

KSR2 is required for store-operated calcium entry (SOCE). KSR2 deficiency affects STIM1/ORAI1 puncta formation and cytoskeleton organization. In addition, KSR2-associated calcineurin is crucial for SOCE. These findings identify the KSR2-calcineurin complex to be crucial for store-dependent STIM1-ORAI1 dynamics.
Abstract
Store-operated calcium entry (SOCE) is the predominant Ca2+ entry mechanism in nonexcitable cells and controls a variety of physiological and pathological processes. Although significant progress has been made in identifying the components required for SOCE, the molecular mechanisms underlying it are elusive. The present study provides evidence for a direct involvement of kinase suppressor of Ras 2 (KSR2) in SOCE. Using lymphocytes and fibroblasts from ksr2−/− mice and shKSR2-depleted cells, we find that KSR2 is critical for the elevation of cytosolic Ca2+ concentration. Specifically, our results show that although it is dispensable for Ca2+-store depletion, KSR2 is required for optimal calcium entry. We observe that KSR2 deficiency affects stromal interaction molecule 1 (STIM1)/ORAI1 puncta formation, which is correlated with cytoskeleton disorganization. Of interest, we find that KSR2-associated calcineurin is crucial for SOCE. Blocking calcineurin activity impairs STIM1/ORAI1 puncta-like formation and cytoskeleton organization. In addition, we observe that calcineurin activity and its role in SOCE are both KSR2 dependent.
INTRODUCTION
Transient elevation of cytosolic Ca2+ is a crucial event in the initiation and regulation of many biochemical and physiological cell processes (Dolmetsch et al., 1998; Carafoli, 2003). This elevation is triggered by its release from the endoplasmic reticulum (ER). This event also triggers an inflow of Ca2+ across the plasma membrane (Lewis, 2007). The mechanism by which this store-operated Ca2+ entry (SOCE) occurs has been the subject of investigations over many years (Parekh and Putney, 2005). The importance of such Ca2+ entry is also underlined by the fact that SOCE is one of the most important and ubiquitous mechanisms for refilling cytoplasmic Ca2+ stores and provides a major route for receptor-stimulated Ca2+ entry in nonexcitable cells (Parekh and Putney, 2005; Putney, 2007; Feske, 2007).
SOCE is orchestrated by different proteins. Among these, stromal interaction molecule 1 (STIM1; Liou et al., 2007) is a key regulator that resides in ER membrane. It contains an EF-end domain that senses Ca2+ concentration in the lumen of ER and, upon store depletion of Ca2+, oligomerizes (Liou et al., 2007) and relocates to distinct puncta at ER–plasma membrane (PM) junctions (Wu et al., 2007; Luik et al., 2008; Várnai et al., 2009), where it activates Ca2+ channels (Park et al., 2009). The pore-forming subunit of these PM channels is represented by Orai1 (Prakriya et al., 2006; Hogan and Rao, 2007), as confirmed by mutational and electrophysiological analysis (Yeromin et al., 2006). In addition to Orai1, transient receptor potential channel 1 (TRPC1), a member of the TRPC family of channels, has also been reported to be regulated by store depletion and interact with STIM1 to activate SOCE (Singh et al., 2000).
SOCE is regulated primarily by ER Ca2+-store levels. However, the activity of both STIM1 and Orai1 proteins can be regulated by different cellular mechanisms. Orai1 activity is regulated by protein kinase C–mediated phosphorylation on serine residues, which induces SOCE suppression (Kawasaki et al., 2010). Moreover, STIM1 is regulated by Ser/Thr phosphorylation (Rosado et al., 2001; Pozo-Guisado et al., 2010; Smyth and Putney, 2012) and Tyr phosphorylation (Lopez et al., 2012). In addition, recent work has provided evidence for the involvement of the cytoskeleton in SOCE activation (Smyth et al., 2007, 2010; Grigoriev et al., 2008; Vaca, 2010).
Other molecules seemingly involved are “scaffold” proteins, which provide a template to allow these proteins to assemble in the correct manner (Pawson and Scott, 1997; Matza et al., 2008; Jardin et al., 2012; Soundararajan et al., 2012).
An important scaffold known to regulate the extracellular signal-regulated kinase (ERK) signaling cascade is the kinase suppressor of Ras (KSR) family. The best-characterized member of this family is KSR1, which promotes activation of the Raf/MEK/ERK kinase cascade (Kornfeld et al., 1995; Sundaram and Han, 1995; Therrien et al., 1995; Kortum and Lewis, 2004). Similar to KSR-1, the scaffold KSR-2 can interact with a number of signaling components of the Ras/mitogen-activated protein kinase pathway, including Ras, RAF-1, MEK-1, and ERK-1/2 (Ohmachi et al., 2002), and kinase and phosphatase proteins (Costanzo-Garvey et al., 2009; Dougherty et al., 2009; Liu et al., 2009; Fernandez et al., 2012) involved in ubiquitin–proteasome, apoptosis, insulin signaling, cell cycle control, and microtubule association (Lin et al., 2009). Owing to the presence of additional 63 amino acids between CA2 and CA3 domains, KSR2 interacts with the Ser/Thr protein phosphatase calcineurin (CN). Due to this interaction, KSR2 activates Ca2+-mediated ERK signaling (Dougherty et al., 2009). Moreover, in response to cell stimulation, both KSR2 and CN are recruited at the plasma membrane (Dougherty et al., 2009; Li et al., 2011). However, the nature of the involvement of KSR2 in store-operated Ca2+ entry remains unknown.
The availability of mice deficient in KSR2 provides a unique system with which to investigate further the role of KSR2 in cell signaling. Here we report such an investigation. Our experiments provide evidence for a novel mechanism for the regulation of Ca2+ signaling through KSR2-mediated dynamic assembly of a SOCE regulatory complex.
RESULTS
Intracellular Ca2+ elevation is defective in KSR2-knockout cells
To determine the effect of KSRs deficiency on Ca2+ signaling, we monitored cytosolic Ca2+ concentration ([Ca2+]i) in nonexcitable cells (including T and B-cells) from wild-type (wt), KSR1-knockout (ksr1−/−), and KSR2-knockout (ksr2−/−) mice in response to immunoreceptor stimulation. First, we found that, in addition to expressing KSR1 (Nguyen et al., 2002), purified mouse T- and B-cells from spleen also expressed the KSR2 protein (Figure 1A). Purified T-cells from wt, ksr1−/−, and ksr2−/− mice (Supplemental Figure S1) were stimulated with anti-CD3 antibody (2C11), followed by anti–immunoglobulin G (IgG) antibody to induce receptor cross-linking. As shown in Figure 1B, in the presence of extracellular Ca2+, the [Ca2+]i elevation in KSR1-deficient T-cells (Figure 1B, green line) was similar to that of wt cells (Figure 1B, black line). Of interest, KSR2-deficient T-cells (Figure 1B, red line) exhibited a significant reduction in Ca2+ rise compared with wt cells, indicating that, in contrast to KSR1, scaffold KSR2 may be involved in the intracellular Ca2+ elevation. Similar results were observed in purified B-cells stimulated with anti-IgM (Figure 1C). In particular, B-cell receptor (BCR) cross-linking–induced intracellular Ca2+ elevation was reduced in B cells from ksr2−/−, whereas we did not observe a significant difference between wt and ksr1−/− B cells. These data indicate that, in contrast to KSR1, KSR2 is involved in intracellular Ca2+ rise in both T- and B-lymphocytes.
FIGURE 1:
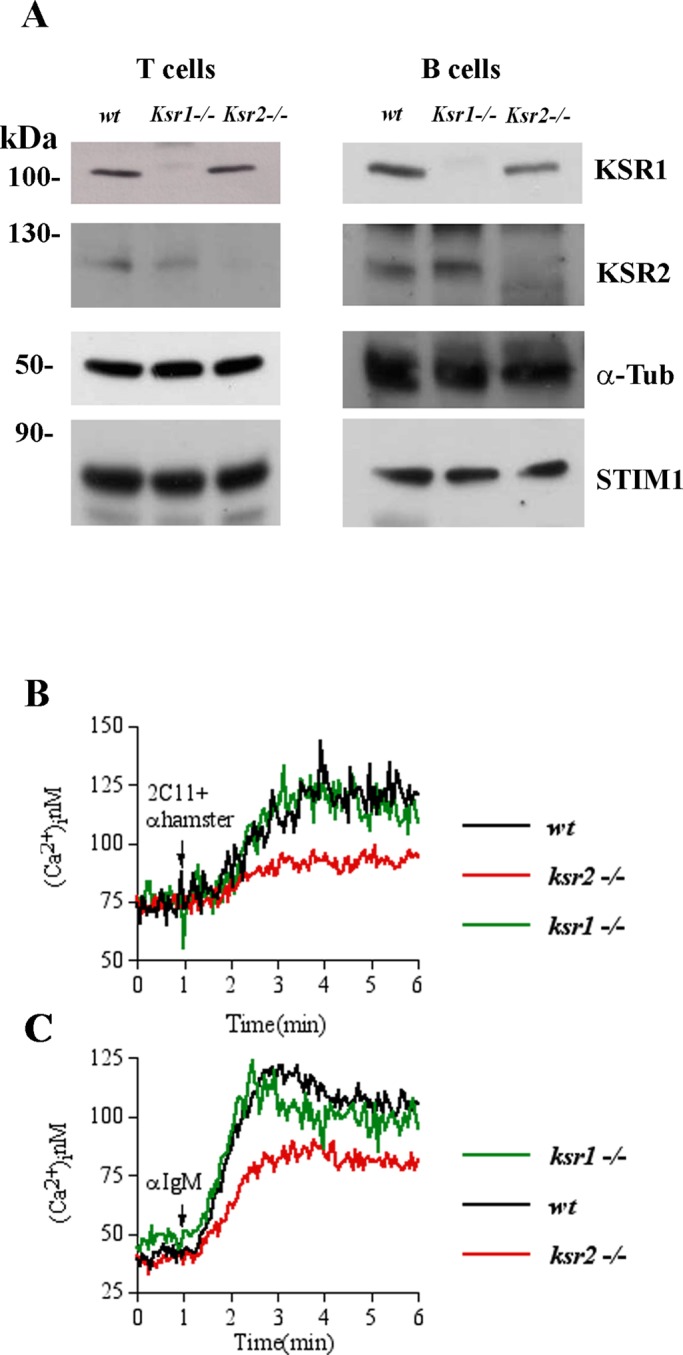
Defective [Ca2+]i elevation in ksr2−/− lymphocytes. (A) Immunoblot analysis of KSR1, KSR2, and STIM1 protein expression in purified T- and B-lymphocytes (15 × 106 cells/condition) from wt, ksr1−/−, and ksr2−/− mice. α-Tubulin (α-Tub) was used as loading control. Left, relative molecular mass (kilodaltons). (B) Elevation of [Ca2+]i induced by anti-CD3 (2C11) followed by anti-IgG antibody (anti-hamster) stimulation was assessed by spectrofluorimetric analysis with Fura-2 in 3 × 106 T-cells purified from wt (black line), ksr1−/− (green line), and ksr2−/− mice (red line). Cells were kept in 1.0 mM Ca2+-containing medium. (C) Elevation of [Ca2+]i induced by anti-IgM stimulation performed in 3 × 106 purified B-cells, as described in B. Traces represent one of three independent experiments.
KSR2 is required for store-operated Ca2+ influx
Because elevation of intracellular Ca2+ concentration is due to two distinct events—intracellular store depletion and extracellular Ca2+ influx—we sought to assess which phase was affected in KSR2-deficient cells. To address this point, we analyzed cytosolic Ca2+ elevation in purified wt and ksr2−/− T-cells in response to stimulation with thapsigargin (Tg; a sarco endoplasmic reticulum Ca2+-ATPase inhibitor) in Ca2+-free conditions (0.2 mM ethylene glycol tetraacetic acid [EGTA]), followed by Ca2+ influx induced by restoration of the extracellular Ca2+ concentration to 1.2 mM. As shown in Figure 2A (left), after Ca2+-store depletion with Tg, wt and ksr2−/− T-cells responded to the same extent. Surprisingly, when extracellular Ca2+ was added, ksr2−/− T-cells showed a significant reduction of Ca2+ influx compared with the fast and wide influx in wt cells. Similar results were observed in purified ksr2−/− B cells (Figure 2A, right). To examine the effect of KSR2 on SOCE induced by a physiological agonist, purified T- and B-cells were stimulated with anti-CD3 antibody or anti-IgM, respectively. As shown in Figure 2B, although store depletion was not affected, the agonist-induced calcium influx was reduced in T-cells (left) and B-cells (right) from ksr2−/− mice. A magnification of the Tg- or agonist-induced response in the absence of external calcium showed that Ca2+-store depletion was not affected (Supplemental Figure S2). Similar results were obtained using the Ca2+ ionophore ionomycin (unpublished data). In addition, to determine the effect of KSR2 deficiency on agonist-induced calcium release–activated channel (CRAC)–mediated Ca2+ influx, we used the Mn2+-quenching approach (Hirata et al., 2006). Quenching of intracellular Fura-2 fluorescence by the entry of extracellular Mn2+ (0.1 mM) via store-operated Ca2+ channel showed that, in contrast to wt cells, the progressive quenching of Fura-2 fluorescence was slowed in ksr2−/− T- and B-cells (Figure 2C), supporting a role for KSR2 in agonist-induced CRAC-mediated Ca2+ entry. The effect of KSR2 on SOCE was not due to altered expression of ORAI1-3 and STIM1-2, as no difference in their mRNA expression was found in purified T- and B-cells from wt and ksr2−/− (unpublished data). Taken together, these data indicate that whereas in the absence of KSR2, ER Ca2+ content is not altered and physiological agonist-induced calcium release is not affected, KSR2 is necessary for optimal Ca2+ influx, induced by store depletion in both T- and B-cells.
FIGURE 2:

KSR2 is required for SOCE. T- and B-lymphocytes (3 × 106 cells) purified from wt (black line) or ksr2−/− (red line) mice were loaded with Fura-2. The experiments were done in a spectrofluorimeter cuvette in a nominally Ca2+-free medium containing 0.2 mM EGTA. (A) Intracellular Ca2+ stores were depleted with 0.2 μM thapsigargin (Tg); subsequently 1.2 mM Ca2+ was added to the medium to reveal Ca2+ influx. (B) Elevation of [Ca2+]i was induced by anti-CD3 (2C11) followed by anti-IgG antibody (anti-hamster) or anti-IgM stimulation for T- and B-lymphocytes, respectively. (C) Store-dependent Ca2+ influx was also evaluated from the rate of Fura-2 fluorescence quenching by Mn2+ (see Materials and Methods). Fura-2–loaded cells were resuspended in cuvette containing Ca2+-free medium and treated or not with agonists (2C11 + anti-hamster or anti-IgM for T- and B-cells, respectively). After 4 min, 0.1 mM MnCl2 was added. Data (shown after MnCl2 addition) were normalized as percentage of fluorescence values obtained immediately before addition of MnCl2. Traces represent one of three independent experiments.
Direct role of KSR2 in SOCE
To further investigate the role of KSR2 in modulating [Ca2+]i, we measured SOCE in HEK 293T cells upon short hairpin RNA (shRNA)–mediated depletion of KSR2 or KSR1 (Figure 3, A–C). As shown, SOCE was reduced in KSR2-depleted cells, whereas no significant effect was observed in shRNA KSR1 cells. As observed in T- and B-cells from wt and ksr2−/− mice, KSR2 suppression had no effect on ER Ca2+-store depletion. Similar results were obtained in HeLa cells, for which KSR2 suppression significantly reduced Tg-induced Ca2+ influx (Figure 3, D and F) without any effects on Ca2+-store depletion (Supplemental Figure S3). To prove a direct role of KSR2 on SOCE, we ectopically reexpressed KSR2 in HeLa cells stably depleted of KSR2 by shRNA. Expression of a low level of KSR2 (Figure 3E) restored Tg-induced Ca2+ influx (Figure 3F), indicating a direct role of KSR2 in SOCE. KSR2 depletion by shRNA did not alter the mRNA levels of ORAI1-3 and STIM1-2, indicating that reduction of SOCE was not due to defective expression of its essential components (unpublished data).
FIGURE 3:

KSR2 depletion reduces SOCE. Immunoblot analysis showing the reduction of KSR2 (A) or KSR1 (B) in lysates from shRNA KSR2 HEK-293T, and shRNA control HEK-293T (Scr) cells. α-Tubulin (α-Tub) was used as loading control. (C) Spectrofluorimeter analysis of Ca2+ influx in indicated shRNA HEK-293T cells loaded with Fura-2. Ca2+ stores were depleted with 0.2 μM Tg (in Ca2+-free medium), and subsequently 1.2 mM Ca2+ was added to the medium to reveal Ca2+ influx. (D) Immunoblot analysis of KSR2 and STIM1 in lysates from shRNA KSR2 HeLa and shRNA control HeLa (Scr) cells. (E) Immunoblot analysis of KSR2 and STIM1in lysates from shRNA control HeLa (Scr) transfected with 0.2 μg of Flag or shRNA KSR2 HeLa transfected with 0.2 μg of KSR2-Flag. Lysates were blotted for KSR2 and STIM1. (F) Spectrofluorimeter analysis of Ca2+ influx in indicated shRNA HeLa cells loaded with Fura-2. Ca2+ influx was analyzed as in C. (G) Immunoblot analysis showing the presence of KSR2 in lysates from HEK-293T cells transfected as indicated. Lysates were blotted for KSR2. ERK1/2 was used as loading control. (H) Spectrofluorimeter analysis of 1 × 106 HEK-293T cells transfected with expression vectors encoding KSR2-Flag at different doses (0.2, 0.4, 0.8 μg; top) or Flag alone as control (bottom). Elevation of [Ca2+]i was measured with Fura-2 as described in C. Traces represent one of three independent experiments.
As is typical for scaffold molecules, KSR proteins levels need to be carefully regulated and kept below a certain limit in order for the proteins to properly carry out their functions (Cacace et al., 1999). To further investigate the role of KSR2 in modulating [Ca2+]i, we analyzed the effect of different expression levels of KSR2 on Ca2+ influx. To address this issue, we transfected HEK 293T cells naturally expressing KSR2 with three different doses of the expression vector encoding the KSR2-Flag fusion protein or Flag alone (as control; Figure 3G) and measured Ca2+ influx by fluorimetry. As shown in Figure 3H, although ectopic expression of a low level of KSR2 (0.2 μg) did not alter any phase of [Ca2+]i elevation (Figure 3H, black line) compared with Flag-transfected control cells (Figure 3H, bottom), cells transfected with 0.4 μg (Figure 3H, green line) or 0.8 μg (Figure 3H, red line) of KSR2 expression construct showed a dose-dependent decrease in Ca2+ influx. No effect on [Ca2+]i elevation was observed when different doses of Flag alone (as control) were expressed. Of note, Tg-induced Ca2+ release from the ER was independent of KSR2 expression level, further indicating a specific role of KSR2 in SOCE.
Taken together, these results indicate that, although store Ca2+ depletion was not affected, KSR2 suppression strongly impaired the influx of extracellular Ca2+. Furthermore, KSR2 reexpression at low level in KSR2-depleted cells restored SOCE. Of note, the KSR2 effect on SOCE was not due to altered expression of STIM1. In addition, KSR2 overexpression reduced SOCE in a dose-dependent manner, suggesting direct involvement of KSR2 in store-operated Ca2+ entry.
KSR2 is required for STIM1/ORAI1 puncta formation
Because the defect of SOCE in KSR2-deficient cells does not involve intracellular Ca2+-store depletion, we focused our attention on the extracellular Ca2+ influx phase. It is known that, upon store depletion, STIM1 proteins sense the decrease in ER Ca2+ levels and relocalize with ORAI1 proteins into puncta closely associated with the plasma membrane (Liou et al., 2005; Parekh and Putney, 2005; Frischauf et al., 2008). Because the STIM1/ORAI1 association into puncta at ER-PM junctions is strictly required for optimal Ca2+ influx (Muik et al., 2008), we investigated whether KSR2 suppression might affect STIM1/ORAI1 puncta formation. HeLa stably depleted of KSR2 (sh KSR2) and control shRNA HeLa cells (sh Scr) were cotransfected with expression vectors encoding yellow fluorescent protein (YFP)–STIM1 and ORAI1–red fluorescent protein (RFP) fusion proteins. After 24 h, cells were stimulated either without (basal) or with Tg for 10 min. As shown in Figure 4, A and E, Tg-mediated Ca2+-store depletion induced aggregation of STIM1 with ORAI1 at the plasma membrane in control shRNA cells (yellow dots). In contrast, STIM1/ORAI1 association was severely impaired in shRNA KSR2 HeLa cells, even though Tg induced STIM1 clustering (Figure 4, C and E). These data were also confirmed by immunoprecipitation experiments. ORAI1 immunoprecipitates were prepared from shRNA control and shRNA KSR2 HeLa cells cotransfected with the expression constructs just described. We found that STIM1 coimmunoprecipitated with ORAI1 after Tg stimulation and that association was partially reduced in shRNA KSR2 HeLa cells (Supplemental Figure S4A).
FIGURE 4:

STIM1-ORAI1 puncta-like formation is impaired in KSR2-depleted HeLa cells. (A–C) ShRNA control and shRNA KSR2 HeLa cells were grown on coverslips and cotransfected with expression vectors encoding YFP-STIM1 (0.2 μg) and ORAI1-RFP (0.2 μg). After starvation for 2 h, cells were stimulated either without (basal) or with 1 μM Tg. Distribution of Tg-induced STIM1-ORAI1 puncta formation (yellow dots) was visualized under confocal microscopy. Bar, 5 μm. (B–D) shRNA KSR2–depleted HeLa cells were cotransfected with expression vectors encoding YFP-STIM1 (0.2 μg) and ORAI1-RFP (0.2 μg) and Flag alone 0.2 μg (B) or KSR2-Flag 0.2 μg (D). After starvation, cells were stimulated as in A–C. Cells were fixed, permeabilized, and incubated with anti-Flag antibody, followed by Alexa Fluor 647–labeled secondary antibody. Tg-induced STIM1-ORAI1 puncta formation (yellow dots) was visualized under confocal microscopy. Bar, 5 μm. One representative cell of >30 cells in three experiments. White squares show enlarged images. Bar, 1 μm. The graph bars illustrate the mean per cell ± SEM of puncta in response to Tg treatment in >30 cells/condition. Significance of values is compared with control Tg-treated cells. *p < 0.05.
To test the direct involvement of KSR2 in the puncta-like structures formation, we reconstituted KSR2-Flag (or Flag alone as control) in shRNA KSR2 HeLa cells cotransfected with the expression constructs encoding the YFP-STIM1 and ORAI1-RFP fusion proteins. As shown in Figure 4, D and F, reexpression of KSR2 at low level (0.2 μg) resulted in restoration of the ability of the cells to form puncta after Tg stimulation.
These data were confirmed in COS-7 cells that lack endogenous KSR2 (Dougherty et al., 2009; Supplemental Figure S4B). COS-7 cells were transfected with the expression construct encoding STIM1 and ORAI1 fusion proteins and Flag (0.2 μg) as control, and puncta formation was analyzed as described. Although Tg induced STIM1 clustering, STIM1/ORAI1 association was severely impaired (Figure 5, A and C). Reexpression of low level of KSR2 in COS-7 cells cotransfected with the expression constructs encoding the YFP-STIM1 and ORAI1-RFP fusion proteins resulted in restoration of the ability of the cells to form puncta after Tg stimulation (Figure 5, B and C). We observed that KSR2 expression alone at low level was able to increase SOCE in KSR2-deficient COS-7 cells compared with high KSR2 level (Supplemental Figure S4C), suggesting that a positive effect of KSR2 on SOCE could be dependent on a defined stoichiometry between KSR2 and endogenous ORAI1 and STIM1. Pertinent to this point, expression of low level of KSR2 (0.2 μg) with the expression constructs encoding the YFP-STIM1 and ORAI1-RFP fusion proteins significantly increased calcium influx compared with Flag-transfected control cells without any particular effect on Tg-induced store-Ca2+ release (Figure 5D, green line). In contrast, reexpression of high levels of KSR2 (0.8 μg) in cotransfected COS-7 cells impaired Tg-induced calcium influx (Figure 5D, red line), suggesting that a defined stoichiometry between KSR2, Orai1, and STIM1 is required for optimal SOCE.
FIGURE 5:

KSR2 is required for puncta formation and colocalizes with STIM1-ORAI1. (A, B) COS-7 cells were cotransfected with expression vectors encoding YFP-STIM1 (0.2 μg), ORAI1-RFP (0.2 μg), and 0.2 μg of Flag alone (A) or 0.2 μg of KSR2-Flag (B). After starvation, cells were either stimulated without (basal) or with 1 μM Tg. Cells were fixed, permeabilized, and incubated with anti-Flag antibody, followed by Alexa Fluor 647–labeled secondary antibody. Tg-induced STIM1-ORAI1 puncta formation (yellow dots) was visualized under confocal microscopy. Bar, 5 μm. One representative cell of >30 cells in three experiments. White squares show enlarged images. The colocalization of KSR2 with puncta (as indicated by white arrows) was evaluated by performing fluorescence intensity profiles along a straight line where fluorescent signals were colocalized. Bar, 1 μm. (C) The graph bars illustrate the mean per cell ± SEM of puncta in response to Tg treatment in >30 cells/condition. Significance of values is compared with control Tg-treated cells. *p < 0.05. (D) Intracellular Ca2+ concentration was monitored by video imaging experiments in COS-7 cells transfected with STIM1 and ORAI1 (black dotted line) as in B or cotransfected with KSR2-Flag 0.2 μg (low, green line) or KSR2-Flag 0.8 μg (high, red line). As control, COS-7 cells were cotransfected with Flag alone (black line). Ca2+ stores were depleted with 0.5 μM Tg in the absence of extracellular Ca2+ and the presence of 0.5 mM EGTA. Subsequently the medium was changed with one that contains 1.5 mM Ca2+ to reveal Ca2+ influx. Each trace represents the average response of all the fluorescent cells on a single coverslip. (E) The graph bars illustrate the percentage of KSR2 colocalized with STIM1-ORAI1 puncta per cell ± SEM in response to Tg treatment in >30 cells/condition. Significance of values is compared with control Tg-treated cells. *p < 0.05. (F) Western blot illustrating coimmunoprecipitation of KSR2 with STIM1-ORAI1 puncta. COS-7 cells were cotransfected as described in B and stimulated with 1 μM Tg at the indicated times (in minutes). ORAI1 immunoprecipitates (IPs) were prepared and analyzed by immunoblotting using anti-KSR2. The membrane was then stripped and reprobed with anti-STIM1 and then anti-ORAI1. One representative experiment of three is shown. Left, relative molecular mass (kilodaltons). (G) Inverse coimmunoprecipitation of KSR2 with STIM1-ORAI1. COS-7 cells were cotransfected and stimulated as described in B. KSR2 IPs were prepared and analyzed by immunoblotting as in F. One representative experiment of three is shown.
Of interest, KSR2 was found to colocalize with several STIM1/ORAI1 puncta (Figure 5B, white arrows). The association of KSR2 with STIM1-ORAI1 complexes was confirmed by immunoprecipitation experiments. ORAI1 immunoprecipitates were prepared from COS-7 cells cotransfected with expression constructs as described. We found that, after Tg stimulation, KSR2 coimmunoprecipitated with ORAI1 and STIM1 (Figure 5F). These data were confirmed with reverse coimmunoprecipitations (using anti KSR2 antibodies; Figure 5G). In particular, KSR2 coassociated with STIM1-ORAI1 puncta after Tg-induced store depletion and was not preassociated with STIM1 and Orai1.
Taken together, these data demonstrate direct involvement of KSR2 in store-operated Ca2+ influx, as its expression is required for proper formation of STIM1/ORAI1 puncta and Ca2+ influx.
Calcineurin is involved in SOCE, and its role is related to KSR2 expression
It has been reported that KSR2 contains a unique 63–amino acid sequence located between the CA2 and CA3 domains (Dougherty et al., 2009). Given that several of the KSR2-specific binding partners are proteins known to be involved in Ca2+ signaling (Dougherty et al., 2009), we reasoned that insights into KSR2’s role in SOCE might be gained by investigating KSR2 binding partners. Because Ca2+/calmodulin-regulated Ser/Thr phosphatase calcineurin (CN) is the most abundant of KSR2-specific interactors (Dougherty et al., 2009), we decided to test the hypothesis that CN may have a role in SOCE. To address this point, we treated HeLa cells with cypermethrin (Cyper; a specific calcineurin inhibitor) and analyzed Ca2+ influx. As shown in Figure 6A, whereas Tg-induced Ca2+ depletion was not affected, Cyper pretreatment greatly reduced Ca2+ influx, suggesting that CN is required for SOCE. Similar results were obtained after inhibition with another common CN inhibitor, cyclosporin A (CsA). As shown in Supplemental Figure S5, A and B, in cells expressing KSR2 (HeLa and Jurkat cells), CsA treatment at low concentrations (such as 1 μM) induced a remarkable decrease in Ca2+ influx without affecting Ca2+ release from intracellular Ca2+ stores.
FIGURE 6:

SOCE is reduced by Cyper pretreatment only in KSR2-expressing cells. Left: (A) HeLa cells loaded with Fura-2 were analyzed by spectrofluorimeter as described in Figure 3F. Black and red traces represent, respectively, cells pretreated without or with 5 μM Cyper for 15 min at 37°C. The traces show a representative experiments that was repeated at least in triplicate. (B) Elevation of [Ca2+]i was measured in shRNA KSR2 HeLa as described in A. (C) Analysis of SOCE in COS-7 cells loaded with Fura-2 as described in A. Right: Cells were pretreated for 15 min at 37°C with the indicated Cyper concentrations, and peak values of Tg-dependent Ca2+ entry after Ca2+ readmission were calculated. Percentage of residual Ca2+ influx after Cyper treatment compared with maximal Ca2+ peak of their respective vehicle-pretreated controls. Values are mean ± SEM of at least three independent experiments.
Of interest, the inhibitory effect of Cyper on SOCE was less prominent in shRNA KSR2 HeLa cells (Figure 6B), suggesting that scaffold KSR2 is necessary for Cyper-mediated SOCE inhibition. Because KSR2 was not completely suppressed in shRNA KSR2 cells, we tested the effect of CN inhibition on SOCE in KSR2-deficient COS-7 cells. As shown in Figure 6C, Cyper-mediated CN inhibition did not seem to have an inhibitory effect on Ca2+ influx, even at high concentrations (10 μM), indicating that the role of CN on Ca2+ influx is correlated with KSR2 expression. These data were confirmed in COS-7 cells treated with CsA (Supplemental Figure S5C), indicating that CN activity is required in store-operated calcium entry and that its role in Ca2+ influx is correlated with KSR2 expression.
Calcineurin-induced nuclear translocation of nuclear factor of activated T-cells is impaired in ksr2−/−
It was reported that CN is required for KSR2 dephosphorylation and contributes to Ca2+-mediated ERK activation (Dougherty et al., 2009). However, whether KSR2 affects CN activity remain to be established. Our data show that KSR2 is required for CsA-mediated SOCE inhibition, suggesting that the scaffold KSR2 might regulate CN activity. Given that nuclear factor of activated T-cells (NFAT) nuclear translocation is dependent on dephosphorylation of the NFAT-regulatory domain by CN, nuclear translocation of green fluorescent protein (GFP)–NFAT expressed in fibroblasts from wt and ksr2−/− was used to determine the role of KSR2 in CN activation. As shown in Figure 7, Tg-dependent Ca2+-store depletion induced NFAT translocation into the nucleus in ∼85% of wt cells. Of importance, in the absence of KSR2, the percentage of cells with nuclear translocation of NFAT was drastically reduced after Tg-induced elevation of [Ca2+]i, indicating that KSR2 is required for CN-mediated NFAT nuclear translocation.
FIGURE 7:
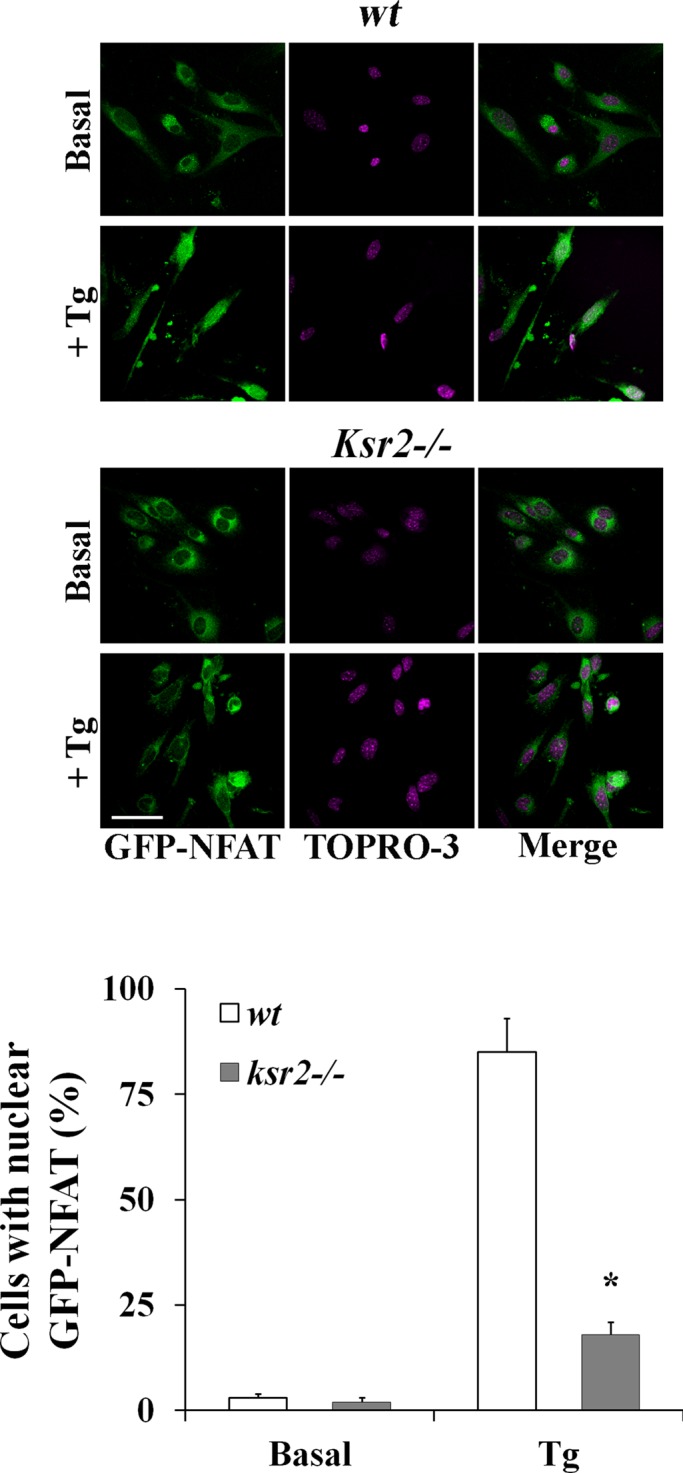
KSR2 is required for CN activation. (A) Confocal microscopy of NFAT nuclear translocation in wt (top) and ksr2−/− (bottom) fibroblasts expressing GFP-NFAT (green) and stimulated without (basal) or with 1 μM Tg for 30 min followed by TO-PRO-3 staining of the nucleus (purple). The merge of NFAT and TO-PRO-3 staining (right) shows the NFAT translocation into the nucleus in wt cells. Representative cells for each condition of three experiments is shown. Quantitative analysis (mean ± SD) of wt and ksr2−/− cells with nuclear GFP-NFAT before and after stimulation with Tg from three experiments is shown in the graph bars. *p < 0.01. Bar, 5 μm.
Calcineurin inhibition affects Tg-induced STIM-ORAI puncta formation
The role of CN and KSR2 in STIM-ORAI puncta formation had not been assessed. We therefore tested the effect of CN inhibition on STIM-ORAI puncta formation, using the KSR2-expressing cell line HeLa. Cells were cotransfected with the expression constructs encoding YFP-STIM1 and ORAI1-RFP fusion proteins. Transfectants were pretreated or not with 10 μM Cyper or CsA and then stimulated either without (basal) or with Tg. As shown in Figure 8, Tg-mediated Ca2+-store depletion induced aggregation of STIM1 with ORAI1 into puncta-like structures (yellow dots). Of interest, STIM1-ORAI1 puncta formation was impaired after CN inhibition, with the mean number of puncta per cell significantly different from non-pretreated cells stimulated with Tg. These results indicate that CN activity is necessary for STIM1-ORAI puncta formation.
FIGURE 8:
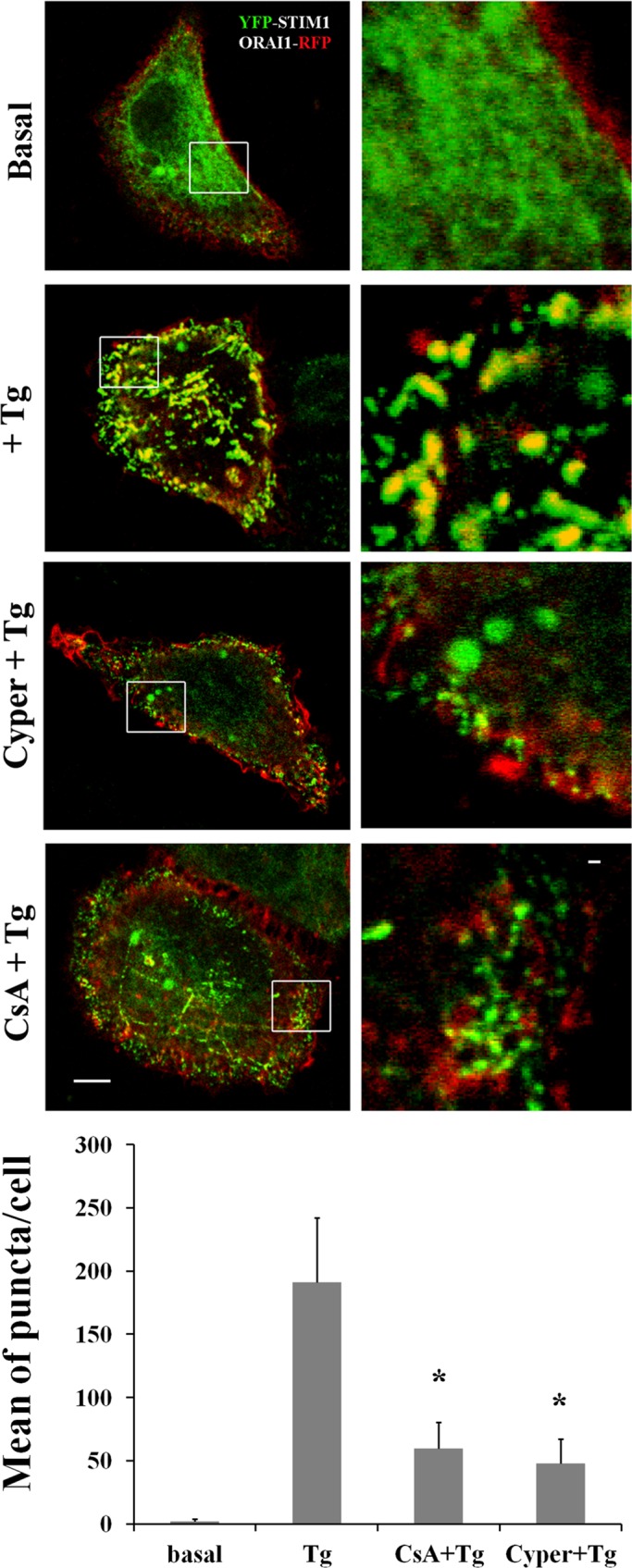
Calcineurin inhibition reduces STIM-ORAI puncta formation. Representative images of HeLa cells coexpressing YFP-STIM1 (0.2 μg) and ORAI1-RFP (0.2 μg). Cells were starved overnight and then stimulated without (basal) or with 1 μM Tg (+Tg) and pretreated or not with 10 μM Cyper or CsA. Bar, 5 μM. White squares show enlarged images. Bar, 1 μM. The quantification of puncta, expressed as mean per cell ± SEM, is based on the number of cells indicated in parentheses: basal (20); Tg (25); CsA+Tg (28); Cyper+Tg (26). The number of puncta formed in response to Tg treatment and the effect of inhibition of CN are shown in the graph bars. Significance of values is compared with control Tg-treated cells. *p < 0.01.
KSR2-deficient cells have altered cytoskeleton organization
We demonstrated that KSR2 is required for puncta formation, which is necessary for Ca2+ influx after intracellular Ca2+-store depletion. The question arises as to the mechanism underlying the role of KSR2 in puncta formation. On the basis of the strong impairment of STIM1-ORAI1 puncta-like structure in the absence of KSR2, we focused our attention on possible mechanisms that mediate STIM1/ORAI1 association. Among these, cytoskeleton rearrangements appear to play a key role in allowing STIM1 to associate with ORAI1 (Galán et al., 2011). Thus we hypothesized that in the absence of KSR2 the cytoskeleton lacks either the organization or the plasticity required to allow puncta formation. To address this point, we compared the organization of F-actin and α-tubulin filaments in fibroblasts from wt and ksr2−/− mice both in resting conditions and after stimulation with Tg (Supplemental Figure S6). To visualize the two key components of the cytoskeleton, F-actin and α-tubulin, at steady state, we incubated wt and ksr2−/− mice fibroblasts in low-serum–containing medium and stained with anti–α-tubulin and phalloidin. Although the intensity of F-actin and α-tubulin was very similar in ksr2−/− and wt cells, the organization and the distribution of cytoskeleton components at steady state were very different between wt and ksr2−/− cells. As shown in Supplemental Figure S6, actin stress fibers and tubulin filaments were formed within the cell body, which indicated correct integrity for both cytoskeletal components in wt cells. In contrast, ksr2−/− fibroblasts contained fewer actin stress fibers and tubulin filaments, which appear to be disassembled. After stimulation, we observed an increase in the number of actin stress fibers and well-organized tubulin filaments in wt cells. On the other hand, the organization of F-actin fibers and tubulin filaments was severely affected in the absence of KSR2. These data indicate that KSR2 deficiency affects the cytoskeleton rearrangements that are required for SOCE. Given that the Ser/Thr phosphatase CN is involved in the regulation and structural plasticity of the cytoskeleton (Faul et al., 2008; Kume et al., 2011), we decided to investigate the effect of CN inhibition on cytoskeleton organization. We observed that treatment of wt fibroblasts with 10 μM CsA severely impaired the organization of F-actin fibers and α-tubulin filaments after Tg stimulation (Supplemental Figure S6), indicating that CN activity is necessary for the organization and rearrangement of the cytoskeleton.
DISCUSSION
In this study, we identified the scaffold molecule KSR2 as a new player in store-operated Ca2+ entry in nonexcitable cells. Although common functions and regulatory mechanisms have been identified for the scaffold KSR proteins (Dougherty et al., 2009), we define here a specific function for KSR2 in SOCE. Using lymphocytes from ksr1−/− or ksr2−/− mice and shRNA-mediated KSR2 depleted cells, we find that, in contrast to KSR1, the scaffold KSR2 plays a role in elevation of [Ca2+]i after stimulation (Figures 1–3). The increase in cytosolic Ca2+ is due to two distinct mechanisms: Ca2+ release from ER stores and extracellular Ca2+ influx across the plasma membrane. Of interest, we find that KSR2 is required for optimal Ca2+ influx, but its absence does not affect Ca2+-store depletion (Supplemental Figures S2 and S3). In agreement with a concentration-dependent effect of scaffold proteins in several signaling pathways (Cacace et al., 1999), here we find that KSR2 overexpression reduces Ca2+ influx in a dose-dependent manner. Taken together, these data indicate that KSR2, although dispensable for Ca2+-store depletion, acts as a regulator for influx of extracellular calcium.
It has been shown that the depletion of Ca2+ from the ER causes the oligomerization of STIM1 and further triggers the opening of ORAI1 (Park et al., 2009). A crucial step for SOCE is the association of STIM1 with ORAI1 in puncta-like complexes (Zhang et al., 2005; Luik et al., 2008). Here, using confocal microscopy, we observe that, although STIM1 multimerization is not altered, STIM1-ORAI1 puncta formation is impaired in shRNA KSR2 cells in response to Ca2+-store depletion (Figure 4). These data are corroborated by immunoprecipitation experiments in which the amount of STIM1 coassociated with ORAI1 after Tg-induced store depletion is partially reduced in shRNA KSR2 cells (Supplemental Figure S4). This observation might explain the reduced SOCE observed in Tg-induced KSR2-depleted cells (Figure 3). Similar results were observed in COS-7 cells not expressing endogenous KSR2. Of importance, reexpression of low levels of KSR2 in shRNA KSR2 HeLa (Figure 4D) or COS-7 cells (Figure 5B) restored STIM1/ORAI1 puncta structures and induced intracellular Ca2+ elevation after Tg stimulation (Figure 5D). In addition, coimmunoprecipitation and colocalization experiments suggest that KSR2 forms a ternary complex with STIM1 and Orai1 upon Ca2+-store depletion (Figure 5, F and G). This is reminiscent of the CRACR2A sensor, which seems to act as stabilizer for the interaction between ORAI1 and STIM1 in T-cells (Srikanth et al., 2010). Whether KSR2 interacts directly with both STIM1 and Orai1 and have an active role in translocation of ORAI1 and STIM1 remains to be elucidated. Taken together, these data demonstrate that KSR2 is required for STIM1-ORAI1 puncta formation and optimal Ca2+ influx in response to store depletion. Given that TRPC channels are involved in SOCE (Ong et al., 2007), it is reasonable to suggest that, although STIM1-ORAI1 puncta formation is impaired in the absence of KSR2, STIM1 cluster might couple with TRPC, thereby partially mediating Ca2+ entry.
Because KSR1 deficiency did not affect SOCE, we investigated the molecular mechanism involved in KSR2-dependent Ca2+ influx. Previous studies showed that, in contrast to KSR1, KSR2 has a unique domain that specifically binds several proteins known to be involved in Ca2+ signaling (Dougherty et al., 2009; Liu et al., 2009). Given that Ca2+/calmodulin-regulated Ser/Thr phosphatase CN is the most abundant of the KSR2-specific interactors (Dougherty et al., 2009), we tested whether CN could have a role in SOCE. Using the specific CN inhibitor cypermethrin, we found that CN activity is required for SOCE (Figure 6). As observed in KSR2-depleted cells, inhibition of CN reduced Ca2+ influx but had no effect on release of Ca2+ from ER stores. Of interest, SOCE reduction induced by inhibition of CN activity was not observed in KSR2-deficient cells, indicating that KSR2 is required for CN-mediated Ca2+ influx. Similar results were obtained using another CN inhibitor, cyclosporin A (Supplemental Figure S5).
It has been reported that CN dephosphorylates KSR2, allowing its translocation to the plasma membrane and facilitating ERK activation in response to Ca2+ (Dougherty et al., 2009). Furthermore, our data indicate that KSR2, besides being a CN target required for ERK activation (Dougherty et al., 2009), might in turn regulate CN after ER Ca2+ depletion. Using a GFP-NFAT reporter, we observed indeed that Tg-induced NFAT nuclear translocation is impaired in ksr2−/− fibroblasts (Figure 7), suggesting that KSR2 is involved in CN phosphatase activity in response to SOCE. This implies a new regulatory mechanism in calcineurin-NFAT signaling. Because CN regulates KSR2 (Dougherty et al., 2009), a small CN activity (e.g., induced by the calcium released from the ER stores) is required for KSR2 activation. Our data suggest that, once activated by CN, KSR2, by allowing sustained SOCE, is required for strong CN activation and NFAT nuclear translocation, supporting positive feedback regulation between KSR2 and calcineurin.
The question arises as to how the scaffold KSR2 affects STIM1/ORAI1 puncta-like formation. Because cytoskeletal dynamics is crucial for SOCE (Galán et al., 2011), we analyzed the organization of F-actin and α-tubulin filaments in ksr2−/− fibroblasts. We found a high degree of disorganization of both F-actin and α-tubulin filaments (Supplemental Figure S6), indicating that KSR2 is required for cytoskeleton organization and rearrangement. Because our data indicate that KSR2 is required for CN activity, and given that CN is involved in the regulation and structural plasticity of the cytoskeleton (Faul et al., 2008; Kume et al., 2011), we tested the effect of CN inhibition on cytoskeleton. Our findings show that F-actin and tubulin organization in response to Ca2+ was severely affected after CsA treatment (Supplemental Figure S6).
It seems reasonable that, by regulating cytoskeleton organization, the KSR2-CN complex facilitates the STIM1/ORAI1 puncta formation necessary for Ca2+ entry. Although proteomic studies report that KSR2 binds several cytoskeletal proteins, including α- and β-tubulin and F-actin capping proteins (Liu et al., 2009), and that CN affects microtubule dynamics (Kume et al., 2011), additional work is needed to understand the molecular mechanism by which the KSR2-CN complex affects cytoskeleton organization.
Our observation that KSR2 colocalizes with STIM1-ORAI1 puncta and that KSR2 is recruited to STIM1-ORAI1 complex after Tg treatment (Figure 5) also suggests that the KSR2-CN complex might directly affect ORAI1 function. It is possible that recruitment of KSR2 to the plasma membrane (Dougherty et al., 2009) might assist CN recruitment to puncta-like structures. Additional studies are required to prove a direct functional link between calcineurin and the ORAI1 channel.
In summary, our results define a new regulatory role of KSR2 in Ca2+ signaling. Indeed, by regulating the STIM1-ORAI1 puncta formation, the KSR2-CN complex modulates the store-dependent calcium influx.
MATERIALS AND METHODS
Mice
Mice C57BL/6 deficient in KSR1 (ksr1−/−) and KSR2 (ksr2−/−) were previously described (Nguyen et al., 2002; Revelli et al., 2011) and were a gift from Andrey S. Shaw (Washington University, St. Louis, MO). All experiments were conducted in 12- to 15-wk-old mice. Mice were housed under specific pathogen-free conditions at the University of Siena, with animal facilities in accordance with institutional guidelines.
Splenocyte isolation
Spleens from wt, ksr1−/−, and ksr2−/− mice were removed aseptically, and single-cell suspensions were prepared using cell strainer filters (BD Falcon; BD Biosciences Europe, Erembodegem, Belgium). Mononuclear cells were isolated from single-cell suspensions from spleen by density centrifugation on Ficoll-Hypaque (Amersham Biosciences Europe, Milan, Italy) and subsequently depleted of monocytes by adherence. Fluorescence-activated cell sorting analysis was performed on mononuclear single-cell suspensions of spleen. Cell suspensions were pelleted, washed twice, and resuspended in MACS solution (phosphate-buffered saline [PBS] containing 10% fetal bovine serum [FBS] and 1 mM EDTA) and counted using trypan blue exclusion viability test. Cells were then incubated in FcR blocking solution (BioLegend, London, UK) for 20 min at 4°C. To determine the percentage of B- and T-lymphocytes, mononuclear cells were then stained with the following antibodies from BD Biosciences, conjugated to either fluorescein isothiocyanate (FITC) or phycoerythrin (PE): CD3-PE (1:50) and B220-FITC (1:30). Samples were run on a BD FACScan instrument (Becton Dickinson, San Jose, CA) and analyzed using CellQuest software.
B- and T-cell purification
Isolation of B-cells from spleens or lymph nodes was made by using the B Cell Isolation Kit (Miltenyi Biotechnology, Bologna, Italy). After removal of erythrocytes by osmotic shock, B-cells were isolated by depletion of non–B-cells. Non–B-cells were indirectly magnetically labeled with a cocktail of biotin-conjugated monoclonal antibodies as primary labeling reagent and anti-biotin monoclonal antibodies conjugated to MicroBeads as secondary labeling reagent. The magnetically labeled non–B-cells were depleted by retaining them within a MACS column in the magnetic field of a MACS separator, whereas the unlabeled B-cells ran through the column and were collected for the examination. All passages were made at 4°C. Isolation of T-cells was by the Pan T Cell Isolation Kit (Miltenyi Biotechnology) using MACS separator as described. After purification, T- and B-cells were washed and immediately used.
Isolation of mouse lung fibroblasts
Murine fibroblasts were isolated and purified by slight modifications of procedures described by Cantini et al. (2002). Wild-type, ksr1−/−, or ksr2−/− mice were killed, and the lungs were removed, washed with PBS, and cut to 1-mm3-sized tissue masses. Tissues were then distributed evenly along the bottom of culture plates and covered with DMEM supplemented with 15% FBS (Euroclone, Milan, Italy), penicillin (50 U/ml), and streptomycin (50 μg/ml). The plates were cultured at 37°C in a humidified 5% CO2 incubator (Heraeus Instruments, Germany), and DMEM was changed every 3 d. When the cultures reached 60–80% confluence, adherent cells were detached by exposure to trypsin 0.05% and EDTA 0.02% in PBS for 5 min and then diluted one-third. Cells grew to a typical fusiform shape after three or four generations. Immunohistochemistry indicated that >90% of cells were vimentin positive but negative for endothelial cell marker (CD31; Santa Cruz Biotechnology, Heidelberg, Germany). Fibroblasts and fibroblast-like cells were used from passages 5 and 7.
Cell cultures, transfection, and shRNA
Mouse fibroblast, COS-7, HeLa, and HEK-293T cells were grown in DMEM supplemented with 10% FBS; Jurkat cells were grown in RPMI-1640 supplemented with 10% FBS. Cells were passaged every 2–3 d as required to maintain log-phase growth for all experiments. Fibroblast, COS-7, HeLa, and HEK-293T cells were transfected with the indicated DNA using Effectene Transfection Reagent (Qiagen, Milan, Italy). Human STIM1 and ORAI1 were tagged with YFP and RFP, respectively, as reported (Várnai et al., 2007). To generate GFP-KSR2 vector, mouse KSR2 full-length cDNA (a gift from A. S. Shaw) was digested with EagI and HindIII and subcloned into the pEGFP-C2 vector (Clontech Laboratories). Mouse KSR2-Flag cDNA, provided by A. S. Shaw, was digested with NheI and HindIII and subcloned in pcDNA 3.1 plasmid (Invitrogen, Life Technologies).
For the shRNA-mediated depletion of KSR2, a lentivirus-based pLKO.1 plasmid expressing human KSR2 shRNA was used. The sequence of shRNA targeting the nucleotides for amino acids 183–189 of human KSR2 (CCGGGCCTCAGGAATGTCCACATGTCTCGAGACATGTGGACATTCCTG AGGCTTTTTTG) was provided by Sigma-Aldrich, Milan, Italy. Viral particles were generated with the Mission lentiviral packaging mix (Sigma-Aldrich) and used to generate populations of HeLa or HEK293T cells stably expressing KSR2-shRNA. To generate the shKSR1 stable cell line, we used a multifunctional lentivirus system (Giurisato et al., 2009).
Confocal imaging and immunofluorescence staining
Transfected cells placed on glass precoated with gelatin (2% in water solution) were fixed in 4% paraformaldehyde (PFA) for 20 min and permeabilized with HEPES-Triton (20 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid [HEPES], 300 mM sucrose, 50 mM NaCl, 3 mM MgCl2, 0.5% Triton X-100) for 3 min at room temperature. To investigate the effect of KSR2 overexpression on puncta formation, transfected cells were incubated with mouse anti-Flag (Sigma-Aldrich) overnight, followed by incubation with Alexa Fluor 647–labeled secondary antibody (Molecular Probes, Thermo Fisher Scientific). To examine the cytoskeleton organization, cells were starved in 0.1% FBS-containing medium for 18 h and stimulated without (basal) or with 1 μM Tg at 37°C for 10 min. Cells were fixed and permeabilized as described. After blocking with 0.2% BSA in PBS for 30 min, cells were stained with mouse anti–α-tubulin (1:4000; Sigma-Aldrich) overnight, followed by incubation with Cy-2–conjugated anti-mouse IgG antibody (Jackson Laboratories, West Grove, PA). After washing, cells were incubated with rhodamine-conjugated phalloidin (Invitrogen). The coverslips were mounted on microscope slides in DAKO fluorescence mounting medium (DAKO Corporation, Milan, Italy). Images were acquired at 23°C by a confocal microscope (LSM 510 META; Carl Zeiss, Jena, Germany) using a Plan-Apochromat 63× oil immersion/1.4 numerical aperture (NA) objective. Images were analyzed by using the LSM 510 software program (Carl Zeiss). Distribution of YFP-STIM1 and ORAI1-RFP signals was evaluated by obtaining fluorescence intensity profiles along a straight line and considering as puncta (yellow dots) only those elements where both signals colocalized. For quantification of STIM1-ORAI1 puncta formation, the puncta were selected as yellow spots of high fluorescence intensity ranging from ∼0.5 to 1.0 μm in diameter and quantified by independent blind counting.
Immunoprecipitation and Western blot analysis
The expression level of KSR2 protein was assessed by immunoblotting. Cells were resuspended in ice-cold lysis buffer with the composition 20 mM Tris base, 137 mM NaCl, 0,2% Triton X-100, 1 mg/ml apoprotenin, and 1 mM phenylmethylsulfonyl fluoride and incubated on ice for 20 min. After centrifugation, proteins from cell lysates were resolved by SDS–PAGE. Proteins were blotted onto activated nitrocellulose membranes (Amersham Pharmacia Biotechnology) and probed with antibodies as specified in each experiment. Cell lysates were analyzed by immunoblotting with anti-KSR2 antibody (Santa Cruz Biotechnology), anti-Flag antibody (Sigma-Aldrich), anti-STIM1 (BD Transduction Laboratories, Milan, Italy), and anti–α-tubulin antibody (Sigma-Aldrich). The primary antibody was detected by incubating the membranes for 1 h with horseradish peroxidase–conjugated anti-mouse or anti-rabbit antibodies (Promega, Madison, WI) followed by enhanced chemiluminescence system for detection (Bio-Rad, Milan, Italy). In the immunoprecipitation experiments, transfected shRNA control HeLa cells, shRNA KSR2 HeLa cells, or COS-7 cells were starved in 0.1% FBS-containing medium for 18 h and stimulated with 1 μM Tg at 37°C for indicated time. Cells were resuspended in ice-cold lysis buffer and centrifuged as described. Nucleus-free supernatant was incubated with 2 μg/ml monoclonal anti-ORAI1 (Sigma-Aldrich) or monoclonal anti-Flag (Sigma-Aldrich) at 4°C for 1 h and then incubated with protein G–Sepharose beads (Invitrogen) at 4°C for 1 h. After washing, ORAI1 or KSR2-Flag immunoprecipitates were resolved by SDS–PAGE, transferred to a membrane, and analyzed by immunoblotting with anti-GFP (Santa Cruz Biotechnology), anti-STIM1 (BD-Biosciences), anti-ORAI1 (Sigma-Aldrich), and anti-KSR2 as described.
Cytosolic Ca2+ concentration measurement
Cells were loaded with 3 μM Fura-2 acetoxymethyl ester (Fura-2 AM; Life Technologies) for 30 min at 37°C in N-2-hydroxythylpiperazine-N-2-ethanesulfonic acid–buffered salt solution (HEPES solution; 140 mM NaCl, 5.4 mM KCl, 1 mM MgCl2, 1 mM CaCl2, 15 mM HEPES, pH 7.4) plus 1% BSA. After loading, the cells were kept at room temperature in the same medium, except for BSA, until used. Just before the experiment, a 1-ml aliquot of the cell suspensions was rapidly centrifuged and resuspended in fresh medium containing 1 mM CaCl2 or in nominally Ca2+-free medium (no added CaCl2).
For anti-CD3–induced activation, purified T-cells were stimulated with hamster anti-mouse CD3 (2C11, 2 μg/ml) followed by anti-hamster (10 μg/ml; Jackson Laboratory) to have receptor cross-linking. For BCR stimulation, purified B-cells were incubated with anti-IgM (1.3 μg/ml; eBioscience, Milan, Italy). In some experiments the cells were treated with thapsigargin (0.2 μM; Sigma-Aldrich) or Ca2+ ionophore ionomycin (0.5 μM; Sigma-Aldrich) to induce maximal ER Ca2+-store depletion and subsequent maximal SOCE activation.
Fluorescence was measured with a Varian Cary Eclipse fluorescence spectrophotometer (Palo Alto, CA; excitation wavelengths, 340 and 380 nm; emission wavelength, 510 nm) equipped with magnetic stirring and temperature control. To minimize leakage of trapped Fura-2, the assay temperature was set at 30°C, and 200 μM sulfinpyrazone was included in the medium. At the end of each incubation, digitonin (50 μg/ml) and EGTA (20 mM, from a solution of 0.5 M EGTA in 3.0 M Tris, pH 9.0) were added in order to measure maximal (Rmax) to minimal (Rmin) ratio (340/380) fluorescence values, respectively. The Kd for the Ca2+–Fura-2 complex was assumed to be 185 at 30°C. Values of [Ca2+]i were calculated by using CA Cricket Graph III software according to the formula [Ca2+]i = Kd(Sf380/Sb380)[(R − Rmin)/(Rmax − R)]. Sf380 is the fluorescence at 380-nm excitation under Ca2+-free conditions, and Sb380 is the fluorescence at 380-nm excitation under Ca2+-saturating conditions. In some experiments, before Ca2+ measurement, different cell lines were pretreated with different concentrations of the Cyper (0.1-10 μM; Sigma-Aldrich) or CsA (1–20 μM; Sigma-Aldrich) at 37°C for 30 min. Reduction of Ca2+ influx by different CN inhibitor concentrations was calculated as percentage of residual, ∆[Ca2+]i ([Ca2+]i peak after Ca2+ addition − basal [Ca2+]i) compared with control (vehicle).
For adherent cells, [Ca2+]i measurements was performed using video imaging microscopy. Briefly, HeLa or COS-7 cells (24 h after transfection) were grown on 13-mm-diameter coverslips (1 × 104 cells/coverslip) in 10% serum RPMI 1640. When cells reached semiconfluence they were loaded with Fura-2 AM as described. Before Ca2+ analysis, GFP-positive cells were identified and acquired with a cooled charge-coupled device (CCD) camera controlled by a MetaMorph imaging system.
Ca2+ measurements were carried out at room temperature. The digital fluorescence imaging microscopy system was mounted on a Nikon Diaphot 300 inverted microscope (Nikon, Tokyo, Japan). The images were collected through a Nikon oil immersion 40×/1.3 NA objective, acquired by a cooled CCD camera (Photometrics, Roper Scientific, Tucson, AZ), and calculated by a Metafluor imaging system (Universal Imaging Corporation, Downingtown, PA).
Mn2+-uptake assay
SOCE was also evaluated from the rate of Fura-2 fluorescence quenching by Mn2+, which enters the cell as a Ca2+ surrogate. Fluorescence excitation is 360 nm, corresponding to the isosbestic point at which changes in fluorescence can be assumed to be caused by Mn2+ alone. Fura2-loaded cells (see description of Ca2+ measurement) were resuspended in a fluorescence spectrophotometer cuvette in nominally Ca2+-free medium and treated or not with agonists (2C11 + anti-hamster or anti-IgM for T- and B-cells, respectively). After 0.1 mM MnCl2 addition, fluorescence (F) was monitored at excitation and emission wavelengths of 360 and 510 nm, respectively. Data were normalized as a percentage of fluorescence (F0) values obtained immediately before addition of MnCl2, F/F0.
NFAT nuclear translocation
Wild-type and ksr2−/− fibroblasts (5 × 103/coverslip) were placed on glass precoated with gelatin (2%), transfected with 0.8 μg of GFP-NFAT (provided by C. T. Baldari, University of Siena) using Effectene Transfection Reagent (Qiagen), starved with DMEM without serum for 1 h, stimulated with 1 μM Tg for 30 min at 37°C, and fixed in 4% PFA for 10 min at room temperature. Cells were washed four times with PBS and stained with TO-PRO-3 (Molecular Probes, Thermo Fisher Scientific) for 10 min at room temperature to investigate the effect of KSR2 on calcineurin-mediated NFAT nuclear translocation. After five washings with PBS, the coverslips were mounted on microscope slides in mounting medium and analyzed by fluorescence microscopy with a Nikon Eclipse Ti (Nikon, Japan) using a 40×/oil objective (Plan Fluor ELWD 0.60 DIC M/N1). The experiment was performed in duplicate.
Statistical analysis
In the Ca2+ measurements, p values were obtained by paired two-tailed Student's t test in Prism 4 (GraphPad). All error bars represent mean ± SEM based on several independent experiments. In the fluorescence experiments, statistical analyses were performed using a paired Student's t test. Statistically significant differences are indicated in the figure legends.
Supplementary Material
Acknowledgments
We thank A. S. Shaw for providing ksr1−/− and ksr2−/− mice and T. Balla for the YFP-STIM1 and ORAI1-RFP plasmids. We also thank C. T. Baldari and F. Bygrave for comments on the manuscript and I. Russo for invaluable advice and technical assistance. This research was supported by an Istituto Toscano Tumori 2008 Grant and Italian Ministry for University and Research PRIN 2009 to E.G.
Abbreviations used:
- BCR
B-cell receptor
- [Ca2+]i
cytosolic Ca2+ concentration
- CN
calcineurin
- CsA
cyclosporin A
- GFP
green fluorescent protein
- KSR
kinase suppressor of Ras
- ksr1−/−
KSR1 knockout
- ksr2−/−
KSR2 knockout
- NFAT
nuclear factor of activated T-cells
- PM
plasma membrane
- RFP
red fluorescent protein
- SOCE
store-operated calcium entry
- STIM1
stromal interaction molecule 1
- Tg
thapsigargin
- TRPC
transient receptor potential channel
- wt
wild type
- YFP
yellow fluorescent protein
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E13-05-0292) on March 26, 2014.
E.G. designed the research; E.G., A.G., and C.U. analyzed and interpreted the data and wrote the manuscript; E.G., A.G., C.U., E.R., S.M., E.G., D.R., and V.S. performed the research and contributed reagents.
REFERENCES
- Cacace AM, Michaud NR, Therrien M, Mathes K, Copeland T, Rubin GM, Morrison DK. Identification of constitutive and ras-inducible phosphorylation sites of KSR: implications for 14-3-3 binding, mitogen-activated protein kinase binding, and KSR overexpression. Mol Cell Biol. 1999;19:229–240. doi: 10.1128/mcb.19.1.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantini M, Giurisato E, Radu C, Tiozzo S, Pampinella F, Senigaglia D, Zaniolo G, Mazzoleni F, Vitiello L. Macrophage-secreted myogenic factors: a promising tool for greatly enhancing the proliferative capacity of myoblasts in vitro and in vivo. Neurol Sci. 2002;23:189–194. doi: 10.1007/s100720200060. [DOI] [PubMed] [Google Scholar]
- Carafoli E. The calcium-signalling saga: tap water and protein crystals. Nat Rev Mol Cell Biol. 2003;4:326–332. doi: 10.1038/nrm1073. [DOI] [PubMed] [Google Scholar]
- Costanzo-Garvey DL, et al. KSR2 is an essential regulator of AMP kinase, energy expenditure, and insulin sensitivity. Cell Metab. 2009;10:366–378. doi: 10.1016/j.cmet.2009.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolmetsch RE, Keli X, Lewis RS. Calcium oscillations increase the efficiency and specificity of gene expression. Nature. 1998;392:933–936. doi: 10.1038/31960. [DOI] [PubMed] [Google Scholar]
- Dougherty MK, Ritt DA, Zhou M, Specht SI, Monson DM, Veenstra TD, Morrison DK. KSR2 is a calcineurin substrate that promotes ERK cascade activation in response to calcium signals. Mol Cell. 2009;34:652–662. doi: 10.1016/j.molcel.2009.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faul C, et al. The actin cytoskeleton of kidney podocytes is a direct target of the antiproteinuric effect of cyclosporine A. Nat Med. 2008;14:931–938. doi: 10.1038/nm.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez MR, Henry MD, Lewis RE. Kinase suppressor of Ras 2 (KSR2) regulates tumor cell transformation via AMPK. Mol Cell Biol. 2012;32:3718–3731. doi: 10.1128/MCB.06754-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feske S. Calcium signalling in lymphocyte activation and disease. Nat Rev Immunol. 2007;7:690–702. doi: 10.1038/nri2152. [DOI] [PubMed] [Google Scholar]
- Frischauf I, Schindl R, Derler I, Bergsmann J, Fahrner M, Romanin C. The STIM/Orai coupling machinery. Channels (Austin) 2008;2:261–268. doi: 10.4161/chan.2.4.6705. [DOI] [PubMed] [Google Scholar]
- Galán C, Dionisio N, Smani T, Salido GM, Rosado JA. The cytoskeleton plays a modulatory role in the association between STIM1 and the Ca2+ channel subunits Orai1 and TRPC1. Biochem Pharmacol. 2011;82:400–410. doi: 10.1016/j.bcp.2011.05.017. [DOI] [PubMed] [Google Scholar]
- Giurisato E, Lin J, Harding A, Cerutti E, Cella M, Lewis RE, Colonna M, Shaw AS. The mitogen-activated protein kinase scaffold KSR1 is required for recruitment of extracellular signal-regulated kinase to the immunological synapse. Mol Cell Biol. 2009;6:1554–1564. doi: 10.1128/MCB.01421-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigoriev I, et al. STIM1 is a MT-plus-end-tracking protein involved in remodeling of the ER. Curr Biol. 2008;18:177–182. doi: 10.1016/j.cub.2007.12.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirata Y, et al. Uncoupling store-operated Ca2+ entry and altered Ca2+ release from sarcoplasmic reticulum through silencing of junctophilin genes. Biophys J. 2006;90:4418–4427. doi: 10.1529/biophysj.105.076570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogan PG, Rao A. Dissecting ICRAC, a store-operated calcium current. Trends Biochem Sci. 2007;32:235–245. doi: 10.1016/j.tibs.2007.03.009. [DOI] [PubMed] [Google Scholar]
- Jardin I, Albarrán L, Bermejo N, Salido GM, Rosado JA. Homers regulate calcium entry and aggregation in human platelets: a role for Homers in the association between STIM1 and Orai1. Biochem J. 2012;445:29–38. doi: 10.1042/BJ20120471. [DOI] [PubMed] [Google Scholar]
- Kawasaki T, Ueyama T, Lange I, Feske S, Saito N. Protein kinase C-induced phosphorylation of Orai1 regulates the intracellular Ca2+ level via the store-operated Ca2+ channel. J Biol Chem. 2010;285:25720–25730. doi: 10.1074/jbc.M109.022996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornfeld K, Hom DB, Horvitz HR. The ksr-1 gene encodes a novel protein kinase involved in Ras-mediated signaling in C. elegans. Cell. 1995;83:903–913. doi: 10.1016/0092-8674(95)90206-6. [DOI] [PubMed] [Google Scholar]
- Kortum RL, Lewis RE. The molecular scaffold KSR1 regulates the proliferative and oncogenic potential of cells. Mol Cell Biol. 2004;24:4407–4416. doi: 10.1128/MCB.24.10.4407-4416.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kume K, Koyano T, Kanai M, Toda T, Hirata D. Calcineurin ensures a link between the DNA replication checkpoint and microtubule-dependent polarized growth. Nat Cell Biol. 2011;13:234–242. doi: 10.1038/ncb2166. [DOI] [PubMed] [Google Scholar]
- Lewis RS. The molecular choreography of a store-operated calcium channel. Nature. 2007;446:284–287. doi: 10.1038/nature05637. [DOI] [PubMed] [Google Scholar]
- Lin J, Harding A, Giurisato E, Shaw AS. KSR1 modulates the sensitivity of mitogen-activated protein kinase pathway activation in T cells without altering fundamental system outputs. Mol Cell Biol. 2009;29:2082–2091. doi: 10.1128/MCB.01634-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liou J, Fivaz M, Inoue T, Meyer T. Live-cell imaging reveals sequential oligomerization and local plasma membrane targeting of stromal interaction molecule 1 after Ca2+ store depletion. Proc Natl Acad Sci USA. 2007;104:9301–9306. doi: 10.1073/pnas.0702866104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liou J, Kim ML, Heo WD, Jones JT, Myers JW, Ferrell JE, Jr, Meyer T. STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr Biol. 2005;15:1235–1241. doi: 10.1016/j.cub.2005.05.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Rao A, Hogan PG. Interaction of calcineurin with substrates and targeting proteins. Trends Cell Biol. 2011;21:91–103.. doi: 10.1016/j.tcb.2010.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Channavajhala PL, Rao VR, Moutsatsos I, Wu L, Zhang Y, Lin LL, Qiu Y. Proteomic characterization of the dynamic KSR-2 interactome, a signaling scaffold complex in MAPK pathway. Biochim Biophys Acta. 2009;1794:1485–1495. doi: 10.1016/j.bbapap.2009.06.016. [DOI] [PubMed] [Google Scholar]
- Lopez E, Jardin I, Berna-Erro A, Bermejo N, Salido GM, Sage SO, Rosado JA, Redondo PC. STIM1 tyrosine-phosphorylation is required for STIM1-Orai1 association in human platelets. Cell Signal. 2012;24:1315–1322. doi: 10.1016/j.cellsig.2012.02.012. [DOI] [PubMed] [Google Scholar]
- Luik RM, Wang B, Prakriya M, Wu MM, Lewis RS. Oligomerization of STIM1 couples ER calcium depletion to CRAC channel activation. Nature. 2008;454:538–542. doi: 10.1038/nature07065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matza D, Badou A, Kobayashi KS, Goldsmith-Pestana K, Masuda Y, Komuro A, McMahon-Pratt D, Marchesi VT, Flavell RA. A scaffold protein, AHNAK1, is required for calcium signaling during T cell activation. Immunity. 2008;28:64–74. doi: 10.1016/j.immuni.2007.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muik M, et al. Dynamic coupling of the putative coiled-coil domain of ORAI1 with STIM1 mediates ORAI1 channel activation. J Biol Chem. 2008;283:8014–8022. doi: 10.1074/jbc.M708898200. [DOI] [PubMed] [Google Scholar]
- Nguyen A, et al. Kinase suppressor of Ras (KSR) is a scaffold which facilitates mitogen-activated protein kinase activation in vivo. Mol Cell Biol. 2002;22:3035–3045. doi: 10.1128/MCB.22.9.3035-3045.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohmachi M, Rocheleau CE, Church D, Lambie E, Schedl T, Sundaram MV. C. elegans ksr-1 and ksr-2 have both unique and redundant functions and are required for MPK-1 ERK phosphorylation. Curr Biol. 2002;12:427–433. doi: 10.1016/s0960-9822(02)00690-5. [DOI] [PubMed] [Google Scholar]
- Ong HL, et al. Dynamic assembly of TRPC1-STIM1-Orai1 ternary complex is involved in store-operated calcium influx. Evidence for similarities in store-operated and calcium release-activated calcium channel components. J Biol Chem. 2007;282:9105–9116. doi: 10.1074/jbc.M608942200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parekh AB, Putney JW., Jr Store-operated calcium channels. Physiol Rev. 2005;85:757–810. doi: 10.1152/physrev.00057.2003. [DOI] [PubMed] [Google Scholar]
- Park CY, Hoover PJ, Mullins FM, Bachhawat P, Covington ED, Raunser S, Walz T, Garcia KC, Dolmetsch RE, Lewis RS. STIM1 clusters and activates CRAC channels via direct binding of a cytosolic domain to Orai1. Cell. 2009;136:876–890. doi: 10.1016/j.cell.2009.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawson T, Scott JD. Signaling through scaffold, anchoring, and adaptor proteins. Science. 1997;278:2075–2080. doi: 10.1126/science.278.5346.2075. [DOI] [PubMed] [Google Scholar]
- Pozo-Guisado E, Campbell DG, Deak M, Álvarez-Barrientos A, Morrice NA, Álvarez IS, Alessi DR, Martín-Romero FJ. Phosphorylation of STIM1 at ERK1/2 target sites modulates store-operated calcium entry. J Cell Sci. 2010;123:3084–3093. doi: 10.1242/jcs.067215. [DOI] [PubMed] [Google Scholar]
- Prakriya M, Feske S, Gwack Y, Srikanth S, Rao A, Hogan PG. Orai1 is an essential pore subunit of the CRAC channel. Nature. 2006;443:230–233. doi: 10.1038/nature05122. [DOI] [PubMed] [Google Scholar]
- Putney JW., Jr New molecular players in capacitative Ca2+ entry. J Cell Sci. 2007;120:1959–1965. doi: 10.1242/jcs.03462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revelli JP, et al. Profound obesity secondary to hyperphagia in mice lacking kinase suppressor of ras 2. Obesity (Silver Spring) 2011;19:1010–1018. doi: 10.1038/oby.2010.282. [DOI] [PubMed] [Google Scholar]
- Rosado JA, Porras T, Conde M, Sage SO. Cyclic nucleotides modulate store-mediated calcium entry through the activation of protein-tyrosine phosphatases and altered actin polymerization in human platelets. J Biol Chem. 2001;276:15666–15675. doi: 10.1074/jbc.M009217200. [DOI] [PubMed] [Google Scholar]
- Singh BB, Liu X, Ambudkar IS. Expression of truncated transient receptor potential protein 1alpha (Trp1alpha): evidence that the Trp1 C terminus modulates store-operated Ca2+ entry. J Biol Chem. 2000;275:36483–36486. doi: 10.1074/jbc.C000529200. [DOI] [PubMed] [Google Scholar]
- Smyth JT, DeHaven WI, Bird GS, Putney JW., Jr Role of the microtubule cytoskeleton in the function of the store-operated Ca2 +channel activator STIM1. J Cell Sci. 2007;120:3762–3771. doi: 10.1242/jcs.015735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth JT, Hwang SY, Tomita T, DeHaven WI, Mercer JC, Putney JW. Activation and regulation of store-operated calcium entry. J Cell Mol Med. 2010;14:2337–2349. doi: 10.1111/j.1582-4934.2010.01168.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth JT, Putney JW. Regulation of store-operated calcium entry during cell division. Biochem Soc Trans. 2012;40:119–123. doi: 10.1042/BST20110612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soundararajan R, Ziera T, Koo E, Ling K, Wang J, Borden SA, Pearce D. Scaffold protein connector enhancer of kinase suppressor of Ras isoform 3 (CNK3) coordinates assembly of a multiprotein epithelial sodium channel (ENaC)-regulatory complex. J Biol Chem. 2012;287:33014–33025. doi: 10.1074/jbc.M112.389148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srikanth S, Jung HJ, Kim KD, Souda P, Whitelegge J, Gwack Y. A novel EF-hand protein, CRACR2A, is a cytosolic Ca2+ sensor that stabilizes CRAC channels in T cells. Nat Cell Biol. 2010;5:436–446. doi: 10.1038/ncb2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundaram M, Han M. The C. elegans ksr-1 gene encodes a novel Raf-related kinase involved in Ras-mediated signal transduction. Cell. 1995;83:889–901. doi: 10.1016/0092-8674(95)90205-8. [DOI] [PubMed] [Google Scholar]
- Therrien M, Chang HC, Solomon NM, Karim FD, Wassarman DA, Rubin GM. KSR, a novel protein kinase required for RAS signal transduction. Cell. 1995;83:879–888. doi: 10.1016/0092-8674(95)90204-x. [DOI] [PubMed] [Google Scholar]
- Vaca L. SOCIC: the store-operated calcium influx complex. Cell Calcium. 2010;47:199–209. doi: 10.1016/j.ceca.2010.01.002. [DOI] [PubMed] [Google Scholar]
- Várnai P, Hunyady L, Balla T. STIM and Orai: the long-awaited constituents of store-operated calcium entry. Trends Pharmacol Sci. 2009;30:118–128. doi: 10.1016/j.tips.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Várnai P, Tóth B, Tóth DJ, Hunyady L, Balla T. Visualization and manipulation of plasma membrane-endoplasmic reticulum contact sites indicates the presence of additional molecular components within the STIM1-Orai1 Complex. J Biol Chem. 2007;282:29678–29690. doi: 10.1074/jbc.M704339200. [DOI] [PubMed] [Google Scholar]
- Wu MM, Luik RM, Lewis RS. Some assembly required: constructing the elementary units of store-operated Ca2+ entry. Cell Calcium. 2007;42:163–172. doi: 10.1016/j.ceca.2007.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeromin AV, Zhang SL, Jiang W, Yu Y, Safrina O, Cahalan MD. Molecular identification of the CRAC channel by altered ion selectivity in a mutant of Orai. Nature. 2006;443:226–229. doi: 10.1038/nature05108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang SL, Yu Y, Roos J, Kozak JA, Deerinck TJ, Ellisman MH, Stauderman KA, Cahalan MD. STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature. 2005;437:902–905. doi: 10.1038/nature04147. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.