

Nicotine up-regulates NeuroD1 in bronchial epithelial cells and certain undifferentiated carcinomas. NeuroD1 enhances expression of nicotinic acetylcholine receptor subunits. Increased invasion in Matrigel depends on these receptor subunits. Nicotine may induce positive feedback through NeuroD1 and increased expression of its own receptor.
Abstract
Cigarette smoking is a major risk factor for acquisition of small cell lung cancer (SCLC). A role has been demonstrated for the basic helix-loop-helix transcription factor NeuroD1 in the pathogenesis of neural and neuroendocrine lung cancer, including SCLC. In the present study we investigate the possible function of NeuroD1 in established tumors, as well as actions early on in pathogenesis, in response to nicotine. We demonstrate that nicotine up-regulates NeuroD1 in immortalized normal bronchial epithelial cells and a subset of undifferentiated carcinomas. Increased expression of NeuroD1 subsequently leads to regulation of expression and function of the nicotinic acetylcholine receptor subunit cluster of α3, α5, and β4. In addition, we find that coordinated expression of these subunits by NeuroD1 leads to enhanced nicotine-induced migration and invasion, likely through changes in intracellular calcium. These findings suggest that aspects of the pathogenesis of neural and neuroendocrine lung cancers may be affected by a nicotine- and NeuroD1-induced positive feedback loop.
INTRODUCTION
Signature characteristics of tumor pathogenesis include the acquisition of qualities that enable unrestrained growth and metastasis. Many of the genes expressed during tumorigenesis regulate developmental gene programs initiated during embryogenesis and organogenesis (Wong et al., 2008; Ben-David and Benvenisty, 2011; Ben-David et al., 2011). Small cell lung cancers (SCLCs) and other neuroendocrine carcinomas display increased expression of the neuronal basic helix-loop-helix transcription factors neurogenic differentiation 1 (NeuroD1) and achaete-scute homologue (ASCL1; Ball et al., 1993; Rostomily et al., 1997; Hu et al., 2004; Syder et al., 2004; Fratticci et al., 2007; Jiang et al., 2009; Osborne et al., 2013b).
SCLC accounts for 15–20% of all lung cancers. It is a highly aggressive form of lung cancer, is strongly associated with cigarette smoking, and is characterized by neuroendocrine morphological features, distinguished by large nuclei, little cytoplasm, and expression of classical neuroendocrine markers such as synaptophysin and chromogranin A, fast growth, early dissemination, and a high frequency of metastasis, especially to brain (Quan et al., 2004; Jackman and Johnson, 2005; van Meerbeeck et al., 2011). Tumors of patients who develop lung cancer and SCLC in particular as a result of smoking have a significantly higher number of mutations per megabase of DNA in specific genes (such as TP53) than do nonsmokers with the same subtypes of lung cancer (Pleasance et al., 2010; Govindan et al., 2012).
Nicotine induces factors that participate in multiple signal transduction pathways throughout the body, including brain and lung (Hukkanen et al., 2005). Nicotine exposure through cigarette smoking has been linked to lung cancer pathogenesis via mechanisms mediated by the pentameric ligand-gated ion channels nicotinic acetylcholine receptors (nAChRs; Lam et al., 2007; Improgo et al., 2010a). Originally studied as neuronal and muscle ion channels, these receptors, when activated by their ligand, acetylcholine or nicotine, are selectively permeable to calcium. Formed from a combination of nine α (2–10) and three β (2–4) subunits of specific homomeric and heteromeric combinations, these pentameric nAChRs are also expressed in many nonneuronal and nonmuscle cells, such as macrophages, keratinocytes, and normal lung epithelia (Tournier et al., 2006; Kalamida et al., 2007; Lam et al., 2007; Improgo et al., 2010a).
It has been suggested that nicotine induces ASCL1 expression, and in turn ASCL1 induces expression of the nAChR gene cluster of CHRNA5/A3/B4, specifically α3 and β4 (Linnoila, 2006; Improgo et al., 2010a). During development, expression of the transcription factors ASCL1 and NeuroD1 is temporally separated, with the expression of ASCL1 preceding that of NeuroD1, specifically in hypothalamic neuroendocrine cells, olfactory neuronal progenitors, and neuroendocrine cells located in pulmonary epithelia (Cau et al., 1997; Ito et al., 2000; McNay et al., 2006; Neptune et al., 2008). We demonstrated that NeuroD1 is essential for survival and metastasis of a subset of lung cancers with neuroendocrine features, acting primarily through its downstream targets, the receptor tyrosine kinase TrkB and the neural cell adhesion molecule (Osborne et al., 2013b). In this study in lung epithelial cells and several tumors, we found that nicotine induces NeuroD1 and TrkB in a cell type–specific manner, leading to up-regulation of the nAChR gene cluster of CHRNA5/A3/B4. Our findings support the conclusion that NeuroD1 is induced by nicotine. One consequence is increased expression of the nAChR gene cluster of CHRNA5/A3/B4, ultimately contributing to nicotine-induced migration.
RESULTS
Nicotine induces expression of NeuroD1 in immortalized bronchial epithelial cells and some lung cancers
To gain perspective on the mechanism of NeuroD1 action, we examined its expression in several neural/neuroendocrine cancer cell lines, focusing predominantly on lung cancer and neuroblastoma models (Table 1). In addition to patient-derived SCLC cell lines, we used several human bronchial epithelial cell (HBEC) lines. The cells were immortalized by expressing cyclin-dependent kinase 4 and human telomerase and were from different donors (indicated by numbers, HBEC3KT, HBEC4KT, and HBEC30KT). We also used an isogenic manipulated tumor-forming clone (HBEC3KTRL53-Clone 5, referred to hereafter as Clone 5) derived from HBEC3KT by stable depletion of p53 and expression of oncogenic KRAS, because it displayed an increase in NeuroD1 relative to HBEC3KT parent cells (Sato et al., 2006, 2013; Osborne et al., 2013b; Figure 1A). Previously, we characterized actions of NeuroD1 in Clone 5 and found that loss of the protein decreased migration and formation of colonies in soft agar (Osborne et al., 2013b). In addition, we used several non–small cell lung cancer (NSCLC) lines, including HCC4017, which was taken from the contralateral lung of the HBEC30KT donor. For the neural cancer model, we used the human neuroblastoma cell line SHSY5Y, previously noted to express NeuroD1, and the human embryonal carcinoma cell line NTERA2 clone D1 (referred to hereafter as NTERA2), which we confirmed does not (Figure 1A). NTERA2 is reported to have stem cell characteristics and can be differentiated along a neuroectodermal lineage with retinoic acid (RA), which causes expression of NeuroD1 (Andrews, 1984; Mavilio, 1993; Przyborski et al., 2000; Huang et al., 2011). NeuroD1 expression was also detected in several of the patient-derived neuroendocrine lung cancer cell lines, as well as in the rodent pheochromocytoma PC12 cell; its expression was undetectable in the undifferentiated germ cell tumor NTERA2 and low in the immortalized normal HBEC lines (Figure 1, A and B).
TABLE 1:
Mutational status of cell lines.
| Cell line | Cell type | p53 status | KRAS mutation | Additional mutation |
|---|---|---|---|---|
| H69 | SCLC-V | Mutant | No | PIK3CA/RB1 |
| H82 | SCLC-V | Mutant | No | RB1 |
| H358 | NSCLC | Null | Yes (G12V) | a |
| H460 | NSCLC | Wild type | Yes (Q61H) | PIK3CA/CDKN2A/STK11 |
| H524 | SCLC-V | Mutant | No | RB1 |
| H526 | SCLC-V | Mutant | No | RB1 |
| H889 | SCLC-V/C | Mutant | No | – |
| H1155 | NSCLC-NE | Mutant | Yes (Q61H) | PTEN/MSH6 |
| H1184 | SCLC-C | Mutant | No | a |
| H2171 | SCLC-V | Mutant | No | a |
| H2227 | SCLC-C | Mutant | No | RB1 |
| SHSY5Y | Neuroblastoma | Wild type | No | ALK |
| NTERA2 | Germ cell tumor | Wild type | No | SF3B1 |
| HCC4017 | NSCLC | Unknown | Yes (G12C) | – |
| HBEC3KTRL53- Clone 5 | NSCLC-NE | Constitutive knockdown | Overexpress (G12V) | – |
| HBEC3KT-53 | Immortalized bronchial epithelial | Constitutive knockdown | No | – |
| HBEC3KT | Immortalized bronchial epithelial | Wild type | No | – |
| HBEC4KT | Immortalized bronchial epithelial | Wild type | No | – |
| HBEC30KT | Immortalized bronchial epithelial | Wild type | No | – |
C, classic; K, CDK4; T, hTERT; V, variant.
aMany more in COSMIC database (http://cancer.sanger.ac.uk/cancergenome/projects/cosmic/).
FIGURE 1:

Effect of nicotine on expression of NeuroD1 in patient-derived lung cancer, bronchial epithelial, and neuroendocrine cell lines. (A) Patient-derived SCLC, HBEC3KT, Clone 5, SHSY5Y, NTERA2, and rodent PC12 cell lines were lysed, and 25 μg of total protein was resolved on gels and immunoblotted for NeuroD1; GAPDH was the loading control. (B) Immortalized bronchial epithelial cells HBEC3KT, HBEC4KT, and HBEC30KT were treated with 0.25, 0.5, 1, and 2 μM nicotine for 24–48 h. Cells were fixed and immunostained for NeuroD1 and 4′,6-diamidino-2-phenylindole (DAPI). (C) qRT-PCR analysis of NeuroD1 expression in HBEC3KT, Clone5, HBEC30KT, and NSCLC HCC4017 with or without exposure to 0.25 μM nicotine for 48 h, plotted as relative expression. The control was 18s RNA. One of two independent experiments in duplicate. (D) HBEC3KT and Clone 5 cells were treated with 1 μM nicotine for 48 h, and 25 μg of lysate protein was immunoblotted as in A with tubulin as control. Relative expression quantified using LI-COR infrared imaging in three independent experiments; *p <0.05.
Cigarettes contain various carcinogens, including the addictive component nicotine, which has been linked to the increased onset of lung cancer (Hecht, 1999). Of all the subtypes of lung cancer, SCLC has the highest occurrence in patients with a history of tobacco use (Jackman and Johnson, 2005). Nicotine has a half-life in the circulation of 6–8 h and a measured average blood concentration (depending on consumption, type, etc.) ranging from 25 to 1000 nM (Russell et al., 1980; Hukkanen et al., 2005; Pillai et al., 2011). To investigate potential effects of nicotine action on NeuroD1 early in tumor pathogenesis, we treated normal immortalized bronchial epithelial cell lines HBEC3KT, HBEC4KT, and HBEC30KT with increasing concentrations of nicotine for 24–48 h. An increase in NeuroD1 expression was observed after exposure of cells to concentrations of nicotine achieved in smokers (Figure 1B). Increased NeuroD1 mRNA and protein expression was detected at the lowest concentration of nicotine in both HBEC3 and 30KT lines and HCC4017 cells; however, further increase was not observed in Clone 5 (Figure 1, C and D).
Nicotine-induced NeuroD1 expression is ERK1/2 pathway independent in SCLC, NSCLC, clone 5, HBECs, and SHSY5Y neuroblastoma cells
Phosphorylation of serine residues in NeuroD1 by ERK1/2 is required for proper transactivation activity on the insulin promoter in pancreatic β-cells; however, the importance of these phosphorylation events for protein accumulation and activity seems to be cell type specific (Khoo et al., 2003; Dufton et al., 2005). Activating mutations of the MAPK-ERK1/2 pathway in SCLC are rarely observed; in addition, activation of this pathway via overexpression of Raf-1 in several SCLC cell lines results in growth arrest (Mitsudomi et al., 1991; Alessi et al., 1993; Ravi et al., 1998; Sun et al., 2007). The term “variant” refers to a subclass of SCLC cells with morphological features similar to those of NSCLC large cells, which in culture are floating, loosely bound, multicell aggregates that contrast with classic SCLC cells, which form tightly packed spheres (Carney et al., 1985a, b; Gazdar et al., 1985; Figure 2A). We observed that variant SCLC (e.g., H69 and H82) had higher NeuroD1 expression than classic SCLC (Figures 1A and 2A). The variant SCLC cell line H82 had lower basal phosphorylated ERK1/2 than the classic SCLC cell line H1184 (Figure 2B). Treatment of the SCLC cell lines H69 and H82 with the MEK1/2 inhibitor PD0325901 did not decrease anchorage-independent growth; however, growth inhibition was observed in the NSCLC lines H358 and H460 (Figure 2C). In addition, we found that nicotine caused an increase in phosphorylated ERK1/2 within minutes in several of the SCLC cell lines (Figure 2D), consistent with previous work. (Dajas-Bailador et al., 2002b; Jin et al., 2004; Xu and Deng, 2004; Chen et al., 2008).
FIGURE 2:

Effect of nicotine and common signaling inhibitors on NeuroD1 expression and colony formation in lung cancer and immortalized bronchial epithelial cells. (A) Differential interference contrast images at 10× magnification of SCLC and NSCLC-neuroendocrine (NE) lines. (B) ERK1/2 and pERK1/2 were immunoblotted in 25 μg of lysate protein from HBEC, SCLC, and NSCLC cells as in Figure 1. (C) Formation of colonies in soft agar by SCLC (H69, H82) and NSCLC (H358, H460) cells after 4-wk exposure to 0, 0.1, 1, and 10 μM PD0325901. Colony number plotted as percentage of control. (D) SCLC H82, H69, and H2171 lines were starved for 18 h in 0.1% FBS and then untreated (NT) or treated with 1 μM nicotine for 15 and 120 min. ERK1/2 and pERK1/2 were immunoblotted as in B. (E) HBEC30KT and HCC4017 cells in normal medium were treated for 2 h as follows: A, control; B–E, 1 μM nicotine; C, +0.5 μM PD0325901; D, +10 μM LY294002; E, +10 μM mecamylamine. Lysates were immunoblotted for NeuroD1 and ERK1/2 as before. One of three independent experiments.
Nicotine acts through nicotinic acetylcholine receptors (nAChRs). These ion channels have many possible subunit compositions and have been shown to be expressed in normal lung and lung cancer cells, specifically SCLC (Lam et al., 2007; Improgo et al., 2010a). Thus we tested the effects of mecamylamine, a nonselective and noncompetitive antagonist of several nAChRs. Mecamylamine caused >50% reduction in nicotine-induced NeuroD1 expression in HBEC30KT and HCC4017 cells (Figure 2E), confirming that nAChRs can mediate actions of nicotine on the transcription factor. We also asked whether inhibition of ERK1/2 or phosphatidylinositol 3-kinase (PI3K) affected nicotine-induced NeuroD1 expression. Effects of inhibitors of the MEK1/2 or PI3K pathway on nicotine-induced NeuroD1 expression varied (Supplemental Figures S1 and S2), suggesting that neither pathway is essential for NeuroD1 expression, but instead multiple pathways can affect nicotine-induced NeuroD1 expression.
Nicotine can up-regulate NeuroD1 without causing differentiation of SHSY5Y and NTERA2 cells
Because NeuroD1 was originally identified as a gene required for neuronal differentiation, we investigated whether the increase in NeuroD1 expression was also accompanied by neuronal differentiation (Lee et al., 1995). To examine this possibility, we used the embryonal carcinoma cell line NTERA2 and the neuroblastoma cell line SHSY5Y. These cell lines were previously shown to differentiate along neuronal lineages after treatment with RA. The resulting cells have biochemical and morphological similarities to neurons (Andrews, 1984; Lopez-Carballo et al., 2002). Treatment of these cell lines with RA induced expression of NeuroD1 and one of its key targets, TrkB, as well as of a marker of mature neurons, NeuN. In contrast, treatment with nicotine caused an increase in NeuroD1 and TrkB but no significant increase in NeuN (Figure 3, A and B), suggesting that it was not able to induce a differentiated state. We examined expression of transcription factors ASCL1 and NGN3, which lie upstream of NeuroD1. NGN3, like ASCL1, is a bHLH transcription factor found in brain and pancreas during development (Cau et al., 1997; Huang et al., 2000; Ito et al., 2000; McNay et al., 2006; Neptune et al., 2008). RA caused decrease or no change in expression of ASCL1 or NGN3 in either cell line (Figure 3B), whereas nicotine caused increased ASCL1 and NGN3 in the NTERA2 cell line, supporting the idea that nicotine is not promoting a more differentiated state.
FIGURE 3:

Nicotine can increase NeuroD1 expression without causing neuroendocrine differentiation. (A, B) NTERA2 and SHSY5Y cells were treated with 25 μM nicotine or 25 μM retinoic acid for 14 d to induce differentiation. (A) Cells were fixed and immunostained for NeuroD1 and NeuN (mature neuronal marker) and DAPI. Representative of three independent experiments. (B) Cell lysates were immunoblotted for NeuroD1, TrkB, ASCL1, NGN3, and p53. GAPDH was the loading control. (C) HBEC3KT cells were treated with 0.63, 1.25, 2.5, and 5 μM nicotine over 24–48 h. Cell lysates were immunoblotted for p53; GAPDH was the loading control.
Nicotine decreases p53 expression in a cell type–specific manner
We demonstrated that loss of p53 led to increased NeuroD1 expression in a cell type–specific manner (Osborne et al., 2013a). Nicotine induces proliferation and leads to down-regulation of p53 (Pfeifer et al., 2002; Dasgupta and Chellappan, 2006; Dasgupta et al., 2006; Sato et al., 2008; Puliyappadamba et al., 2010). Treatment of SHSY5Y and the NTERA2 cells with nicotine did not suppress p53 expression (Figure 3B). However, p53 expression was reduced by nicotine in HBEC3KT (Figure 3C). This leads to the idea that suppression of p53 in nonepithelial tissues may not be required to increase NeuroD1 expression.
NeuroD1 regulates the expression of nAChR α3 and α5 but not β4 subunits in SHSY5Y neuroblastoma, SCLC, and HBEC3KT
Several genome-wide studies mapped a specific region on chromosome 15q25 as a locus for lung cancer susceptibility, and this locus contains the genes that encode the nAChR subunit cluster of α3, α5, and β4 (Amos et al., 2008; Hung et al., 2008). ASCL1 can regulate the α3 and β4 subunits (Improgo et al., 2010a). We detected higher expression of the nAChR subunits and NeuroD1 in the cancer lines than in the HBEC lines (Figure 4A). Several of the subunits are heavily glycosylated and acetylated to enable proper localization to the plasma membrane, which may explain the extra bands visualized (Albuquerque et al., 2009). Using low concentrations of nicotine, we found that NeuroD1 and all three AChR subunit mRNAs increased in SHSY5Y cells (Figure 4B). An increase in each of the subunits was also detected in the HBEC3KT cells with exposure to nicotine (Supplemental Figure S3A). Increase in neither NeuroD1 (Supplemental Figure S3C) nor subunit expression was observed in the HBEC3KT53, an HBEC cell line with constitutive knockdown of p53 (Supplemental Figure S3A). The inability of nicotine to cause a significant increase in NeuroD1 and AChR subunit expression was also observed in the NSCLC cell lines Clone 5 and H358 (both of which have oncogenic KRAS and have lost p53; Supplemental Figure S3B; see Table 1 for cell-line oncogenotype).
FIGURE 4:

NeuroD1 regulates the expression of α3 and α5, but not β4, nAChR subunits in SHSY5Y, Clone 5, SCLC, and NSCLC-NE lines. (A) NeuroD1 and α3, α5, and β4 subunits were immunoblotted in 25 μg of lysate protein from SCLC, NSCLC, HBEC3KT, Clone 5, and SHSY5Y cells as in Figure 1A. (B) SHSY5Y was treated with 0.1, 0.5, or 2.5 μM nicotine for 48 h. qRT-PCR of NeuroD1 and CHRNA3, A5, and B4 subunits; 18s RNA control. One of three independent experiments. *** p < 0.001, **p < 0.005. (C, D) Knockdown of NeuroD1 in H82, H1155, and Clone 5 lung and SHSY5Y neuroblastoma lines. H82 and H1155 cells were stably infected with shNeuroD1 or shControl vectors. Cells were selected by puromycin resistance and sorted for expression of green fluorescent protein. Then (C) NeuroD1 and CHRNA3, A5, A7, and B4 expression was analyzed by qRT-PCR and plotted relative to 18s RNA, or (D) lysate proteins were immunoblotted for NeuroD1 and α3, α5, and β4 with GAPDH as control.
Next we knocked down NeuroD1 in the cancer cells and examined expression of the nAChR subunits. In the neuroendocrine lung cancer cell lines, knockdown of NeuroD1 led to a significant decrease in both α3 and α5, but less so in β4 or α7, nAChR receptor subunits (Figure 4C). Similarly, knockdown of NeuroD1 in SHSY5Y and Clone 5 cells led to a significant decrease in both α3 and α5 but not β4 subunits (Figure 4D). This suggests that increased expression of the α3 and α5 in response to nicotine may be mediated by NeuroD1.
NeuroD1 regulates nicotine-induced invasion by SHSY5Y neuroblastoma and Clone 5 via regulation of the α3 and α5 subunits
NeuroD1 regulates survival and the metastatic capabilities of several neural/neuroendocrine carcinomas (Huang et al., 2011; Osborne et al., 2013b). Nicotine acts through various combinations of nAChR subunits to promote epithelial-to-mesenchymal transition and metastasis (Xu and Deng, 2004; Tournier et al., 2006; Guo et al., 2008; Dasgupta et al., 2009; Pillai et al., 2011). Thus we sought to investigate the possibility that NeuroD1 could modulate nicotine-induced invasion in our cell lines. Nicotine exposure was able to increase invasion of Clone 5 and SHSY5Y lines through Matrigel, whereas knockdown of NeuroD1 led to >50% reduction in nicotine-induced migration in both cell lines (Figure 5, A and B). Loss of either α3 or α5 expression reduced invasion with or without nicotine treatment. The α5 results were not anticipated, because this receptor subunit is thought of as an accessory or an orphan subunit that is unable to yield functional receptors when expressed alone and speculated to increase receptor sensitivity to ligand (Ramirez-Latorre et al., 1996; Gerzanich et al., 1998; Jackson et al., 2010). Taken together, these data suggest that nicotine acts though nAChR subunits to mediate cell invasion and migration by a NeuroD1-dependent mechanism.
FIGURE 5:
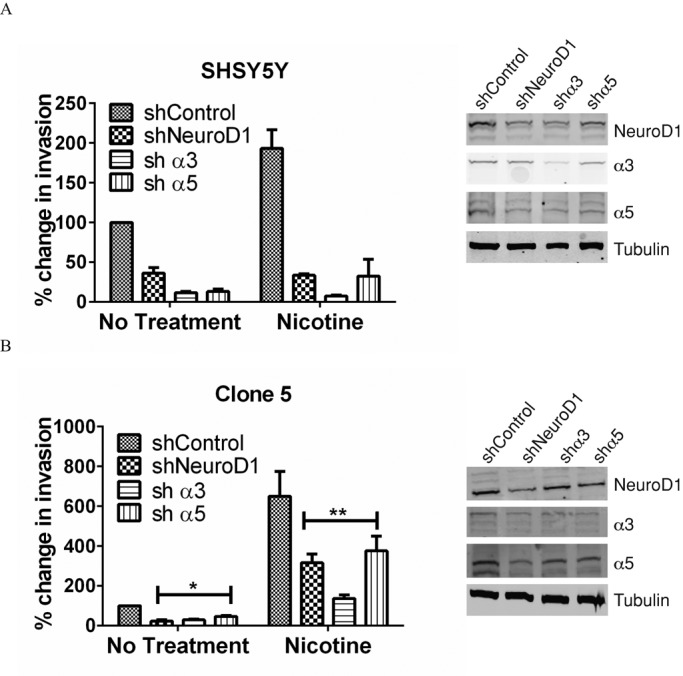
Acetylcholine receptor subunits α3 and α5 contribute to invasion induced by nicotine in SHSY5Y and clone 5 cells. (A) SHSY5Y and (B) clone 5 cells were infected with shNeuroD1, shα3, shα5, or shControl vectors to deplete the targeted proteins. Cells were dissociated, and 25,000 cells were placed in 0.2 ml of growth factor–reduced Matrigel. Invasion was scored as described in Materials and Methods. SHSY5Y, N = 2; Clone 5, N = 3. *p <0.05, **p <0.005. Right, proteins remaining 48 h after treatment with shRNAs, with tubulin as the loading control.
The NeuroD1 target TrkB regulates expression of several nAChR subunits in H69 and H82 SCLC
In addition to increased expression of NeuroD1, we observed that nicotine increased expression of TrkB in NTERA2 cells (Figure 3B). Nicotine via nAChR receptor subunit α7 was reported to stimulate proliferation in the SHSY5Y cell line via release of brain-derived neurotrophic factor (BDNF), the ligand for TrkB (Serres and Carney, 2006). On the basis of this idea, we asked whether phosphorylated TrkB bound the nAChR subunit complex. Immunoprecipitation experiments failed to detect interaction between TrkB and any of the complexes containing α5 (Supplemental Figure S4). We then examined whether there was a relationship between TrkB expression and expression of any other nAChR receptor subunits. Loss of TrkB led to decrease in expression of not only the cluster subunits of α3, α5, and β4, but also of α7, in three of the neuroendocrine lung cancer cell lines examined—H69, H82, and the neuroendocrine NSCLC H1155 (Figure 6A). Next we investigated whether TrkB activity was required for changes in AChR expression. The pan-Trk inhibitor lestaurtinib decreased expression of the nAChR cluster receptor subunits, with the most pronounced effects on α3, α5, and β2 subunits (Figure 6B). Nicotine combined with BDNF increased expression of the α3 and to some extent the α5 subunit (Figure 6C and Supplemental Figure S4A, lanes 1 and 4), consistent with decreased expression of both subunits with lestaurtinib (Figure 6C and Supplemental Figure S4A, lanes 1, 4, and 7). Taken together, these data suggest that nicotine may regulate expression of nAChR subunits by both NeuroD1-dependent and TrkB-dependent mechanisms.
FIGURE 6:

TrkB, a downstream target of NeuroD1, regulates expression of several nAChR subunits in H69 and H82 SCLC and H1155 NSCLC-NE lines. (A) H69, H82, and H1155 cells were stably infected with shTrkB or shControl vectors. Cells were selected by sorting as in Figure 5. qRT-PCR analysis of TrkB and CHRNA3, A5, A7, and B4, plotted as expression relative to the 18s RNA control. (B) H69 and H82 cells were treated with 10 μM lestaurtinib for 8 h, followed by qRT-PCR analysis of TrkB and CHRNA3, A5, A7, B4, and B2 plotted as described. (C) H82 cells were starved for 8 h in 0.1% FBS. Cells were treated with 1 μM nicotine and 50 ng/ml BDNF for 1 h with or without lestaurtinib. Left, cell lysates immunoblotted with indicated antibodies. Right, blots quantified using LI-COR infrared imaging.
NeuroD1 regulates changes in intracellular calcium in SHSY5Y neuroblastoma cells
The function of nAChR receptors upon ligand binding is to elevate cytoplasmic free calcium. Nicotine is believed to promote the influx of calcium predominantly through the α7 subunit, based on effects of a selective antagonist (Dajas-Bailador et al., 2002a). Calcium influx into cells has a plethora of roles exerting effects on many proteins, including those that enhance cell motility. We determined which of the cell lines displayed changes in intracellular calcium in response to nicotine. Nicotine induced acute changes in intracellular calcium only in SHSY5Y cells (Figure 7A). Segregation of the precise source of nicotine-induced changes in intracellular calcium has been complex. Prior work examining the contributions of α7, α3, and β2 subunits revealed that in addition to influx through the nAChR, voltage-gated calcium channels and intracellular stores mediate a large percentage of the nicotine-induced response (Dajas-Bailador et al., 2002b). To ascertain whether the changes we observed were dependent on an extracellular source of calcium, we treated cells with both the general nAChR antagonist mecamylamine and nifedipine, a blocker of L-type voltage-gated calcium channels. Both compounds prevented nicotine-induced changes in intracellular calcium to a similar extent, consistent with the idea that the majority of the response we observed was due to calcium influx (Figure 7B).
FIGURE 7:

Depletion of NeuroD1 decreases nicotine-induced increases in intracellular free calcium in SHSY5Y cells. (A) The indicated cell lines were loaded with fura-2AM and then stimulated with 12.5 μM nicotine. Fluorescence was monitored every 0.74 s after stimulation. (B) SHSY5Y cells loaded with fura-2AM were stimulated with 12.5 μM nicotine plus mecamylamine, nifedipine, or both at 10 μM, and fluorescence was monitored as described. N = 2. (C) SHSY5Y cells were infected with shNeuroD1, shα3, shα5, or shControl vectors for 48 h and then stimulated with 12.5 μM nicotine. Fluorescence was monitored as described. Representative of (A) four, (B) two, and (C) three independent experiments. Bottom, immunoblots showing effectiveness of depletion.
We next examined whether knockdown of NeuroD1, in addition to α3 or α5 subunits, would have an effect on the function of the receptors. Knockdown of α3 or α5 subunits prevented changes in intracellular calcium observed upon short-term exposure of cells to nicotine (Figure 7C). Knockdown of NeuroD1 also led to reduction in nicotine-induced intracellular calcium (Figure 7C). These data support the idea that NeuroD1 can regulate the function of the nAChR receptor subunits via regulation of their expression. These findings suggest the presence of feedback mechanisms that may occur early or throughout neuroendocrine tumorigenesis.
DISCUSSION
Nicotine induces cell survival and metastasis and also has been shown to inhibit differentiation (Sato et al., 2008). We find that up-regulation of NeuroD1 by nicotine occurs in some but not all cell types. In normal immortalized HBEC, exposure to nicotine causes an increase in NeuroD1; however, this was not the case in several nonneuroendocrine cells that lack p53 alone or in combination with KRAS mutations. This is consistent with previous accounts demonstrating that nicotine does not enhance tumorigenesis in KRAS-driven mouse models of lung cancer (Maier et al., 2011). Our findings also suggest that long-term exposure to nicotine does not induce differentiation but does induce NeuroD1 in neural/neuroendocrine models. Consistent with their roles in maintenance of progenitor states during differentiation of stem cells, we found that transcription factors such as ASCL1 and NGN3 decrease with exposure to retinoic acid but increase with nicotine (Cau et al., 1997; Gu et al., 2002; McNay et al., 2006; Xu et al., 2008; Magenheim et al., 2011). It is possible that loss of p53 as result of nicotine exposure could lead to increase in NeuroD1 expression during the development of lung cancer. Similarly, overexpression of p53 in embryonic stem cells has also been shown to promote differentiation and limit proliferation potential, consistent with suppression of NeuroD1 (Li et al., 2012; Menendez et al., 2012).
The ERK1/2-MAPK pathway regulates both transactivation activity and accumulation of NeuroD1 protein in a context-dependent manner. Nicotine activates both AKT and ERK1/2, most likely by calcium-dependent mechanisms (Dajas-Bailador et al., 2002b; West et al., 2003; Tsurutani et al., 2005; Chen et al., 2008; Pillai et al., 2011); however, we find that the increase in NeuroD1 expression as a consequence of nicotine exposure can be independent of both ERK1/2 and PI3K. Calcium influx stimulates other pathways that may control NeuroD1 expression; for example, in neural stem cells this is believed to occur indirectly through calcium, calmodulin-dependent protein kinase II (Schneider et al., 2008). Therefore other pathways need to be investigated that may ultimately be activated by nicotine-induced calcium influx leading directly or indirectly to NeuroD1 expression.
The use of multiple cell lines in this study attempts to direct our understanding of the mechanisms by which nicotine induces NeuroD1 during neuroendocrine carcinogenesis of the lung, as well as in other neuroendocrine tumor types. Although we recently published results indicating that NeuroD1 may enhance metastasis of neuroendocrine carcinomas via regulation of the proto-oncogene TrkB, the link connecting NeuroD1 to active TrkB was not conserved in all cell lines tested (Osborne et al., 2013a). We found that conservation of the regulation of metastatic capability by TrkB depended on the likely cell of origin (i.e., whether the cell expressed neuroendocrine characteristics). This relationship may also be relevant to the actions of nicotine on NeuroD1 in various tissues. Further studies on regulation of NeuroD1 and TrkB by nicotine in other tissues will be valuable to a more thorough examination of this possibility.
We demonstrate that NeuroD1, like ASCL1, regulates the nAChR subunit cluster, specifically α3 and α5, whereas ASCL1 has been suggested to regulate α3 and β4 (Improgo et al., 2010b). Preliminary data suggest direct binding of NeuroD1 to the promoter regions of the nAChR subunit cluster (unpublished data); further characterization of the exact mode of regulation is necessary. Based on assays of intracellular free calcium, regulation of α3 and α5 affects the function of the receptors as ion channels. Thus we hypothesize that both NeuroD1 and ASCL1 regulate expression of the nAChR subunit cluster of α3, α5, and β4, with different relative action on specific subunits. Considering our present and previous findings, we suggest that NeuroD1 lies in a positive feedback loop through which nicotine enhances the expression of its own receptors (Figure 8). Increased expression of NeuroD1 by nicotine presumably leads to increased expression of the receptor subunits; effects may be both dependent on and independent of TrkB activity. This combinatorial regulation may ultimately lead to changes in the levels of calcium and increased migration and invasion. The exact composition of the receptors and the ultimate consequence of regulation of each subunit remain to be defined.
FIGURE 8:
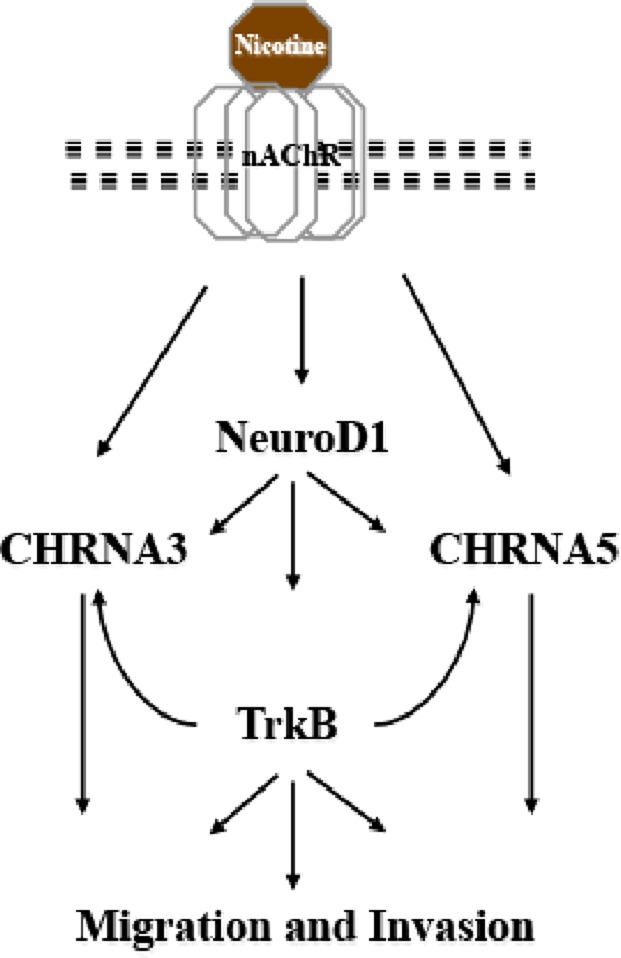
Model. Nicotine induces activation of NeuroD1 and the nAChR subunits. NeuroD1 then regulates its downstream targets, TrkB and the nAChR subunits, which control invasion and migration in cancer cells.
MATERIALS AND METHODS
Cell culture
NTERA2 clone D1 cells (American Type Culture Collection [ATCC, Manassa, VA]) were grown in DMEM with 10% fetal bovine serum (FBS). SCLC and NSCLC lines were from the Hamon Cancer Center Collection (UT Southwestern). SCLC and HBEC3KTRL53-Clone 5 and cell lines were cultured in RPMI 1640 with 10% FBS. SHSY5Y from ATCC were grown in DMEM/Ham's F12 (1:1) with 10% FBS. Immortalized HBECs (except HBEC3KTRL53-Clone 5; Sato et al., 2006) were cultured in Keratinocyte-SFM (Invitrogen, Grand Island, NY) with 5 ng/ml epidermal growth factor and 50 μg/ml bovine pituitary extract. The lung cancer cell lines were DNA fingerprinted using the PowerPlex 1.2 kit (Promega, Madison, WI) and confirmed to be the same as the DNA fingerprint library maintained by either ATCC or the Hamon Cancer Center. The lines were mycoplasma free, as confirmed by the e-Myco kit (Boca Scientific, Boca Raton, FL).
Antibodies and reagents
Immunoblot analyses were as previously described, using equal amounts of protein from each sample (Lawrence et al., 2005). The following antibodies were used for blotting and immunoprecipitation: goat NeuroD1 (N-19), rabbit pan–phospho-Trk (E-6), p53 (DO-1), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH; FL-335; Santa Cruz Biotechnology, Santa Cruz, CA); mouse ASCL1 (BD Biosciences, San Jose, CA); rabbit TrkB (Chemicon, Temecula, CA); mouse NGN3 (R&D Systems, Minneapolis, MN); rabbit nAChR subunits (Millipore, Billerica, MA); and mouse pERK1/2, mouse AKT, and rabbit pAKT-S473 (Cell Signaling [Danvers, MA], Sigma-Aldrich [St. Louis, MO], respectively). α-Tubulin hybridoma was purchased from the Developmental Studies Hybridoma Bank (University of Iowa, Iowa City, IA). Lestaurtinib was purchased from LC labs, Woburn, MA; BDNF from R&D Systems; nicotine, PD0325901, and mecamylamine from Sigma-Aldrich; and LY294002 was from Cell Signaling. Band intensities were quantified using a LI-COR Odyssey Infrared Imaging System.
Invasion assays
For invasion assays, 2.5 × 104 cells were embedded in growth factor–reduced Matrigel in the presence or absence of 1 μM nicotine 48 h after knockdown. Transwell migration was assayed in Transwell permeable supports (3422; Corning, Corning, NY). Cells were seeded in the top chamber in either RPMI with 1% FBS or DMEM/Ham's F12 with 1% FBS and allowed to migrate along a concentration gradient through a polycarbonate membrane with 8-μm pores toward the bottom chambers, which contained medium with 25% FBS. Cells were fixed, stained with methylene blue, and counted.
Transfection of short hairpin RNAs and plasmids
Virus was generated for infection of human pGIPZ lentiviral short hairpin RNA (shRNA) plasmids against (NeuroD1, CHRNA3/A5/B4) created by the RNAi Consortium. These were purchased by UT Southwestern as a library (TRC-Hs1.0 [Human]) from Open Biosystems (sequences are available online at the Open Biosystems website http://dharmacon.gelifesciences.com/rnai-and-custom-rna-synthesis/shrna/gipz-lentiviral-shrna/libraries/). NeuroD1 sh RNA, V2LHS_152218 (shRNA-1); TrkB shRNA, V2LHS_63731.
Calcium assays
Cells were washed twice with phosphate-buffered saline (PBS) and incubated with the fluorescent dye Fura-2AM diluted in keratinocyte serum-free medium with 2.5 mM calcium for 1 h. Cells were washed twice and equilibrated for 30 min. Nicotine (25 μM) was added using a Synergy microplate reader. Changes in intracellular calcium were assessed by dual excitation of Fura-2 at 340/11 and 380/20 nm and emission at 508/20 nm using Gen5 software.
Immunofluorescence and nicotine/differentiation protocol
Cells were treated with nicotine (Sigma-Aldrich; dose response, time course, or long term) or retinoic acid (Invitrogen; long term), washed with PBS fixed with 4% paraformaldehyde (vol/vol) in PBS for 10 min, and washed with PBS. Cells were permeabilized with 0.1% Triton X-100 and washed. After incubation with 10% donkey serum/bovine serum albumin at room temperature for 1 h, cells were incubated with the indicated antibodies at 4°C overnight. Cells were washed with PBS, incubated with Alexa Fluor–conjugated secondary antibody at room temperature for 1 h, washed with PBS, and imaged. Fluorescent Z-stacks (0.2 mm) were acquired and deconvolved using a DeltaVision RT deconvolution microscope.
Colony formation assay
Soft agar colony assays were as described previously (Sato et al., 2006). As indicated, PD0325901 was added once per day after seeding and solidification of agar.
Quantitative real-time PCR
Total RNA from cell lines was isolated with TRI Reagent. cDNA was synthesized using iSCRIPT cDNA Synthesis Kit (Bio-Rad). RNA were quantified by real-time PCR (RT-PCR) with iTaq (Bio-Rad) master mix using TaqMan probes for 18s rRNA, NeuroD1, and TrkB (Applied Biosystems) on an ABI 7500 thermocycler. Relative transcript levels were normalized to 18s rRNA. Transcript amounts in knockdown cells were plotted as fold change relative to control. Data were analyzed using ABI 7500 system software.
Quantitative RT-PCR (qRT-PCR) primers were as follows: α3, reverse, TTGCAGAAACAATCCTGCTG, and forward, ATGCTGTGCTGTCCCTCTCT; α5, forward, CCAAACTGCTTTGCATGAGA, and reverse, TCCACAGAAACATCCGATCA; α7, forward, CCCAAGTGGACCAGAGTCAT, and reverse, GCCACACACTACCCCAGAGT; β4, forward TCCCTGGTCCTTTTCTTCCT, and reverse, TGCAGCTTGATGGAGATGAG; and β2, forward, GGCATGTACGAGGTGTCCTT, and reverse, CACCTCACTCTTCAGCACCA.
Supplementary Material
Acknowledgments
We thank current and former members of the Cobb and Minna laboratories, including A-Young Lee, Jill Larsen, Michael Kalwat, Aroon Karra, and Aileen Klein, for comments on the manuscript and Dionne Ware for administrative assistance. We thank the Huber lab for the NeuN antibody and the Albanesi lab for use of the Synergy microplate reader. This work was supported by grants from the National Institutes of Health (R01DK55310 to M.H.C. and P50CA70907 to J.D.M.), grants from the Cancer Prevention and Research Institute of Texas to M.H.C. and J.D.M., and support from Department of Defense PROSPECT and the Longenbaugh Foundation to J.D.M. J.K.O. was supported by National Institute of General Medical Sciences Pharmacological Sciences Training Grant 5-T32 GM007062. The content of this article is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Abbreviations used:
- ERK2
extracellular signal-regulated kinase 2
- MAPK
mitogen-activated protein kinase
- nAchR
nicotinic acetylcholin receptors
- NeuroD1
neuronal differentiation 1
- NSCLC
non–small cell lung cancer
- SCLC
small cell lung cancer
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E13-06-0316) on April 9, 2014.
REFERENCES
- Albuquerque EX, Pereira EF, Alkondon M, Rogers SW. Mammalian nicotinic acetylcholine receptors: from structure to function. Physiol Rev. 2009;89:73–120. doi: 10.1152/physrev.00015.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alessi DR, Smythe C, Keyse SM. The human CL100 gene encodes a Tyr/Thr-protein phosphatase which potently and specifically inactivates MAP kinase and suppresses its activation by oncogenic ras in Xenopus oocyte extracts. Oncogene. 1993;8:2015–2020. [PubMed] [Google Scholar]
- Amos CI, et al. Genome-wide association scan of tag SNPs identifies a susceptibility locus for lung cancer at 15q25.1. Nat Genet. 2008;40:616–622. doi: 10.1038/ng.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews PW. Retinoic acid induces neuronal differentiation of a cloned human embryonal carcinoma cell line in vitro. Dev Biol. 1984;103:285–293. doi: 10.1016/0012-1606(84)90316-6. [DOI] [PubMed] [Google Scholar]
- Ball DW, Azzoli CG, Baylin SB, Chi D, Dou S, Donis-Keller H, Cumaraswamy A, Borges M, Nelkin BD. Identification of a human achaete-scute homolog highly expressed in neuroendocrine tumors. Proc Natl Acad Sci USA. 1993;90:5648–5652. doi: 10.1073/pnas.90.12.5648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-David U, Benvenisty N. The tumorigenicity of human embryonic and induced pluripotent stem cells. Nat Rev. Cancer. 2011;11:268–277. doi: 10.1038/nrc3034. [DOI] [PubMed] [Google Scholar]
- Ben-David U, Mayshar Y, Benvenisty N. Large-scale analysis reveals acquisition of lineage-specific chromosomal aberrations in human adult stem cells. Cell Stem Cell. 2011;9:97–102. doi: 10.1016/j.stem.2011.06.013. [DOI] [PubMed] [Google Scholar]
- Carney DN, Bepler G, Gazdar AF. The serum-free establishment and in vitro growth properties of classic and variant small cell lung cancer cell lines. Recent Results Cancer Research. 1985a;99:157–166. doi: 10.1007/978-3-642-82533-0_16. [DOI] [PubMed] [Google Scholar]
- Carney DN, Gazdar AF, Bepler G, Guccion JG, Marangos PJ, Moody TW, Zweig MH, Minna JD. Establishment and identification of small cell lung cancer cell lines having classic and variant features. Cancer Res. 1985b;45:2913–2923. [PubMed] [Google Scholar]
- Cau E, Gradwohl G, Fode C, Guillemot F. Mash1 activates a cascade of bHLH regulators in olfactory neuron progenitors. Development. 1997;124:1611–1621. doi: 10.1242/dev.124.8.1611. [DOI] [PubMed] [Google Scholar]
- Chen RJ, Ho YS, Guo HR, Wang YJ. Rapid activation of Stat3 and ERK1/2 by nicotine modulates cell proliferation in human bladder cancer cells. Toxicol Sci. 2008;104:283–293. doi: 10.1093/toxsci/kfn086. [DOI] [PubMed] [Google Scholar]
- Dajas-Bailador FA, Mogg AJ, Wonnacott S. Intracellular Ca2+ signals evoked by stimulation of nicotinic acetylcholine receptors in SH-SY5Y cells: contribution of voltage-operated Ca2+ channels and Ca2+ stores. J Neurochem. 2002a;81:606–614. doi: 10.1046/j.1471-4159.2002.00846.x. [DOI] [PubMed] [Google Scholar]
- Dajas-Bailador FA, Soliakov L, Wonnacott S. Nicotine activates the extracellular signal-regulated kinase 1/2 via the alpha7 nicotinic acetylcholine receptor and protein kinase A, in SH-SY5Y cells and hippocampal neurones. J Neurochem. 2002b;80:520–530. doi: 10.1046/j.0022-3042.2001.00725.x. [DOI] [PubMed] [Google Scholar]
- Dasgupta P, et al. Nicotine induces cell proliferation, invasion and epithelial-mesenchymal transition in a variety of human cancer cell lines. Int. J Cancer. 2009;124:36–45. doi: 10.1002/ijc.23894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasgupta P, Chellappan SP. Nicotine-mediated cell proliferation and angiogenesis: new twists to an old story. Cell Cycle. 2006;5:2324–2328. doi: 10.4161/cc.5.20.3366. [DOI] [PubMed] [Google Scholar]
- Dasgupta P, Rastogi S, Pillai S, Ordonez-Ercan D, Morris M, Haura E, Chellappan S. Nicotine induces cell proliferation by beta-arrestin-mediated activation of Src and Rb-Raf-1 pathways. J Clin Invest. 2006;116:2208–2217. doi: 10.1172/JCI28164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dufton C, Marcora E, Chae JH, McCullough J, Eby J, Hausburg M, Stein GH, Khoo S, Cobb MH, Lee JE. Context-dependent regulation of NeuroD activity and protein accumulation. Mol Cell Neurosci. 2005;28:727–736. doi: 10.1016/j.mcn.2004.12.004. [DOI] [PubMed] [Google Scholar]
- Fratticci A, et al. Differential expression of neurogenins and NeuroD1 in human pituitary tumours. J Endocrinol. 2007;194:475–484. doi: 10.1677/JOE-07-0020. [DOI] [PubMed] [Google Scholar]
- Gazdar AF, Carney DN, Nau MM, Minna JD. Characterization of variant subclasses of cell lines derived from small cell lung cancer having distinctive biochemical, morphological, and growth properties. Cancer Res. 1985;45:2924–2930. [PubMed] [Google Scholar]
- Gerzanich V, Wang F, Kuryatov A, Lindstrom J. alpha 5 Subunit alters desensitization, pharmacology, Ca++ permeability and Ca++ modulation of human neuronal alpha 3 nicotinic receptors. J Pharmacol Exp Ther. 1998;286:311–320. [PubMed] [Google Scholar]
- Govindan R, et al. Genomic landscape of non-small cell lung cancer in smokers and never-smokers. Cell. 2012;150:1121–1134. doi: 10.1016/j.cell.2012.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu G, Dubauskaite J, Melton DA. Direct evidence for the pancreatic lineage: NGN3+ cells are islet progenitors and are distinct from duct progenitors. Development. 2002;129:2447–2457. doi: 10.1242/dev.129.10.2447. [DOI] [PubMed] [Google Scholar]
- Guo J, Ibaragi S, Zhu T, Luo LY, Hu GF, Huppi PS, Chen CY. Nicotine promotes mammary tumor migration via a signaling cascade involving protein kinase C and CDC42. Cancer Res. 2008;68:8473–8481. doi: 10.1158/0008-5472.CAN-08-0131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hecht SS. Tobacco smoke carcinogens and lung cancer. J Natl Cancer Inst. 1999;91:1194–1210. doi: 10.1093/jnci/91.14.1194. [DOI] [PubMed] [Google Scholar]
- Huang HP, Liu M, El-Hodiri HM, Chu K, Jamrich M, Tsai MJ. Regulation of the pancreatic islet-specific gene BETA2 (neuroD) by neurogenin 3. Mol Cell Biol. 2000;20:3292–3307. doi: 10.1128/mcb.20.9.3292-3307.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang P, Kishida S, Cao D, Murakami-Tonami Y, Mu P, Nakaguro M, Koide N, Takeuchi I, Onishi A, Kadomatsu K. The neuronal differentiation factor NeuroD1 downregulates the neuronal repellent factor Slit2 expression and promotes cell motility and tumor formation of neuroblastoma. Cancer Res. 2011;71:2938–2948. doi: 10.1158/0008-5472.CAN-10-3524. [DOI] [PubMed] [Google Scholar]
- Hukkanen J, Jacob P, 3rd, Benowitz NL. Metabolism and disposition kinetics of nicotine. Pharmacol Rev. 2005;57:79–115. doi: 10.1124/pr.57.1.3. [DOI] [PubMed] [Google Scholar]
- Hu Y, Wang T, Stormo GD, Gordon JI. RNA interference of achaete-scute homolog 1 in mouse prostate neuroendocrine cells reveals its gene targets and DNA binding sites. Proc Natl Acad Sci USA. 2004;101:5559–5564. doi: 10.1073/pnas.0306988101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung RJ, et al. A susceptibility locus for lung cancer maps to nicotinic acetylcholine receptor subunit genes on 15q25. Nature. 2008;452:633–637. doi: 10.1038/nature06885. [DOI] [PubMed] [Google Scholar]
- Improgo MR, Schlichting NA, Cortes RY, Zhao-Shea R, Tapper AR, Gardner PD. ASCL1 regulates the expression of the CHRNA5/A3/B4 lung cancer susceptibility locus. Mol Cancer Res. 2010a;8:194–203. doi: 10.1158/1541-7786.MCR-09-0185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Improgo MR, Scofield MD, Tapper AR, Gardner PD. The nicotinic acetylcholine receptor CHRNA5/A3/B4 gene cluster: dual role in nicotine addiction and lung cancer. Prog Neurobiol. 2010b;92:212–226. doi: 10.1016/j.pneurobio.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito T, Udaka N, Yazawa T, Okudela K, Hayashi H, Sudo T, Guillemot F, Kageyama R, Kitamura H. Basic helix-loop-helix transcription factors regulate the neuroendocrine differentiation of fetal mouse pulmonary epithelium. Development. 2000;127:3913–3921. doi: 10.1242/dev.127.18.3913. [DOI] [PubMed] [Google Scholar]
- Jackman DM, Johnson BE. Small-cell lung cancer. Lancet. 2005;366:1385–1396. doi: 10.1016/S0140-6736(05)67569-1. [DOI] [PubMed] [Google Scholar]
- Jackson KJ, Marks MJ, Vann RE, Chen X, Gamage TF, Warner JA, Damaj MI. Role of alpha5 nicotinic acetylcholine receptors in pharmacological and behavioral effects of nicotine in mice. J Pharmacol Exp Ther. 2010;334:137–146. doi: 10.1124/jpet.110.165738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang T, Collins BJ, Jin N, Watkins DN, Brock MV, Matsui W, Nelkin BD, Ball DW. Achaete-scute complex homologue 1 regulates tumor-initiating capacity in human small cell lung cancer. Cancer Res. 2009;69:845–854. doi: 10.1158/0008-5472.CAN-08-2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Z, Gao F, Flagg T, Deng X. Tobacco-specific nitrosamine 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone promotes functional cooperation of Bcl2 and c-Myc through phosphorylation in regulating cell survival and proliferation. J Biol Chem. 2004;279:40209–40219. doi: 10.1074/jbc.M404056200. [DOI] [PubMed] [Google Scholar]
- Kalamida D, Poulas K, Avramopoulou V, Fostieri E, Lagoumintzis G, Lazaridis K, Sideri A, Zouridakis M, Tzartos SJ. Muscle and neuronal nicotinic acetylcholine receptors. Structure, function and pathogenicity. FEBS J. 2007;274:3799–3845. doi: 10.1111/j.1742-4658.2007.05935.x. [DOI] [PubMed] [Google Scholar]
- Khoo S, Griffen SC, Xia Y, Baer RJ, German MS, Cobb MH. Regulation of insulin gene transcription by ERK1 and ERK2 in pancreatic beta cells. J Biol Chem. 2003;278:32969–32977. doi: 10.1074/jbc.M301198200. [DOI] [PubMed] [Google Scholar]
- Lam DC, et al. Expression of nicotinic acetylcholine receptor subunit genes in non-small-cell lung cancer reveals differences between smokers and nonsmokers. Cancer Res. 2007;67:4638–4647. doi: 10.1158/0008-5472.CAN-06-4628. [DOI] [PubMed] [Google Scholar]
- Lawrence MC, McGlynn K, Park BH, Cobb MH. ERK1/2-dependent activation of transcription factors required for acute and chronic effects of glucose on the insulin gene promoter. J Biol Chem. 2005;280:26751–26759. doi: 10.1074/jbc.M503158200. [DOI] [PubMed] [Google Scholar]
- Lee JE, Hollenberg SM, Snider L, Turner DL, Lipnick N, Weintraub H. Conversion of Xenopus ectoderm into neurons by NeuroD, a basic helix-loop-helix protein. Science. 1995;268:836–844. doi: 10.1126/science.7754368. [DOI] [PubMed] [Google Scholar]
- Li M, He Y, Dubois W, Wu X, Shi J, Huang J. Distinct regulatory mechanisms and functions for p53-activated and p53-repressed DNA damage response genes in embryonic stem cells. Mol Cell. 2012;46:30–42. doi: 10.1016/j.molcel.2012.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linnoila RI. Functional facets of the pulmonary neuroendocrine system. Lab Invest. 2006;86:425–444. doi: 10.1038/labinvest.3700412. [DOI] [PubMed] [Google Scholar]
- Lopez-Carballo G, Moreno L, Masia S, Perez P, Barettino D. Activation of the phosphatidylinositol 3-kinase/Akt signaling pathway by retinoic acid is required for neural differentiation of SH-SY5Y human neuroblastoma cells. J Biol Chem. 2002;277:25297–25304. doi: 10.1074/jbc.M201869200. [DOI] [PubMed] [Google Scholar]
- Magenheim J, Klein AM, Stanger BZ, Ashery-Padan R, Sosa-Pineda B, Gu G, Dor Y. Ngn3(+) endocrine progenitor cells control the fate and morphogenesis of pancreatic ductal epithelium. Dev Biol. 2011;359:26–36. doi: 10.1016/j.ydbio.2011.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier CR, Hollander MC, Hobbs EA, Dogan I, Linnoila RI, Dennis PA. Nicotine does not enhance tumorigenesis in mutant K-ras-driven mouse models of lung cancer. Cancer Prev Res (Phila) 2011;4:1743–1751. doi: 10.1158/1940-6207.CAPR-11-0365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mavilio F. Regulation of vertebrate homeobox-containing genes by morphogens. Eur J Biochem. 1993;212:273–288. doi: 10.1111/j.1432-1033.1993.tb17660.x. [DOI] [PubMed] [Google Scholar]
- McNay DE, Pelling M, Claxton S, Guillemot F, Ang SL. Mash1 is required for generic and subtype differentiation of hypothalamic neuroendocrine cells. Mol Endocrinol. 2006;20:1623–1632. doi: 10.1210/me.2005-0518. [DOI] [PubMed] [Google Scholar]
- Menendez S, et al. Increased dosage of tumor suppressors limits the tumorigenicity of iPS cells without affecting their pluripotency. Aging Cell. 2012;11:41–50. doi: 10.1111/j.1474-9726.2011.00754.x. [DOI] [PubMed] [Google Scholar]
- Mitsudomi T, Viallet J, Mulshine JL, Linnoila RI, Minna JD, Gazdar AF. Mutations of ras genes distinguish a subset of non-small-cell lung cancer cell lines from small-cell lung cancer cell lines. Oncogene. 1991;6:1353–1362. [PubMed] [Google Scholar]
- Neptune ER, Podowski M, Calvi C, Cho JH, Garcia JG, Tuder R, Linnoila RI, Tsai MJ, Dietz HC. Targeted disruption of NeuroD, a proneural basic helix-loop-helix factor, impairs distal lung formation and neuroendocrine morphology in the neonatal lung. J Biol Chem. 2008;283:21160–21169. doi: 10.1074/jbc.M708692200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborne JK, et al. NeuroD1 regulates survival and migration of neuroendocrine lung carcinomas via signaling molecules TrkB and NCAM. Proc Natl Acad Sci USA. 2013b;110:6524–6529. doi: 10.1073/pnas.1303932110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborne JK, Larsen JE, Gonzales JX, Shames DS, Sato M, Wistuba II, Girard L, Minna JD, Cobb MH. NeuroD1 regulation of migration accompanies the differential sensitivity of neuroendocrine carcinomas to TrkB inhibition. Oncogenesis. 2013a;2:e63. doi: 10.1038/oncsis.2013.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeifer GP, Denissenko MF, Olivier M, Tretyakova N, Hecht SS, Hainaut P. Tobacco smoke carcinogens, DNA damage and p53 mutations in smoking-associated cancers. Oncogene. 2002;21:7435–7451. doi: 10.1038/sj.onc.1205803. [DOI] [PubMed] [Google Scholar]
- Pillai S, Rizwani W, Li X, Rawal B, Nair S, Schell MJ, Bepler G, Haura E, Coppola D, Chellappan S. ID1 facilitates the growth and metastasis of non-small cell lung cancer in response to nicotinic acetylcholine receptor and epidermal growth factor receptor signaling. Mol Cell Biol. 2011;31:3052–3067. doi: 10.1128/MCB.01311-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pleasance ED, et al. A small-cell lung cancer genome with complex signatures of tobacco exposure. Nature. 2010;463:184–190. doi: 10.1038/nature08629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Przyborski SA, Morton IE, Wood A, Andrews PW. Developmental regulation of neurogenesis in the pluripotent human embryonal carcinoma cell line NTERA-2. Eur J Neurosci. 2000;12:3521–3528. doi: 10.1046/j.1460-9568.2000.00230.x. [DOI] [PubMed] [Google Scholar]
- Puliyappadamba VT, Cheriyan VT, Thulasidasan AK, Bava SV, Vinod BS, Prabhu PR, Varghese R, Bevin A, Venugopal S, Anto RJ. Nicotine-induced survival signaling in lung cancer cells is dependent on their p53 status while its down-regulation by curcumin is independent. Mol Cancer. 2010;9:220. doi: 10.1186/1476-4598-9-220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quan AL, Videtic GM, Suh JH. Brain metastases in small cell lung cancer. Oncology. 2004;18:961–972. discussion, 974, 979–980, 987. [PubMed] [Google Scholar]
- Ramirez-Latorre J, Yu CR, Qu X, Perin F, Karlin A, Role L. Functional contributions of alpha5 subunit to neuronal acetylcholine receptor channels. Nature. 1996;380:347–351. doi: 10.1038/380347a0. [DOI] [PubMed] [Google Scholar]
- Ravi RK, et al. Activated Raf-1 causes growth arrest in human small cell lung cancer cells. J Clin Invest. 1998;101:153–159. doi: 10.1172/JCI831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rostomily RC, Bermingham-McDonogh O, Berger MS, Tapscott SJ, Reh TA, Olson JM. Expression of neurogenic basic helix-loop-helix genes in primitive neuroectodermal tumors. Cancer Res. 1997;57:3526–3531. [PubMed] [Google Scholar]
- Russell MA, Jarvis M, Iyer R, Feyerabend C. Relation of nicotine yield of cigarettes to blood nicotine concentrations in smokers. Br Med J. 1980;280:972–976. doi: 10.1136/bmj.280.6219.972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato M, et al. Multiple oncogenic changes (K-RAS(V12), p53 knockdown, mutant EGFRs, p16 bypass, telomerase) are not sufficient to confer a full malignant phenotype on human bronchial epithelial cells. Cancer Res. 2006;66:2116–2128. doi: 10.1158/0008-5472.CAN-05-2521. [DOI] [PubMed] [Google Scholar]
- Sato M, et al. Human lung epithelial cells progressed to malignancy through specific oncogenic manipulations. Mol Cancer Res. 2013;11:638–650. doi: 10.1158/1541-7786.MCR-12-0634-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato T, Abe T, Nakamoto N, Tomaru Y, Koshikiya N, Nojima J, Kokabu S, Sakata Y, Kobayashi A, Yoda T. Nicotine induces cell proliferation in association with cyclin D1 up-regulation and inhibits cell differentiation in association with p53 regulation in a murine pre-osteoblastic cell line. Biochem Biophys Res Commun. 2008;377:126–130. doi: 10.1016/j.bbrc.2008.09.114. [DOI] [PubMed] [Google Scholar]
- Schneider JW, Gao Z, Li S, Farooqi M, Tang TS, Bezprozvanny I, Frantz DE, Hsieh J. Small-molecule activation of neuronal cell fate. Nat Chem Biol. 2008;4:408–410. doi: 10.1038/nchembio.95. [DOI] [PubMed] [Google Scholar]
- Serres F, Carney SL. Nicotine regulates SH-SY5Y neuroblastoma cell proliferation through the release of brain-derived neurotrophic factor. Brain Res. 2006;1101:36–42. doi: 10.1016/j.brainres.2006.05.023. [DOI] [PubMed] [Google Scholar]
- Sun S, Schiller JH, Spinola M, Minna JD. New molecularly targeted therapies for lung cancer. J Clin Invest. 2007;117:2740–2750. doi: 10.1172/JCI31809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syder AJ, Karam SM, Mills JC, Ippolito JE, Ansari HR, Farook V, Gordon JI. A transgenic mouse model of metastatic carcinoma involving transdifferentiation of a gastric epithelial lineage progenitor to a neuroendocrine phenotype. Proc Natl Acad Sci USA. 2004;101:4471–4476. doi: 10.1073/pnas.0307983101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tournier JM, et al. alpha3alpha5beta2-Nicotinic acetylcholine receptor contributes to the wound repair of the respiratory epithelium by modulating intracellular calcium in migrating cells. Am J Pathol. 2006;168:55–68. doi: 10.2353/ajpath.2006.050333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsurutani J, Castillo SS, Brognard J, Granville CA, Zhang C, Gills JJ, Sayyah J, Dennis PA. Tobacco components stimulate Akt-dependent proliferation and NFkappaB-dependent survival in lung cancer cells. Carcinogenesis. 2005;26:1182–1195. doi: 10.1093/carcin/bgi072. [DOI] [PubMed] [Google Scholar]
- van Meerbeeck JP, Fennell DA, De Ruysscher DK. Small-cell lung cancer. Lancet. 2011;378:1741–1755. doi: 10.1016/S0140-6736(11)60165-7. [DOI] [PubMed] [Google Scholar]
- West KA, Brognard J, Clark AS, Linnoila IR, Yang X, Swain SM, Harris C, Belinsky S, Dennis PA. Rapid Akt activation by nicotine and a tobacco carcinogen modulates the phenotype of normal human airway epithelial cells. J Clin Invest. 2003;111:81–90. doi: 10.1172/JCI16147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong DJ, Liu H, Ridky TW, Cassarino D, Segal E, Chang HY. Module map of stem cell genes guides creation of epithelial cancer stem cells. Cell Stem Cell. 2008;2:333–344. doi: 10.1016/j.stem.2008.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Deng X. Tobacco-specific nitrosamine 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone induces phosphorylation of mu- and m-calpain in association with increased secretion, cell migration, and invasion. J Biol Chem. 2004;279:53683–53690. doi: 10.1074/jbc.M409889200. [DOI] [PubMed] [Google Scholar]
- Xu X, et al. Beta cells can be generated from endogenous progenitors in injured adult mouse pancreas. Cell. 2008;132:197–207. doi: 10.1016/j.cell.2007.12.015. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.