 10.4049/jimmunol.177.4.2431. [DOI] [PubMed] [Google Scholar]
- 48.Holikova Z, Hrdlickova-Cela E, Plzak J, Smetana K, Jr, Betka J, Dvorankova B, et al. Defining the glycophenotype of squamous epithelia using plant and mammalian lectins. Differentiation-dependent expression of alpha2,6- and alpha2,3-linked Nacetylneuraminic acid in squamous epithelia and carcinomas, and its differential effect on binding of the endogenous lectins galectins-1 and-3. APMIS. 2002;110:845–856. doi: 10.1034/j.1600-0463.2002.1101202.x. [DOI] [PubMed] [Google Scholar]
- 49.Hedlund M, Ng E, Varki A, Varki NM. alpha 2–6-Linked sialic acids on N-glycans modulate carcinoma differentiation in vivo. Cancer Res. 2008;68:388–394. doi: 10.1158/0008-5472.CAN-07-1340. [DOI] [PubMed] [Google Scholar]
- 50.Lee M, Lee HJ, Bae S, Lee YS. Protein sialylation by sialyltransferase involves radiation resistance. Mol Cancer Res. 2008;6:1316–1325. doi: 10.1158/1541-7786.MCR-07-2209. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.