

Abstract
Human aldo–keto reductases AKR1C1–AKR1C3 are involved in the biosynthesis and inactivation of steroid hormones and prostaglandins and thus represent attractive targets for the development of new drugs. We synthesized a series of N-benzoyl anthranilic acid derivatives and tested their inhibitory activity on AKR1C enzymes. Our data show that these derivatives inhibit AKR1C1–AKR1C3 isoforms with low micromolar potency. In addition, five selective inhibitors of AKR1C3 were identified. The most promising inhibitors were compounds 10 and 13, with IC50 values of 0.31 µM and 0.35 µM for AKR1C3, respectively.
Keywords: Anthranilic acid, AKR1C3 inhibitors, Anticancer agents
Human aldo–keto reductases AKR1C1–AKR1C4 are involved in the biosynthesis and inactivation of steroid hormones, neurosteroids, prostaglandins, products of lipid peroxidation and xenobiotics. These AKR1C isoenzymes reduce carbonyl containing substrates to alcohols and also act as 3-keto, 17-keto and 20-ketosteroid reductases to varying extents. In this manner, they regulate the activity of androgens, estrogens, progesterone and the occupancy and activation of their corresponding nuclear receptors.1,2 They possess a broad spectrum of physiological roles, but when differentially expressed they are also involved in different pathophysiological conditions, such as hormone-dependent and independent cancers for example prostate cancer,3–8 other hormone-dependent diseases,9 depression, epilepsy and neurodegenerative diseases.2 These enzymes, and especially AKR1C1 and AKR1C3, thus represent attractive targets for the development of new drugs. As AKR1C enzymes share more than 86% sequence identity at the amino acid level and interestingly, AKR1C1 and AKR1C2 differ in only one residue in the active site (Leu/Val54), development of specific inhibitors is a challenging task.
Anthranilic acid derivatives have been known as good inhibitors of AKR1C isoenzymes, with Ki values in low micromolar or nanomolar range. Almost 30 years ago, Penning and Talalay showed that indomethacin and mefenamic acid, nonselective NSAIDs, strongly inhibit rat liver hydroxysteroid dehydrogenase AKR1C9, a model for human AKR1C isoenzymes.10 Other nonselective NSA-IDs and selective COX-2 inhibitors also proved to be potent inhibitors of AKR1C isoenzymes.3,11 This inhibition of AKR1C enzymes by NSAIDs may thus explain the known antineoplastic effects of these drugs. To confirm the hypothesis on involvement of AKR1C isoenzymes in COX-independent anti-proliferative effects, Penning et al. developed selective inhibitors of AKR1Cs based on N-phenylanthranilic acid and cholanic acid with IC50 values in low µM range.3 Recently, we synthesized a series of N-benzoyl anthranilic acid derivatives as inhibitors of penicillin binding proteins.12 Due to their structural similarities to several known inhibitors of AKR1C enzymes, we tested their inhibitory activity on AKR1C enzymes.13,14 We demonstrated that these derivatives inhibit AKR1C isoenzymes in the low micromolar range. In addition, 5 compounds were found to be selective inhibitors of AKR1C3.
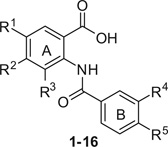
The compounds were synthesized by the formation of the amide bond between carboxylic group of benzoic acid derivatives and the amino moiety of C-protected anthranilic acids. The carboxylic acid groups were converted into acid chlorides using SOCl2, followed by the addition of the corresponding C-protected anthranilic acid derivatives. The esters were subsequently deprotected with alkaline hydrolysis and where necessary an aromatic NO2 group was reduced into their corresponding amine by catalytic hydrogenation to obtain the target compounds 1–16. The general experimental procedures for the synthesis of compounds presented in Table 1 and their spectroscopic properties are described in Ref.12 The detailed synthetic procedure and the spectroscopic data of a representative compound (11) are described in note.15
Table 1.
Inhibitory activities for compounds 1–16a
 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| R1 | R2 | R3 | R4 | R5 | AKR1C1 inhibitionb,c(%) | AKR1C2 inhibitionb,c(%) | AKR1C3 inhibitionb,c(%) | AKR1C4 inhibitionc(%) | |
| 1 | OH | H | H | NO2 | H | NI | 31.6d | 1.9 | n.d. |
| 2 | Cl | H | H | NO2 | H | NI | 23.4 | 3.4 | n.d. |
| 3 | OMe | OMe | H | NO2 | H | 22.2 | NI | 3.5 | n.d. |
| 4 | Cl | H | Me | NO2 | H | NI | 22.5 | NI | n.d. |
| 5 | H | H | H | NH2 | H | 8.0 | NI | NI | n.d. |
| 6 | OMe | OMe | H | NH2 | H | 18.0 | 20.2 | 5.6 | n.d. |
| 7 | OH | H | H | NH2 | H | 9.7 | 28.7 | 5.6 | n.d. |
| 8 | Br | H | H | NO2 | F | 53.2 | 33.6 | 5.6 | n.d. |
| IC50 = 8.4 µM | IC50 = 15.6 µM | ||||||||
| 9 | H | H | H | OH | H | NI | NI | 42.1 | n.d. |
| IC50 = 12.7 µM | |||||||||
| IC50 = >100 µMc (>19)e | IC50 = 48.1 µMc (9)e | IC50 = 5.19 µMc | IC50 = 100 µMc (>19)e | ||||||
| 10 | Cl | H | H | OH | H | 17.4 | 6.7 | 77.8 | n.d. |
| IC50 = 2.2 µM | |||||||||
| IC50 = 50.8 µMc (164)e | IC50 = 50.2 µMc (162)e | IC50 = 0.31 µMc | IC50 = 20.1 µMc (65)e | ||||||
| 11 | OMe | OMe | H | OH | H | 11.8 | 29.4 | 70.2 IC50 = 5.2 µM | n.d. |
| IC50 = 86.3 µMcc (30)e | IC50 = 30.0 µMc (10)e | IC50 = 2.9 µMc | IC50 = 50.9 µMc (18)e | ||||||
| 12 | NO2 | H | H | OH | H | 11.8 | 10.4 | 87.9 IC50 = 2.6 µM | n.d. |
| IC50 = 35.2 µMc (42)e | IC50 = 41.6 µMc (50)e | IC50 = 0.84 µMc | IC50 = 32.3 µMc (38)e | ||||||
| 13 | Br | H | H | OH | H | 21.6 | 16.7 | 89.6 | n.d. |
| IC50 = 1.9 µM | |||||||||
| IC50 = ~100 µMc (286) | IC50 = 63.0 µMc (180)e | IC50 = 0.35 µMc | IC50 = 30.3 µMc (86)e | ||||||
| 14 | Br | H | H | OEt | H | 31.9 | 36.8 | 19.1 | n.d. |
| IC50 = 20.6 µM | IC50 = 17.1 µM | ||||||||
| 15 | Br | H | H | OBu | H | 65.9 | 70.5 | 30.4 | n.d. |
| IC50 = 4.9 µM | IC50 = 4.9 µM | ||||||||
| 16 | H | H | H | OBu | NO2 | 70.8 | 58.3 | 47.2 | n.d. |
| IC50 = 3.2 µM | IC50 = 6.5 µM | IC50 = 7.5 µM | |||||||
NI, No inhibition observed.
n.d., Not determined.
The data represent the mean value of two independent experiments. Standard deviations were within ±10% of these mean values.
The inhibitory activities at 10 µM of each compound and IC50 values were determined with the primary assay, first line.
The IC50 values of the selected 5 compounds were determined with the secondary assay, second line.
The compound precipitated at higher concentrations under assay conditions.
Values in parenthesis, determined on the basis of the secondary assay results, represent fold selectivity for AKR1C3.
Screening of the compounds against recombinant AKR1C1–AKR1C416 gave the results presented in Table 1. We found that compounds that possessed either NO2 (1–4) or NH2 group (5–7) at the meta position of B ring (R4 = NO2 or NH2) and in addition contained different substituents on the anthranilic acid (A) ring were inactive on all AKR1C isoforms. The only exception was compound 8 that had an additional fluorine atom positioned ortho with respect to the NO2 group on the ring B (R4 = NO2, R5 = F). It exhibited quite potent inhibitory activity against AKR1C1 and AKR1C2 with IC50 values of 8.4 µM and 15.6 µM, respectively, whereas no AKR1C3 isoform inhibition was observed.
AKR1C3 inhibition was, however, achieved with compounds 9–13. On the basis of these results we were able to postulate an initial structure–activity relationship for 1C3 isoform inhibition. We found that the presence of the hydroxy group at the meta position on the B ring (R4 = OH, compounds 9–13) was essential for selective AKR1C3 inhibition. However, if the hydroxyl group was transformed into an ethoxy (R4 = OEt, compound 14) or butoxy (R4 = OBu, compound 15) moiety, the inhibition shifted towards AKR1C1–C2 isoforms, whereas inhibition of AKR1C3 was almost completely lost. The importance of the hydroxyl group was also confirmed when we compared the inhibitory activities of compounds 2 (3.4% AKR1C3 inhibition) and 3 (3.5% AKR1C3 inhibition), bearing a NO2 group at the meta position on B ring (R4 = NO2), with their corresponding hydroxy analogs 10 (IC50 = 2.2 µM) and 11 (IC50 = 5.2 µM). The same pattern was observed when inhibitory potency of compounds 5 and 6 were compared to the inhibitory potency of compounds 9 and 11. The latter two compounds possessed a OH group (R4 = OH) instead of a NH2 moiety at the meta position of B ring (R4 = NH2); again this substitution led to increased AKR1C3 inhibitory potency (Table 1). We also found that the substitution on the anthranilic acid (A) ring does not play a significant role in AKR1C3 inhibition as the inhibitory potencies of compounds 9–13 which bear variable substituents on this ring gave comparable IC50 values ranging from 1.9 to 12.7 µM. However, due to some subtle differences in the IC50 values we hypothesized that introduction of a large bromine atom to the meta position on the A ring (R1 = Br) to yield compound 13, would produce a more potent inhibitor. Compound 13 was found to have an IC50 value of 1.9 µM for AKR1C3 (Table 1). Compound 16 inhibited all three isoforms 1C1–1C3, with IC50s of 3.2, 6.5, and 7.5 µM, respectively.
Those compounds that were selective AKR1C3 inhibitors in the primary screen against 1C1–1C3 isoforms, were subjected to a confirmatory screen which included AKR1C4. These compounds showed even better inhibitory potencies (Table 1), which we assign to differences in assay procedures and conditions.16 As the IC50 values for compounds 9–13 were determined on all AKR1C isoforms, we were able to calculate the range of selectivity for the most potent AKR1C3 inhibitors. Here, the most promising compound was 13 with an IC50 value of 0.35 µM on the AKR1C3 isoform and it exhibited 286-, 180- and 86-fold selectivity for AKR1C3 in comparison with isoforms 1C1, 1C2 and 1C4, respectively (Table 1). Also, compound 10 seems very promising as it was similarly selective as compound 13. These two inhibitors of AKR1C3 are amongst the most potent selective non-steroidal inhibitors published so far. The only significantly more potent inhibitors were steroidal lactones, which were active in nanomolar concentrations. However, their selectivity over other AKR1C isoforms has not been demonstrated.17,18
To enhance our understanding of the results of enzymatic assays and to confirm our postulated SAR we used molecular docking19,20 to predict the hypothetical binding pose of compound 13 in the active site of AKR1C3 (PDB code 1S2A).21 The predicted binding pose of 13 showed several important interactions. With its carboxyl group, it was predicted to form H-bonds with the catalytic tetrad members Tyr55 and His117 (Fig. 1). Surprisingly, the other part of compound 13 (ring B) was predicted to bind to the SP3 binding pocket21,22 composed of Tyr24, Glu192, Ser221 and Tyr305 which is similar to the binding mode of indomethacin.23 The 3-hydroxy group of ring B was predicted to form H-bonds with Ser221 and the backbone nitrogen of Gln222. Additional π–π interactions were predicted to form between ring B and Tyr24. The interactions with SP3 binding pocket seem to be crucial for good AKR1C3 inhibitory activity in this series as compounds 14 and 15 with alkylated hydroxy groups exhibited lower AKR1C3 inhibition, most probably due to the loss of H-bonds with Ser221 and Gln222. It is interesting to note that this hypothetical binding pose is different from binding poses of highly related N-phenylaminobenzoate, 22 which were shown to occupy the SP1 binding pocket (formed by Ser118, Asn167, Phe306, Phe311, and Tyr319), similarly as flufenamic acid.23
Figure 1.

The predicted binding conformation of compound 13 (blue) in the active site of AKR1C3. Relevant enzyme residues are shown as green sticks, the catalytic tetrad is presented as yellow sticks, the co-crystallized indomethacin as magenta sticks and the cofactor as orange sticks. Compound 13 occupies a portion of the SP3 pocket which up until now has only been occupied by indomethacin.
To conclude, we have evaluated a series of N-benzoyl anthranilic acid derivatives for their inhibition of aldo–keto reductases AKR1C1–C4 and discovered some new selective inhibitors of AKR1C3, an important drug target. The results represent an important basis for the synthesis of next generation of AKR1C3 inhibitors. As the synthetic procedures to obtain structurally related derivatives are very straightforward, we are certain that further improvements can be achieved. Both phenyl rings (A and B) offer several possibilities for the introduction of new functionalities at different positions. The most promising way forward is to synthesize compounds which retain moieties that seem to play an important role for selective AKR1C3 inhibition and to explore the SAR of different compounds with varied substitution patterns on the anthranilic acid phenyl ring. These optimized compounds will have the potential to be further developed into drug candidates for treatment of hormone dependent and independent forms of prostate and breast cancers.
References and notes
- 1.Barski OA, Tipparaju SM, Bhatnagar A. Drug. Metab. Rev. 2008;40:553. doi: 10.1080/03602530802431439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Penning TM, Drury JE. Arch. Biochem. Biophy. 2007;464:241. doi: 10.1016/j.abb.2007.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bauman DR, Rudnick SI, Szewczuk LM, Jin Y, Gopishetty S, Penning TM. Mol. Pharmacol. 2005;67:60. doi: 10.1124/mol.104.006569. [DOI] [PubMed] [Google Scholar]
- 4.Wang S, Yang Q, Fung KM, Lin HK. Mol. Cel. Endocrinol. 2008;289:60. doi: 10.1016/j.mce.2008.04.004. [DOI] [PubMed] [Google Scholar]
- 5.Adeniji AO, Twenter BM, Byrns MC, Jin Y, Winkler JD, Penning TM. Bioorg. Med. Chem. Lett. 2011;21:1464. doi: 10.1016/j.bmcl.2011.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Desmond JC, Mountford JC, Drayson MT, Walker EA, Hewison M, Ride JP, Loung QT, Hayden RE, Vanin EF, Bunce CM. Cancer Res. 2003;63:505. [PubMed] [Google Scholar]
- 7.Dozmorov MG, Azzarelo JT, Wren JD, Fung KM, Yang Q, Davis JS, Hurst RE, Culkin DJ, Penning TM, Lin HK. BMC Cancer. 2010;10:672. doi: 10.1186/1471-2407-10-672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lewis MJ, Wiebe JP, Heathcote JG. BMC Cancer. 2004;4:27. doi: 10.1186/1471-2407-4-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hevir N, Vovk K, Pucelj MR, Rižner TL. Chem.-Biol. Interact. 2011;191:217. doi: 10.1016/j.cbi.2011.01.003. [DOI] [PubMed] [Google Scholar]
- 10.Penning TM, Talalay P. Med. Sci. 1983;80:4504. doi: 10.1073/pnas.80.14.4504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gobec S, Brožič P, Rižner TL. Bioorg. Med. Chem. Lett. 2005;15:5170. doi: 10.1016/j.bmcl.2005.08.063. [DOI] [PubMed] [Google Scholar]
- 12.Sosič I, Turk S, Sinreih M, Trošt N, Verlaine O, Amoroso A, Zervosen A, Luxen A, Joris B, Gobec S. Acta Chim. Slov. 2012;59:380. [PubMed] [Google Scholar]
- 13.Brožič P, Šmuc T, Gobec S, Rižner TL. Mol. Cell. Endocrinol. 2006;259:30. doi: 10.1016/j.mce.2006.08.001. [DOI] [PubMed] [Google Scholar]
- 14.Brožič P, Turk S, Lanišnik-Rižner T, Gobec S. Curr. Med. Chem. 2011;18:2554. doi: 10.2174/092986711795933713. [DOI] [PubMed] [Google Scholar]
- 15.Experimental procedure for the preparation of methyl 2-(3-hydroxybenzamido)-4,5-dimethoxybenzoate: To a solution of 3-hydroxybenzoic acid (138.12 mg, 1.0 mmol) in CH2Cl2 (10 mL), pyridine (99 mg, 1.25 mmol) and SOCl2 (773 mg, 6.5 mmol) were slowly added. After stirring at 45 °C for 2 h, the solvent was removed under reduced pressure. The reaction mixture was dissolved in toluene, and then methyl 2-amino-4,5-dimethoxybenzoate (264 mg, 1.25 mmol) was added. The reaction mixture was stirred at 100 °C for 3 h. After the reaction was complete (monitored by TLC), the solvent was evaporated, then an aqueous solution of Na2CO3 (10%, 10 mL) was added, and the aqueous layer was extracted with CH2Cl2 (3 × 10 mL). The combined organic phases were washed with brine (2 × 20 mL) and dried over Na2SO4. The solvent was evaporated, and the pure product was obtained by crystallization from EtOH. Yield, 71% (235 mg). Experimental procedure for the preparation of 2-(3-hydroxybenzamido)-4,5-dimethoxybenzoic acid (11): To a stirred solution of the methyl 2-(3-hydroxybenzamido)-4,5-dimethoxybenzoate (165 mg, 0.5 mmol) in dioxane/THF mixture (1:1, 2 mL), 1 M NaOH (1 mL) was added, and the reaction mixture stirred until the starting material had completely reacted (monitored by TLC). The solvent was then evaporated under reduced pressure, the residue diluted with H2O (10 mL), and washed with EtOAc (2 × 10 mL). The aqueous phase was acidified to pH 2 using 1 M HCl, and extracted with EtOAc (3 × 10 mL). The combined organic phases were washed with brine (3 × 10 mL), then dried over Na2SO4, filtered, and evaporated to dryness, to provide compound 11. Yield, 74% (117 mg).Spectroscopic data for 2-(3-hydroxybenzamido)-4,5-dimethoxybenzoic acid (11): White solid; Yield: 51% (2 steps); Rf 0.49 (CH2Cl2/MeOH/AcOH = 9/1/0.1); Mp: 245.0–248.0 °C; 1H NMR (400 MHz, DMSO-d6): δ 3.79 (s, 3H, OCH3), 3.87 (s, 3H, OCH3), 6.95–7.03 (m, 1H, Ar-H), 7.32–7.39 (m, 3H, Ar-H), 7.49 (s, 1H, Ar-H), 8.52 (s, 1H, Ar-H), 9.91 (br s, 1H, NHCO), 13.51 (br s, 1H, COOH); 13C NMR (100 MHz, DMSO-d6): δ 55.54 (2C), 102.98, 107.47, 112.83, 113.84, 117.15, 119.01, 129.95, 135.95, 136.98, 143.58, 153.25, 157.76, 164.41, 169.72; HRMS (ESI) m/z calcd for C16H15N2O5 [M+H]+ 318.0978, found 318.0969; HPLC purity: 95.54%, retention time: 14.06 min. HPLC was performed on an Agilent Eclipse C18 column (4.6 × 50 mm, 5 µm), with a flow rate of 1.0 mL/min, detection at 254 nm, and an eluent system of: A = 0.1% TFA in H2O; B = MeOH. The following gradient was applied: 0–3 min, 40% B; 3–18 min, 40% B →80% B; 18–23 min, 80% B; 23–30 min, 80% B → 40% B. The run time was 30 min, at a temperature of 25 °C.
- 16.The primary inhibition assay: Recombinant enzymes AKR1C1–AKR1C3 were prepared as described previously (13). Inhibition of the individual isoenzyme was monitored spectrophotometrically in the direction of oxidation, with 1-acenaphthenol as a substrate and NAD+ as a coenzyme. Reaction was followed by measuring the increase in NADH absorbance (ελ340 = 6220 M−1cm−1) in the absence and presence of each of the compound. The assays were carried out in a 300 µL volume that included 100 mM phosphate buffer (pH 9.0), 0.005% triton X-114 and 5% DMSO as a co-solvent. A substrate concentration of 30 µM (Km), 60 µM (Km) and 100 µM (<Km) and an enzyme concentration of 0.1 µM, 0.3 µM, and 1.5 µM were used for assays with AKR1C1, AKR1C2 and AKR1C3, respectively, in the presence of 2.3 mM NAD+. Screening was performed at 10 µM concentration of compounds. For compounds that showed more than 30% AKR1C1–AKR1C3 inhibition, IC50 values were determined. The measurements were performed on a Biotek PowerWave XS microplate readers with initial velocities calculated, and the IC50 values were determined graphically, using GraphPad Prism Version 4.00 (GraphPad Software, Inc.).The secondary inhibition assay: The potency of the compounds was determined also by measuring their ability to inhibit the NADP+ dependent oxidation of S-(+)-1,2,3,4-tetrahydro-1-naphthol (S-tetralol) catalyzed by AKR1C1–AKR1C4 enzymes. The continuous assay was conducted using a 96-well plate format and the reaction measured the appearance of NADPH fluorimetrically (Exc/Emi, 340/460 nm) on a BIOTEK Synergy 2 Multimode plate reader. The assay mixture consisted of 100 mM phosphate buffer, pH 7.0, S-tetralol (in DMSO), inhibitor (in DMSO), 200 µM NADP+, and purified recombinant enzyme to give a total volume of 200 µl, and 4% DMSO. The concentration of S-tetralol used in the assays for each AKR1C isoform was equal to the Km value for the respective enzyme. The Km value obtained for S-tetralol for AKR1C1, AKR1C2, AKR1C3 and AKR1C4 under the same experimental conditions are 8 µM, 22.5 µM, 165 µM, and 25 µM, respectively. Enzyme concentrations used for AKR1C1, AKR1C2, AKR1C3 and AKR1C4 were 111 nM, 86 nM, 95 nM and 184 nM, respectively. Initial velocity from progress curves was calculated with the GEN5 software. Inhibition data were fit using Grafit 5.0[y = (range)/(1+I/IC50)s) + background] to give the IC50 values.
- 17.Bydal P, Luu-The V, Labrie F, Poirier D. Eur. J. Med. Chem. 2009;44:632. doi: 10.1016/j.ejmech.2008.03.020. [DOI] [PubMed] [Google Scholar]
- 18.Qiu W, Zhou M, Mazumdar M, Azzi A, Ghanmi D, Luu-The V, Labrie F, Lin S-X. J. Biol. Chem. 2007;282:8368. doi: 10.1074/jbc.M606784200. [DOI] [PubMed] [Google Scholar]
- 19.For docking experiments, FlexX 3.1 (BiosolveIT GmbH) (20) was used together with the crystal structure of AKR1C3 (PDB code 1S2A (21). The active site was defined as the volume of the enzyme within 6 Å from co-crystallized ligand. The cofactor NADP+ was retained in the active site. For the base placement, Triangle Matching was used: this programme generated the maxima of 200 solutions per iteration and 200 per fragmentation. The docking procedure was validated since FlexX was able to reproduce the conformation of cocrystallized ligand.
- 20.Rarey M, Kramer B, Lengauer T, Klebe G. J. Mol. Biol. 1996;261:470. doi: 10.1006/jmbi.1996.0477. [DOI] [PubMed] [Google Scholar]
- 21.Byrns MC, Jin Y, Penning TM. J. Steroid. Biochem. Mol. Biol. 2011;125:95. doi: 10.1016/j.jsbmb.2010.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen M, Adeniji AO, Twenter BM, Winkler JD, Christianson DW, Penning TM. Bioorg. Med. Chem. Lett. 2012;22:3492. doi: 10.1016/j.bmcl.2012.03.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lovering AL, Ride JP, Bunce CM, Desmond JC, Cummings SM, White SA. Cancer Res. 1802;2004:64. doi: 10.1158/0008-5472.can-03-2847. [DOI] [PubMed] [Google Scholar]